Kvantitatív proteomikai módszerek fejlesztése és alkalmazása komplex biológiai kérdések tanulmányozására és potenciális
biomarkerek azonosítására
MTA Doktori Értekezés
Csősz Éva
Biokémiai és Molekuláris Biológiai Intézet Általános Orvostudományi Kar
Debreceni Egyetem
2020
TARTALOMJEGYZÉK 1
1. BEVEZETÉS 4
2. IRODALMI ÁTTEKINTÉS 6
2.1. Gél alapú proteomikai módszerek 6
2.1.1. Kétdimenziós elektroforézis 7
2.1.2. Differenciál gélelektroforézis 10
2.2. Tömegspektrometrián alapuló proteomikai módszerek 11 2.2.1. A tömegspektrométer felépítése és működése 11 2.2.1.1. Mátrix által segített lézer deszorpció ionizáció (MALDI) 12
2.2.1.2. Az elektrospray ionizáció (ESI) 13
2.2.2. Kromatográfiával kapcsolt tömegspektrometria 16
2.2.3. Fehérjék azonosítása LC-MS segítségével 17
2.2.4. Adatgyűjtési módok 19
2.2.5. Fehérjék kvantitálása 21
2.2.6. Adatgondozás, adatelemzés 22
2.3. A proteomikai módszerek alkalmazási területei 23
2.3.1. Fehérjék szerepének vizsgálata 24
2.3.2. Betegségek patomechanizmusának vizsgálata 25
2.3.3. Biomarkerek azonosítása 26
2.3.3.1. A kémiai barrier, mint potenciális biomarker forrás 27
3. CÉLKITŰZÉSEK 30
4. ANYAGOK ÉS MÓDSZEREK 31
4.1. Vegyszerek 31
4.2. Mintagyűjtés 31
4.2.1. Könnyminták 31
4.2.2. Csarnokvíz minták 31
4.2.3. Verejtékminták 32
4.2.4. Nyálminták 32
4.2.5. Sejtkultúra minták 32
4.2.6. Candida albicans sejtlizátumok 32
4.2.7. Üvegtest minták 33
4.2.8. HIV-1 transzdukált sejtlizátumok 33
4.3. MRM/SRM módszertervezés 34
4.4. Fehérjék emésztése oldatban 34
4.5. Fehérjék emésztése gélben 35
4.6. Tömegspektrometriás analízis 35
4.7. Kétdimenziós elektroforézis 36
4.7.1. Mintaelőkészítés 36
4.7.2. Izoelektromos fókuszálás 37
4.7.3. Ekvilibrálás 37
4.7.4. SDS-poliakrilamid gélelektroforézis 37
4.7.5. Gélek festése 38
4.7.6. Géldokumentáció és gélkép analízis 39
4.8. SDS-poliakrilamid gélelektroforézis 39
4.9. Immunológiai módszerekkel történő vizsgálatok 40
4.10. Statisztikai analízis 40
4.11. Hálózatok tanulmányozása 41
4.11.1. Súlyozott hálózatok létrehozása 41
4.11.2. A súlyozott hálózatok hálózati paramétereinek vizsgálata 43
5. EREDMÉNYEK ÉS MEGBESZÉLÉSÜK 45
5.1. A Candida albicans PPZ1 szerepének vizsgálata kétdimenziós elektroforézis
segítségével 45
5.2. Proliferatív vitreoretinopátia tanulmányozása kétdimenziós elektroforézis
segítségével 59
5.3. A könnyben, nyálban, verejtékben levő kémiai barrier vizsgálata és potenciális
biomarkerek azonosítása 72
5.3.1. Humán béta defenzinek vizsgálatára alkalmas MRM/SRM-alapú
célzott tömegspektrometriás módszer kifejlesztése 72 5.3.2. A könny, mint potenciális biomarker forrás 78 5.3.3. A könny kémiai barrierjének vizsgálata és Alzheimer kór-specifikus
potenciális könny biomarkerek azonosítása 78
5.3.4. A szemben zajló sebgyógyulási folyamatok tanulmányozása, és olyan potenciális biomarkerek azonosítása, amelyek előrejelezhetik a trabekulektómia
utáni komplikációk kialakulását 84
5.3.4.1. Citokinek mennyiségének vizsgálata könnyben 87 5.3.4.2. Citokinek mennyiségének vizsgálata csarnokvízben 91
5.3.4.3. Könny és csarnokvíz citokin tartalmának összehasonlítása 92 5.3.4.4. Könny fehérje profil változások tanulmányozása trabekulektómia
után bekövetkező sebgyógyulás során 94
5.3.5. A nyál kémiai barrierjének vizsgálata és szájüregi laphámrák-
specifikus potenciális biomarkerek azonosítása 108
5.3.5.1. Az OSCC-re jellemző nyálfehérje profil vizsgálata 110 5.3.5.2. A magyarországi populációban diagnosztikus jelentőségű
potenciális nyálbiomarkerek azonosítása 121
5.3.6. A normál verejték tömegspektrometriás analízise, és a nagy mennyiségben
levő verejtékfehérjék azonosítása 129
5.4. Komplex, rendszerszintű hálózatelemzési módszerek kidolgozása és alkalmazása a minőségi és mennyiségi proteomikai adatok együttes elemzésére 134
5.4.1. Fehérje-fehérje interakciós hálózatok analízise 138
6. ÚJ MEGÁLLAPÍTÁSOK 147
7. IRODALOMJEGYZÉK 149
8. KÖSZÖNETNYILVÁNÍTÁS 190
AZ ÉRTEKEZÉS ALAPJÁT KÉPEZŐ PUBLIKÁCIÓK 192
FÜGGELÉK 194
1. BEVEZETÉS
A biológiai rendszerek bonyolult működésének a megismerése régóta foglalkoztatja a gondolkodó embert. Az évszázadok során különféle módszereket alkalmaztak és alkalmaznak, hogy információt gyűjtsenek, az információt felhasználva modellt alkossanak, amelyet aztán az újabb információk birtokában megerősíthetnek vagy elvethetnek. Az információgyűjtés és modellalkotás elsődleges célja, hogy jobban megértsük az élővilág egyszerű építőkövekből felépülő igen komplex működését.
Régóta kérdés az is, hogy mit vizsgáljunk ahhoz, hogy egy vizsgálattal minél több biológiailag releváns ismeretet nyerhessünk. A géneket vizsgálva információt kaphatunk a genomról, epigenetikai módosításokról, a transzkriptómot vizsgálva megismerhetjük az adott időpillanatban bekapcsolt géneket, viszont ezek még csak közelítő információt adnak a sejtek, biológiai rendszerek tényleges működéséről. Amennyiben a kivitelezőket, a fehérjéket, vizsgáljuk, tisztább képet kaphatunk, de a tényleges működést csak akkor érthetjük meg, ha a termékeket, vagyis a metabolómot is megvizsgáljuk. Ideális esetben a teljes omikai palettát lefedhetjük vizsgálatokkal: tanulmányozhatjuk a genomot és/vagy transzkriptómot, a proteómot és a metabolómot, ez esetben viszont a komplex adatok értelmezése jelenthet problémát. Igen jelentős próbálkozások vannak annak érdekében, hogy megfelelő rendszerbiológiai modelleket dolgozzanak ki, amelyek segítségével egy rendszeren belül értelmezhetők a különböző típusú omikai adatok. Sajnos mindezidáig nem született egységes modell, amely egyben tudná kezelni a különböző omikai eredményeket [1].
Ugyanakkor az is kérdés, hogy milyen módszerrel történjen a vizsgálat. A régóta jól bevált hipotézis-centrikus megközelítéseket alkalmazva egy jól körülhatárolt kérdést próbálunk körüljárni sok esetben úgy, hogy egy változást indukálunk a biológiai rendszerben és az arra adott választ kíséreljük meg minél pontosabban karakterizálni. Tekintve, hogy ebben az esetben egy beavatkozást végzünk a rendszerben, nem tudjuk biztosítani, hogy a megfigyeléssel begyűjtött adatok tényleg a normális biológiai állapotot és nem csak a mesterségesen megzavart rendszer válaszait tükrözik. Ha célunk nem egy előzetesen felállított hipotézis megerősítése vagy megcáfolása, lehetőségünk van a rendszer egészének megfigyelésére, a teljes élőlényre/rendszerre vonatkozó adatok gyűjtésére. Az omikai vizsgálatok általában ezt az ún. hipotézis nélküli megközelítést alkalmazzák, és a vizsgálat alatt begyűjtött több ezer adatpontból származó információ alapján utólag, az adatok feldolgozása révén próbálnak egy modellt létrehozni, vagy egy működési mechanizmust leírni [2]. Tekintve, hogy hipotézis vagy modell itt is születik, a „hipotézis nélküli”
megnevezés nem teljes mértékben helytálló, talán célszerűbb helyette az „adatintenzív”,
„adatvezérelt” vagy „adatalapú” megközelítés megnevezések használata [3]. Tudományos gondolkodók és filozófusok többször kifejtették nézeteiket az egyik vagy a másik megközelítés támogatására, ám ha nem választani akarunk, hanem célunk a minél jobb megismerés, a két módszert akár egymás komplementereként is használhatjuk. Két, eltérő megközelítést használó kutatócsoport lényegileg ugyanarra az eredményre jutott hipotézis- vezérelt kutatást, illetve omikai megfigyelést alkalmazva a koenzim Q szintézisét illetően [4].
Ez a példa is azt sugallja, hogy a kétféle módszer nem egymással versengve, hanem egymást kiegészítve, mindkét módszer erősségeit kiaknázva vezethet a biológiai jelenségek pontosabb megismeréséhez.
A proteomikai vizsgálatok az esetek legnagyobb részében az adatalapú megközelítést részesítik előnyben, amikor két vagy több állapot proteómját hasonlítják össze. A sok adatpontot generáló proteomikai technikák (kétdimenziós elektroforézis, tömegspektrometria alapú fehérje vizsgálatok, multiplex immunesszék stb.) lehetőséget biztosítanak az egyes állapotok között jelentős változást mutató fehérjék vagy fehérje módosulatok azonosítására.
Ez a típusú vizsgálati megközelítés alkalmas beteg- és kontrollcsoportok közötti különbségek vizsgálatára, potenciális biomarkerek azonosítására.
Kutatómunkám során célom volt a nagyműszeres analitika biológiai alkalmazása, a széleskörű proteomikai módszertan fejlesztése és optimalizálása a kétdimenziós elektroforézistől a célzott és shotgun tömegspektrometriás eljárásokon át az átfogó bioinformatikai adatelemzésig. Eközben a számomra ismerősebb kétdimenziós elektroforézistől haladtam a bonyolultabb, tömegspektrometriás módszerfejlesztést és optimalizálást igénylő technikák kifejlesztése és alkalmazása, majd a bioinformatikai adatfeldolgozás felé. A célom az volt, hogy a rendelkezésemre álló átvett és/vagy általunk kifejlesztett módszereket felhasználva ismeretet nyerjek biológiai rendszerekről, fehérjék szerepéről és betegségek patomechanizmusáról. Ugyanakkor kiemelten fontosnak tartottam a könnyen hozzáférhető, nem invazív módon gyűjthető biológiai minták (könny, nyál, verejték) tanulmányozását, hogy betegségekre jellemző biomarkereket azonosítsak.
Céljaim közé tartozott a proteomikai technikák meghonosítása a Debreceni Egyetemen, és egy olyan kutatólabor kiépítése, amely proteomikai technikákat fejleszt és alkalmaz a saját, valamint a kutatóközönség kérdéseinek megválaszolására. Fontos volt számomra a proteomikai technikák fejlesztése, a lehető legszélesebb proteomikai arzenál naprakészen tartása és használata mellett a megszerzett tudás átadása, hallgatók és munkatársak tanítása, illetve olyan kutatók képzése, akik képesek alkalmazni és továbbfejleszteni a proteomikai módszereket.
2. IRODALMI ÁTTEKINTÉS
A proteomikai technikák alkalmazása lehetőséget biztosít elegendő mennyiségű adat gyűjtésére ahhoz, hogy az egyes biológiai kérdések rendszerszinten legyenek vizsgálhatók. A proteomikai technikákat osztályozva, gyakorlatilag két nagy módszercsoportot lehet elkülöníteni. A gél alapú módszerek első körben gélelektroforézist alkalmaznak, míg a tömegspektrometriás módszerek elsősorban tömegspektrometriás és kromatográfiás technikákat használnak. Természetesen a két módszercsoport metodikai szempontból összefonódik, ugyanis a gél alapú módszerekkel elkülönített fehérje foltokban vagy sávokban levő fehérjék azonosítása majdnem minden esetben tömegspektrometriás technikák használatával történik.
A továbbiakban először a gél alapú módszereket fogom bemutatni, külön kiemelve a kétdimenziós elektroforézist, majd ez után térek rá a tömegspektrometriás módszerek ismertetésére, és a kutatómunkám során tanulmányozott biológiai problémákra.
2.1. Gél alapú proteomikai módszerek
A gél alapú proteomikai technikák mindegyike gélben, az esetek igen jelentős részében poliakrilamid gélben történő elválasztást alkalmaz. A fehérjék egymástól történő elválasztása elektroforézissel történik, amely különböző komponensek elválasztására alkalmazható technika. A név a görög „Ηλεκτροφόρηση [elektrophorese]” szóból származik, amelynek jelentése elektronokat birtokolni. Az elválasztás során a töltött részecskék elektromos erőtérben vándorolnak, és méretük és/vagy töltésük szerint elválaszthatók egymástól. A molekuláris biológiában az elektroforézist gyakran alkalmazzák fehérjék és nukleinsavak vizsgálatára [5,6]. Az elválasztó közeg egy térhálós szerkezet, amelyben a pórusok méretét szabályozni lehet. A fehérjék elválasztására általában poliakrilamidot, míg a nukleinsavak elválasztására agarózt használnak. Az esetek nagy részében a fehérjék méret szerinti elválasztására ún. SDS poliakrilamid gélelektroforézist (SDS-PAGE) végeznek;
ekkor egy detergenst, a nátrium dodecil-szulfátot (SDS) alkalmaznak, amely egyenletesen beborítja a fehérjéket, azoknak negatív töltést kölcsönözve. A negatívan töltött fehérjék az elektromos erőtér hatására az anód felé vándorolnak a meghatározott koncentrációjú, térhálós szerkezetű akrilamid gélben és méretük függvényében elválaszthatók egymástól. A kisebb, és emiatt mozgékonyabb fehérjék nagyobb távolságot képesek megtenni az akrilamid polimerben adott időegység alatt, mint a nagyobb és kevésbé mozgékony fehérjék. Az elválasztás mértékét az akrilamid koncentrációjával lehet szabályozni: minél nagyobb az
akrilamid koncentrációja, annál jobban el lehet választani egymástól a kis molekulatömegű fehérjéket, míg alacsony akrilamid koncentráció mellett az elválasztás hatékonysága a magasabb tömegtartományban nagyobb. Az akrilamid koncentráció változtatásával a jobb elválasztást kívánó tartományt pontosan meg lehet határozni [6]. Attól függően, hogy az elektroforézist megelőző minta-előkészítés során alkalmaznak-e denaturáló szereket, megkülönböztetünk natív és denaturáló gélelektroforézist. A natív elektroforézis során nem alkalmaznak detergenst, denaturáló- vagy redukáló szert, így az adott fehérje negyedleges szerkezete is intakt marad, a fehérjét alkotó alegységek együtt vándorolnak a gélben.
Információ nyerhető a vizsgált fehérje térszerkezetéről, a feltekeredés állapotáról (pl.
részlegesen vagy teljesen denaturált), ligandumhoz vagy más fehérjékhez való kötődéséről, oligomerizációjáról vagy aggregációjáról [7]. A denaturáló változatban a minta-előkészítés során redukáló szert alkalmaznak (pl. béta-merkaptoetanolt), amely felszakítja az alegységek stabilizálására létrejött diszulfid hidakat. A redukált minták SDS gélelektroforézise segítségével az adott komplexet vagy fehérjét alkotó alegységek egyenként vizsgálhatók [6].
A natív vagy denaturáló gélelektroforézis segítségével vagy ezek kombinálásával sok információ gyűjthető egy adott fehérjéről vagy fehérje komplexről (molekulatömeg, alegységek száma, alegységek mérete, kötőpartnerek, negyedleges szerkezetet stabilizáló erők típusa stb.), viszont komplex biológiai minták (pl. sejtlizátum, szövet homogenizátum stb.) vizsgálatára kevésbé alkalmazhatók hatékonyan az alacsony felbontóképesség miatt.
Olyan esetekben, amikor nem csak egy vagy néhány fehérjét, hanem komplex minták proteómját kívánják vizsgálni, célszerűbb a kétdimenziós elektroforézis alkalmazása.
2.1.1. Kétdimenziós elektroforézis
A kétdimenziós elektroforézist 1974-ben alkalmazták először, de csak egy évvel később figyeltek fel rá igazán [8–10]. A technika két elválasztási módot kombinál: az első dimenzióban a molekulák a töltésük, míg a második dimenzióban a méretük szerint kerülnek elválasztásra. Az elválasztás eredményeként O’Farrell 1100 foltot azonosított E. coli sejtlizátumban, de véleménye szerint akár 5000 fehérje elválasztása is lehetséges a módszerrel [9]. A módszer nagy jelentőségűnek bizonyult, ugyanis az egyes foltok tulajdonképpen az egyes fehérje változatokra vagy módosulásokra, az ún. proteoformákra vonatkoznak. A kétdimenziós elektroforézis alkalmazásával megkülönböztethetők egy adott fehérje egyazon mintában előforduló különböző poszt-transzlációs módosulatai (pl.
foszforilált, glikozilált, hasított formák), amelyek integrált vizsgálata más módszerek alkalmazásával egy analízis keretében nehézkes lehet [10].
Az első dimenzió az izoelektromos fókuszálás, amely 3-4% akrilamid tartalmú ún.
fókuszáló gélben (angolul strip) történik. Különböző amfolitok segítségével egy immobilizált pH gradienst hoznak létre a gélben, majd a gélre felvitt fehérjéket elektromos erőtérbe helyezik. Az elektromos erőtér hatására a molekulák vándorolni kezdenek, és abban a pH tartományban, ahol nettó töltésük nulla, emiatt nem hat rájuk sem az anód, sem a katód vonzó hatása, megállnak. Attól függően, hogy milyen izoelektromos pontú fehérjék elválasztása a cél többféle, előre elkészített, immobilizált pH gradiens közül lehet választani. A legszélesebb pH tartományt a pH 3-10 jelenti; ebben az esetben az izoelektromos fókuszáló gélen egyenletes eloszlásban helyezkednek el pH 3 – pH 10 tartományban az egyes pH értékeknek megfelelő zónák. Ezzel szemben a nem lineáris pH 3-10 NL jelzésű géleken az egyes pH értékeknek megfelelő zónák nem egyenletesen vannak elosztva, hanem kevesebb terület jut a pH 3-4, illetve a pH 8-10 tartományok számára, és nagyobb területet foglal el a pH 5-7 közötti tartomány. Ez azért fontos, mert a sejtekben levő fehérjék izoelektromos pontja is főként a pH 5-7 tartományba esik, és viszonylag kevés döntően savas vagy döntően bázikus jellegű fehérje található. Amennyiben a nagyobb felbontóképesség a fontos, egy-két pH értéket tartalmazó fókuszáló gélek is használhatók. A mélyreható vizsgálatok általában kétlépcsős stratégiát alkalmaznak, amelynek során először pH 3-10 lineáris pH tartományt átfogó fókuszáló gélen egy átfogó képet kapnak az adott biológiai rendszerre jellemző fehérje változásokról, majd a gélkép alapján fontosabbnak ítélt tartományt csak egy pH egységet tartalmazó fókuszáló gélen vagy géleken vizsgálják [11]. Az izoelektromos fókuszálás során fontos, hogy a fehérjék kitekert és oldott állapotban legyenek, ezért speciális puffereket alkalmaznak a fehérjék feloldására és oldatban tartására. A leggyakrabban alkalmazott puffer az ún. Rabilloud puffer (7% urea, 2% tiourea, 4% CHAPS, 1% ditio-treitol), ami Thierry Rabilloud a kétdimenziós elektroforézis fejlesztése és alkalmazása terén úttörő munkát végző francia kutatóról kapta a nevét [10].
Ahhoz, hogy az első dimenzióban az izoelektromos fókuszáló gélen elválasztott fehérjék méretük szerint is elválaszthatók legyenek a második dimenzióban, egy köztes lépésre, az ún. ekvilibrálásra van szükség. Az ekvilibrálás során a fókuszáló gélen levő fehérjéket ditio-treitol (DTT) jelenlétében redukálják, majd a redukált tiol csoportokat alkiláló szer (általában jódacetamid) segítségével alkilálják, hogy megakadályozzák a diszulfid hidak kontrollálatlan újraalakulását [10,11]. Az ekvilibrált fókuszált gél megfelelő koncentrációjú poliakrilamid gél tetejére kerül rá, majd elektromos erőtérbe helyezve a gélt megtörténik a méret alapján történő elválasztás. A második dimenzióhoz nagyon gyakran ún. gradiens gélt alkalmaznak, amely felső része alacsony (általában 4-5%-os) akrilamid
tartalmú, az alsó része töményebb (általában 18-20%-os) és a kettő közötti átmenet egyenletes. Az ilyen gradiens gélek biztosítják a legjobb elválasztást, mert mind a magas, mind az alacsony mérettartományban megfelelő felbontóképességgel rendelkeznek [11].
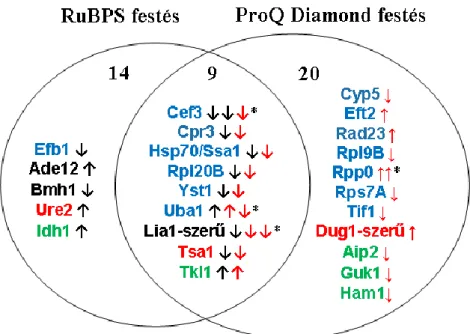
A méret szerinti elválasztást követően a gélben levő fehérjék nem láthatók, ezért valamilyen festési eljárás alkalmazásával lehet őket láthatóvá és/vagy vizsgálhatóvá tenni. A legelterjedtebb módszer talán még mindig a Coomassie Brillant Blue G-250-el történő festés, viszont a fluoreszcens festékek alkalmazása is egyre elterjedtebb. A fehérje foltok vizualizálására alkalmazott festési módszerek közül a legérzékenyebb az ezüst festés (érzékenység kb. 0,1 ng), viszont az összehasonlító mennyiségi elemzésekhez leggyakrabban a fluoreszcens festékeket (pl. Sypro Ruby, Flamingo Red stb.) alkalmazzák, mert ezeknek a legszélesebb a lineáris dinamikus tartománya. A fluoreszcens festékek a poszt-transzlációs módosítások vizsgálatára is alkalmazhatók. Míg a Sypro Ruby és a Flamingo Red a fehérjéken levő poszt-transzlációs módosításoktól függetlenül minden fehérjét megfest, addig a ProQ Diamond csak a foszforilált fehérjéket, a ProQ Emerald pedig csak a glikozilált fehérjéket teszi detektálhatóvá [10–14].
A kétdimeziós elektroforézis alkalmas egy adott mintában levő fehérjék, illetve különböző állapotok vagy proteómok közötti minőségi és mennyiségi különbségek vizsgálatára. A kétdimenziós gélelektroforézissel elválasztott és megfelelő festési eljárással láthatóvá/vizsgálhatóvá tett fehérjéket tartalmazó gélek összehasonlítására komplex képanalízis szükséges. Ezért a gélképeket először megfelelő szkennerek segítségével beszkennelik, és a bennük levő információt digitalizálják, majd bonyolult számítási háttérrel rendelkező szoftverek segítségével elvégzik a gélképek összehasonlító elemzését. A minőségi és mennyiségi analízis révén információ nyerhető pl. egyes organellumok proteómjáról, egy betegségre jellemző proteoformák változásáról, egy adott kezelésre vagy beavatkozásra bekövetkező fehérje szintű változásokról, stb. [15].
A kétdimenziós elektroforézis a sok információt adó, átfogó vizsgálatok közé tartozik, amely betekintést ad az adott mintára jellemző fehérjék minőségi és mennyiségi változásaiba.
A módszer alkalmas a teljes proteóm vizsgálatára, globális képet lehet kapni a fehérjékről és a fehérjék poszt-transzlációs módosításáról, viszont nagyon érzékeny, emiatt nagy technikai tudást és nagyon tiszta, ún. elektroforézis minőségű vegyszereket igényel. Ahhoz, hogy a gélképen tipikusan detektálható 600-700 fehérjét tartalmazó mintázat reprodukálhatóságáról meg lehessen győződni, 3 technikai és 3 biológia párhuzamos alkalmazása javasolt. Tekintve, hogy a kétdimenziós elektroforézis kevéssé automatizálható, és a nagy tisztaságú vegyszerek drágák, nagy erőforrást igényel megfelelő kivitelezése. Továbbá a gélkép elemzés
bonyolultságát és a párhuzamosok számát figyelembe véve az állapotonként több gélkép elemzése jelentős számítógépes erőforrást és sok időt igényelhet [10,11,16].
A kétdimenziós elektroforézis elsősorban sejtlizátumok vizsgálatára alkalmazható a legnagyobb sikerrel, viszont különböző szövetek (pl. izom) vagy más biológiai minták, pl.
szérum, tej, tojás tanulmányozására is használható [17,18]. A membránfehérjék vizsgálata nagy kihívást jelent a módszer alkalmazói számára, ugyanis a membránokban levő fehérjék oldatban tartása az analízis idejére nehezen megoldható feladat [19].
2.1.2. Differenciál gélelektroforézis
A kétdimenziós elektroforézis gyorsítására fejlesztették ki a differenciál gélelektroforézist (DIGE), amely során különböző fluoreszcens festékek segítségével az egyes mintákat megfestik, összekeverik, és egyetlen kétdimenziós elektroforézist végezve a technikai párhuzamosokat kiküszöbölik [16]. A differenciál gélelektroforézis során egyszerre háromféle minta vizsgálható. A háromféle mintát háromféle fluoreszcens festékkel (Cy2, Cy3 és Cy5) jelölik meg, a mintákat összekeverik, és egyszerre végzik el a kétdimenziós elektroforézist. Tekintve, hogy az elválasztás egy gélen megy végbe, nincs különbség az elválasztási körülményekben, ezért nincs szükség technikai párhuzamosokra, ami jelentős erőforrást spórolhat meg. Az egyes állapotokra jellemző fehérjék megjelenítése a géldokumentáció során történik, amikor a gélben lévő jeleket háromféle szűrő segítségével szkennelik be. A módszer alkalmazásával a gélképek analízise is sokkal egyszerűbben és gyorsabban kivitelezhető. A differenciál gélelektroforézis egyetlen hátránya a fluoreszcens festékek magas ára, viszont mindent figyelembe véve, a módszer ár-érték aránya összemérhető a hagyományos kétdimenziós gélelektroforézis ár-érték arányával [16].
A gélképek analízise során statisztikailag szignifikáns különbséget mutató fehérjéket tartalmazó foltokat kivágják, és azokat további analíziseknek vetik alá, amelyeknek általában a célja a fehérjék tömegspektrometriás azonosítása. Ismerve az egyes foltokban megtalálható fehérjéket, össze lehet kapcsolni a gélképen felismert minőségi vagy mennyiségi változást a mintában található fehérje módosulatokkal, a proteoformákkal [20].
2.2. Tömegspektrometrián alapuló proteomikai módszerek
A tömegspektrometria olyan nagyműszeres analitikai platform, amely gázfázisú ionok vizsgálatán alapul. Története az 1890-es évekig nyúlik vissza, amikor Wilhelm Wien, Eugen Goldsein megfigyeléseit alkalmazva egy olyan készüléket épített, amelyben tömeg-töltés arány alapján el tudta egymástól választani az egyes gázok által keltett anódsugarakat, az ún.
„Kanalstrahlen” sugarakat. Ezt a készüléket továbbfejlesztve Joseph John Thompson hozta létre az első tömegspektrográfot. A tömegspektrográfot a tömegspektroszkóp követte, majd 1918-ban Arthur Jeffrey Dempster elkészítette az első modern tömegspektrométert, amely a mai tömegspektrométerek elődje [21].
Felépítését tekintve, a tömegspektrométer ionforrásból, tömeg analizátorból, és detektorból áll. Az ionforrás felelős a gázfázisú ionok létrehozásáért, amelyek aztán a tömeg analizátorban elválasztásra kerülnek egymástól és a detektorba jutnak. A készülékben vákuum uralkodik; a teljes rendszert egy nagyon bonyolult elektronika szabályozza [21].
A tömegspektrometriát kezdetektől előszeretettel alkalmazták különböző fémek, szerves és szervetlen kismolekulák vizsgálatára, viszont fehérjék és nagyméretű biológiailag fontos vegyületek alkalmazására csak néhány évtizede, az ún. kíméletes („lágy”) ionizációs technikák felfedezése után került sor [21]. A tömegspektrometria segítségével kis mintamennyiségből jelentős információ halmazt lehet nyerni, amely felhasználható a mintában levő ismeretlen anyagok azonosítására és mennyiségük meghatározására [22].
Emiatt a technika alkalmazása nagyon elterjedt a természettudományokban; a PubMed adatbázisban több mint 336 ezer publikáció tartalmazza a tömegspektrometria kulcsszót.
Fontos megemlíteni, hogy több Nobel-díj odaítélése is kötődik a tömegspektrometriás fejlesztésekhez és kutatásokhoz. J. J. Thomson a gázokon áthaladó elektromosságra vonatkozó elméleti és kísérleti vizsgálataiért 1906-ban, Francis W. Aston 1922-ben számos nem radioaktív elem izotópjának felfedezéséért és az „egész szám szabály” felállításáért, Ernst O. Lawrence 1939-ben a ciklotron felfedezéséért, 1989-ben Wolfgang Paul és Hans Georg Dehmelt az ioncsapda felfedezéséért, valamint 2002-ben Koichi Tanaka és John Fenn biomakromolekulák vizsgálatát biztosító lágy ionizációs technikák (mátrix által segített lézer deszorpció ionizáció, ill. elektroporlasztásos ionizáció) felfedezéséért kapták meg a legrangosabb tudományos díjat (https://www.nobelprize.org/).
2.2.1. A tömegspektrométer felépítése és működése
A tömegspektrométerek változatos felépítéssel és emiatt széleskörű alkalmazhatósággal rendelkeznek, azonban mindegyik tömegspektrométer tartalmaz három
alapvető elemet: ionforrást, tömeg analizátort és detektort. Az analizátor és a detektor, valamint típustól függően az ionforrás is vákuumban található, amely biztosítja az ionok torzításmentes továbbhaladását az egyes elemeken. A tömegspektrométereket bonyolult elektronika vezérli, minden esetben vezérlő és adatkiértékelő számítógépekkel vannak összekötve, amelyek egyrészt biztosítják a rendszer működését, ill. működtetését, másrészt pedig az adatelemzésben segítenek [22–24].
A tömegspektrométerek lehetnek egyszerű készülékek, amelyek az ionforrás és detektor mellett csak egy analizátort tartalmaznak, vagy lehetnek tandem készülékek, amelyek több analizátorral rendelkeznek. Az analizátorok lehetnek ugyanolyan típusúak pl. a tripla kvadrupól tömegspektrométerek esetében vagy különböző típusúak. A Q-TOF típusú tömegspektrométerek TOF és kvadrupól analizátorokat, a QTRAP típusú készülékek három kvadrupólt tartalmaznak, de a harmadik kvadrupól beállítástól függően kvadrupólként vagy ioncsapdaként is funkcionál, illetve a tribrid készülékek felépítésében ioncsapda, kvadrupól és Orbitrap analizátorok vesznek részt [21,22]. A továbbiakban a proteomikában leggyakrabban használt és legelterjedtebb ionforrás és analizátor típusokat fogom röviden bemutatni.
Az ionforrás felelős a gázfázisú ionok létrehozásáért. Többféle ionizációs technika létezik, mint pl. a kémiai ionizáció, elektron ionizáció, fotoionizáció, gyors atom bombázás, stb. Az ionizáció során energiát közölnek a mintával, amely a mintaionok fragmentációját okozhatja. A kemény ionizációs technikák jelentős mértékű fragmentációval járnak, ezért ezek az ionizációs technikák gyakorlatilag a biológiai makromolekulák vizsgálatára alkalmatlanok [21,24]. A makromolekulák ionizálására az ún. lágy ionizáció alkalmas, ugyanis ebben az esetben a gáz fázisú ionok képződése nem jár jelentős fragmentációval, így akár intakt fehérjék, lipidek vagy nukleinsavak is vizsgálhatók tömegspektrométer segítségével. A biológiai makromolekulák ionizációjának kidolgozása az 1980-as évek közepére tehető, és jelentőségét mutatja, hogy a felfedezést 2002-ben Nobel díjjal jutalmazták [22,24].
2.2.1.1. Mátrix által segített lézer deszorpció ionizáció (MALDI)
A MALDI technika segítségével ionizálandó molekulák szilárd fázisban találhatók, egy fém hordozó felületére kikristályosított formában. A kristályos formában levő mintával energiát közölnek lézer segítségével, és ezzel bírják rá a minta molekulákat a deszorpcióra, a felülettől való elválásra, és gáz fázisba jutásra. A felületre a minta mellé egy segédanyagot, ún. mártrixot is felhelyeznek, akár úgy, hogy előzőleg összekeverik a mintával és együtt
kristályosítják azzal, vagy a már kristályos mintára rétegezik, esetleg szendvics módszerrel a mintát cseppentik egy mátrix kristály rétegre, majd a kikristályosodott mintára ismét mátrixot cseppentenek. A mátrix általában aromás gyűrűt tartalmazó kismolekula pl. 2,5-dihidroxi- benzoésav (DHB), alfa-ciano-4-hidroxifahéjsav (CHCA), szinapinsav, stb., amely a lézer energiájának hatására elválik a felülettől és ionizálódik. Az ionokat ezután iontranszfer segítségével átadja az időközben gáz fázisba kerülő minta molekuláknak, így minta-ionok képződhetnek. Mivel ez a transzfer nem hatékony, a MALDI ionizáció során az esetek nagy részében egyszeresen, esetleg kétszeresen töltött ionok keletkeznek. A megfelelő mátrix kiválasztása a minta típusától függ, a lézer energiájának beállítása pedig az esetek nagy részében empirikus módon, az adott mintára, készülékre, illetve a mérési körülményekre specifikusan történik [21,22,24].
2.2.1.2. Az elektroporlasztásos ionizáció (elektrospray ionizáció - ESI)
Az ESI során a minta folyékony halmazállapotban található, és az ionforrásban a kapilláris elhagyásakor kapja meg a nagyfeszültséget. A nagyfeszültség hatására az kapillárist elhagyó minta cseppek ionizálódnak, majd az ionforrásban a minta áramlási irányával ellentétesen áramló szárító nitrogén gáz hatására az oldószer párolog, és a töltött cseppek mérete csökken.
Amikor a töltött cseppek mérete nem tudja biztosítani a megfelelő távolságot az azonos módon töltött részecskék számára, bekövetkezik az ún. Coulomb féle robbanás, a taszító erő szétveti a cseppet. A cseppek tovább zsugorodnak, ez a folyamat többször megismétlődhet, így mire a tömegspektrométer bemeneti nyílásához, az ún. orifice-hez ér a minta, már alig tartalmaz oldószert, inkább csak többszörösen töltött minta ionokat. A tömegspektrométer bemeneti nyílása fűtött lemezen található, amely biztosítja az oldószer teljes elpárolgását, valamint azt, hogy csak a töltött minta ionok jussanak be a készülékbe [21,22,24,25].
A tömeganalizátor feladata, hogy a bejutott minta ionokat tömeg/töltés (m/z) hányadosuk alapján elválassza egymástól és eljuttassa a detektorba. Többféle típusa ismert, így lehet repülési idő analizátor, kvadrupól, ioncsapda, orbitális ioncsapda stb.
A repülési idő (Time of Flight – TOF) analizátor a bejutott ionokat a méretük és mozgékonyságuk alapján választja el egymástól. Az analizátor gyakorlatilag egy légüres cső, amelyben a kisebb, mozgékonyabb ionok gyorsabban repülnek, így hamarabb bejutnak a detektorba, míg a lomhább, nagyobb ionoknak ugyanez a folyamat több időt vesz igénybe, etért később érik el a detektort. Amennyiben az analizátorba történő belépés és a detektorba jutás között eltelt időt mérik, illetve megfelelő kalibráló anyagokkal rendelkeznek, a repülési idő meghatározásával pontosan meg lehet mondani az adott anyagra jellemző m/z értéket.
Tekintve, hogy a TOF analizátorokat előszeretettel kombinálják MALDI ionizációval, a lézer impulzus kibocsátása és a detektorba jutás közötti időtartamot szokták meghatározni [22].
A TOF analizátor érzékenysége és felbontóképessége igen nagy, ezért a proteomikai alkalmazásokban, peptidek szekvenálására gyakran alkalmazzák. A TOF analizátort gyakran kell kalibrálni, ha MALDI-TOF kombinációban használják, akkor gyakorlatilag minden mérés előtt szükséges elvégezni a kalibrációt [26].
A TOF analizátor felbontóképessége egyenesen arányos az analizátorban megtett út hosszával, vagyis minél hosszabb egy TOF analizátor, annál jobban el tudja választani egymástól a közeli m/z értékkel rendelkező ionokat. A gyártási technológiák alkalmasak lennének igen hosszú TOF analizátorok gyártására is, de a 2-3 m-nél hosszabb analizátorok nem terjedtek el, ugyanis nehezen férnének be a kutatólaboratóriumok többségébe. Azért, hogy növeljék a felbontást, de ez ne járjon az analizátor méretének jelentős növeléséhez a TOF analizátorban egy ún. ion tükröt alkalmaznak, amely potenciál-különbségek sorozatával gyakorlatilag visszafordítja, tükrözi az ionokat. Ily módon az ionok hosszabb utat tudnak bejárni, ezáltal az egymáshoz közeli m/z értékek jobban elkülöníthetők egymástól [21,24].
A kvadrupól analizátor négy fémrúdból áll, amelyekre feszültséget kapcsolnak. A szemben álló fémrúd-párokra kapcsolt váltakozó feszültség a fém rudak között levő töltött részecskéket csigavonal mozgásra készteti. Az rádiófrekvenciás (Rf) és egyenáram (Dc) feszültségek megfelelő megválasztásával stabilizálni lehet egy meghatározott m/z értékkel rendelkező ion pályáját, így csak az jut át az analizátoron, a többi ion pedig eltérül. Az Rf és Dc feszültségek változtatásával különböző ionok pályája stabilizálható, vagyis a kvadrupólra jellemző, hogy szelektíven stabilizálja az ionokat [24].
A kvadrupól felbontóképessége nem nagy, pl. a TOF analizátorral szemben jelentősen alulmarad, viszont az ionok szelektív kiválasztása és más analizátorokkal kombinálva a felvehető pásztázási típusok sokféleségének biztosítása vezethetett a viszonylag széleskörű elterjedéséhez [22].
Egyes tömegspektrométerekben hasonló elven, de precízebben működő hexapólok, oktapólok vagy multipólok biztosítják a nagyobb szelektivitást, de ezek nem terjedtek el széleskörűen [24].
Az ioncsapda felépítését tekintve hasonlít a kvadrupólhoz, viszont itt speciális elektron lencsék segítségével minden iont bent tartanak az analizátorban. A kvadrupólok elhelyezkedését tekintve az ioncsapda lehet lineáris (2D) vagy ún. 3D ioncsapda. Az ioncsapda szelektíven destabilizál, vagyis csak bizonyos m/z értékkel rendelkező ionok
pályáját destabilizálja, ezek kijutnak az ioncsapdából és bejuthatnak egy másik analizátorba vagy a detektorba [24,25].
Fourier-transzformációs ion ciklotron rezonancia (FTICR) analizátor és detektor a jelenleg elérhető legnagyobb felbontású és pontosságú analizátor. Mindezen kiváló tulajdonságai ellenére az FTICR analizátor nem terjedt el a mindennapi használatban, ugyanis nagyon drága, illetve fenntartása is igen nagy költségekkel jár. Az analizátor mágneses térbe elhelyezett négy elektródából áll. Az analizátor belsejében az erős mágneses tér hatására az ionok ciklikus mozgást végeznek és minden egyes ciklus során mikor elhaladnak a detektáló elektród előtt, akkor áramot indukálnak és ezt az áramot rögzítik.
Tehát ebben az esetben, az analizátor tulajdonképpen detektor is. A begyűjtött áramokat bonyolult matematikai műveletek, ún. Fourier transzformáció segítségével alakítják át spektrumokká. Az analizátor felbontóképessége annál jobb minél nagyobb a mágneses tér, ez viszont erős mágneseket, erős hűtést és többek közt jelentős mennyiségű tiszta He és N2
gázokat igényel [22,27].
Orbitális ioncsapda – Orbitrap analizátor a többihez képest a legkisebb méretű nagyfelbontású analizátor, egy néhány cm hosszúságú orsó alakú egység. Az analizátorban a bejutott ioncsomagok a ciklotronra jellemző körpályán mozognak és a mozgásuk során keltett potenciál-változást az analizátor falát képező gyűrű elektródok érzékelik, vagyis az Orbitrap analizátor az FTICR-hez hasonlóan, egyben detektorként is működik. A detektált potenciál- változásokat ez után Fourier-transzformáció segítségével alakítják át spektrumokká. Az Orbitrap analizátor felbontása és pontossága rendkívül nagy, megközelíti az FTICR analizátorok teljesítményét [28]. Tekintve a kis helyigényt, a viszonylag egyszerű üzemeltetést és a kiváló működési paramétereit, manapság nagyon elterjedt a proteomika laboratóriumokban.
Az ütközési cella a tömegspektrométerek nem egy feltétlenül különálló egysége, hanem sokszor a tandem tömegspektrométerek valamelyik analizátora szolgáltat speciális teret a molekulák fragmentációjára. Ebben az egységben vagy üzemmódban általában ütközés indukálta disszociáció (collision induced dissociation – CID) játszódik le. A CID elterjedten használt fragmentációs mód a peptidek azonosításához, ugyanis megfelelően beállítva, a peptid kötés mentén fragmentálja a peptideket. Más fragmentációs módok is használhatók, úgymint az ECD (electron capture dissociation) vagy az ETD (electron transfer dissociation) [29,30], de a peptidek azonosítása során ezeket kevésbé használják.
Elterjedésüket az is limitálja, hogy csak bizonyos tömegspektrométerek speciális tartozékaként lehet alkalmazni.
Az analizátorból érkező ionokat a detektor érzékeli és alakítja át elektromos jellé. A jel intenzitása arányos a detektorba jutó ion mennyiségével. Változatos felépítésű detektorokat alkalmaztak a különböző tömegspektrométerek esetében, ezek jelentős része ma már nem használatos. Az egyik legelterjedtebb detektor az elektron sokszorozó, illetve az utóbbi években megjelent, és az újabb készülékekben található Daly-féle detektorok [31]. Az FTICR, illetve Orbitrap analizátort tartalmazó tömegspektrométerek esetében a tömeganalizátor képezi a detektort is, így az ilyen készülékeknél nincs szükség külön detektor egységre.
A detektor által generált jeleket az adatfeldolgozó rendszer digitális tömegspektrumokká alakítja, amelyeket felhasználva elemezni lehet a mintákban jelen levő anyagok milyenségét és mennyiségét [31].
2.2.2. Kromatográfiával kapcsolt tömegspektrometria (LC-MS)
A tömegspektrométereket alkalmazhatják önmagukban pl. MALDI ionforrással ellátott készülékek esetében, de a leggyakrabban kromatográfiás rendszerrel kapcsolva használják. Az illékony kismolekulák esetében a gázkromatográfia (GC) az elterjedtebb, míg a proteomikában az esetek legnagyobb részében a folyadékkromatográfia (LC) az alkalmazott elválasztási módszer [32,33].
A kromatográfiás elválasztásnak két fő célja van: egyrészt a minta komplexitását hivatott csökkenteni, másrészt pedig a mintában levő anyagok kémiai tulajdonságait kihasználva speciális elválasztási módszereket lehet kidolgozni és alkalmazni a komponensek pontos és reprodukálható elválasztására. A proteomikában az esetek legnagyobb részében a cél a minta komplexitásának csökkentése, és ez általában egydimenziós reverz fázisú elválasztás segítségével történik. Ilyenkor az állófázis kromatográfiás oszlopa C18 alkil láncokkal borított szilika szemcséket tartalmaz, amelyhez a peptidek kikötődnek. A kötődés erőssége a peptidek hidrofobicitásával egyenesen arányos és a mozgó fázisban alkalmazott szerves oldószer mennyiségének a növelésével lehet a peptideket eluálni a töltet szemcséiről [34]. Tekintve, hogy a folyadékkromatográf sorba van kötve a tömegspektrométerrel (ún. on- line kapcsolás) az eluálódó peptidek közvetlenül a tömegspektrométerbe jutnak. A fehérjék folyadékkromatográfiával kapcsolt tömegspektrometriás (LC-MS) vizsgálata során leggyakrabban alkalmazott ionizációs mód az ESI [25].
Azért, hogy megfelelő minőségű elválasztást biztosítsanak az általában rendelkezésre álló kis mintamennyiség mellett is, a töltet szemcséinek méretét, a kromatográfiás oszlop és a mozgófázist hordozó kapillárisok átmérőjét csökkentik. Ilyen körülmények között kisebb
áramlást szükséges alkalmazni, ez az ún. nanoLC [33]. A proteomikában általában nanoLC körülményeket alkalmaznak, tipikusan 200-500 nl/min áramlási sebességgel dolgoznak, szemben a normál LC ml/min nagyságrendű áramlási sebességével.
A nagyon komplex minták esetében gyakran alkalmaznak két- vagy többdimenziós kromatográfiát (MUDPIT), amikor az elválasztási módok kombinálásával választják szét a minta komponenseit. Gyakran a peptideket először ioncserés kromatográfiának vetik alá, majd az egyes frakciókat reverz fázisú kromatográfiával választják szét további frakciókra [35]. A proteomikában többféle frakcionálási technika érhető el (pl. spider frakcionálás), amelyeket lehet a tömegspektrometriás elválasztással on-line vagy offline alkalmazni [36].
A fehérjék azonosítására és/vagy kvantitálására LC-MS-t, pontosabban nanoLC-MS-t használnak az esetek legnagyobb részében [33].
2.2.3. Fehérjék azonosítása LC-MS segítségével
A fehérjék azonosítása alapvetően kétféle módon történhet. Az egyik lehetőség a tömegspektrumok (MS spektrum) alapján az ún. peptidtömeg-térképezés (pepide mass fingerprint – PMF). A PMF során az egyes fehérjékről készült enzimes emésztményben levő peptidek m/z értékeit rögzítik egy tömegspektrometriás analízisben. A keletkezett peptidtömeg listát adatbázisokban levő fehérjék elméleti peptidtömeg listáival vetik össze keresőmotorok segítségével és megfelelő találat esetén a fehérjét azonosítottnak tekintik [33,37–39]. A PMF komplex minták esetén kevésbé jól használható, mert sokszor nehéz a fehérjék pontos és megbízható azonosítása, de fontos előnye, hogy egyszerű tömegspektrométerek esetében is alkalmazható [40].
A pontos és megbízható fehérje azonosítás általában az ún. MS/MS spektrumok alapján történik manuálisan, vagy szoftverek segítségével [41]. Itt minden esetben szükséges a peptideket fragmentálni, ezért ez a módszer leginkább a tandem tömegspektrométerekre jellemző. A folyamat során először egy MS spektrum felvétele történik az enzimes fehérje emésztményről, majd a spektrumban rögzített, általában legintenzívebb csúcsok kiválasztása után a kiválasztott csúcsnak megfelelő peptid, ún. prekurzor ion fragmentációja következik az ütközési cellában. A kiválasztott peptidből keletkező fragmensek adják az ún. MS/MS spektrumot [33,38,40]
A peptidek tömegspektrométerben történő fragmentációja esetében a fragmentációs energiákat úgy állítják be, hogy egyszerre csak egy peptidkötés hasadjon fel. A folyamatban azonban lehetőség nyílik a legtöbb kötés felhasadására. Így ún. „b” ill. „y” ionsorozatokat lehet kapni, amelyekben a sorozat egyik tagja a sorozat következő tagjától 1 aminosav-
maradvány tömegének megfelelő m/z értékkel különbözik (1. ábra). A „b” és „y” ionokat az szerint nevezik el, hogy a peptid melyik része (N- vagy C-terminális) viszi a töltést, és az adott végtől számítva hányadik peptidkötés hasad fel. Pl. a „b4” ion esetében a töltés az N- terminálison van, és az N-terminálistól számított negyedik peptidkötés hasad fel, míg az „y4”
ion esetében a töltés a C-terminálison van és a C-terminálistól számított negyedik peptidkötés hasad fel [33,40].
1. ábra. Az AGFAGDDAPR peptid MS/MS spektruma.
Az ábra bal oldalán a peptid spektrumát és a rögzített „b” és „y” ionokat tüntettük fel, míg a jobb oldalon az elméletileg képződő „b” és „y” ionokat.
Ismerve az aminosavak ún. monoizotópos tömegeit a „b” és „y” ionsorozatokat felhasználva megállapítható a vizsgált peptid szekvenciája [24,42]. Amennyiben nem áll rendelkezésre egyéb információ, csak az MS/MS spektrumot felhasználva lehetőség van a peptid szekvencia egyértelmű azonosítására. Ez az ún. de novo szekvenálás, amely a korszerű keresőszoftverek alkalmazásával is sokszor nagy kihívást jelent [40,43–45].
A keresőmotorok általában más megközelítést alkalmaznak. A felhasználó által kiválasztott adatbázist (UniProt, NCBInr stb.) és paramétereket (hasító enzim típusa, kihagyott hasító helyek száma, alkalmazott tömegspektrométer, ütközés típusa, poszt- transzlációs módosítások stb.) felhasználva egy elméleti spektrum adatbázist generálnak. Az analízis eredményeként kapott spektrumot az elméleti spektrumokhoz hasonlítják, és az összehasonlítás eredményeként egy valószínűségi értéket, valamint az elméleti spektrumnak megfelelő peptidet jelenítenek meg [41]. Amennyiben az eredményben megjelenített valószínűségi értékek megfelelők (legalább 95%), a találat elfogadható. Egy fehérjét akkor tekintünk azonosítottnak, ha legalább 2 olyan peptidet találtunk a mintánkban, ahol a peptid azonosítás valószínűségi értéke 80-99,9%, valamint a fehérjéhez rendelt valószínűségi értéke 0-50% [46]. Annak ellenére, hogy a legalább 2 peptid alapján történő azonosítás széles körben elterjedt, többen megkérdőjelezik a megfelelőségét, és az egy peptid alapján történő
azonosítás mellett érvelnek [47,48]. Kevés találat esetén a spektrumok manuális ellenőrzése elvégezhető, de a nagy pontosságú és nagyfelbontású készülékek által generált nagymennyiségű adatból azonosított több száz fehérje esetében ez a módszer nem alkalmazható. Ebben az esetben a keresést egy random vagy fordított szekvencia adatbázison is célszerű elvégezni, és a kereső- vagy megjelenítő szoftver által kiszámolt hamis felfedezési ráta (false discovery rate – FDR) értékeket is figyelembe véve meghatározni az adott mintában jelen levő fehérjéket [49]. Az elfogadható FDR értékek fehérje szinten általában a 0,01 (1%), aminek elérése sok esetben csak az általános kritériumok (95% peptidekhez és 99% fehérjékhez rendelt valószínűségi értékek és fehérjénként legalább 2 peptid) szigorításával oldható meg.
2.2.4. Adatgyűjtési módok
A leggyakrabban az ún. információfüggő adatgyűjtést (Data/Information Dependent Acquisition – DDA vagy IDA) alkalmazzák a fehérjék azonosítására és kvantitálására. A módszer során a tömegspektrométer gyorsaságától függően az n (tömegspektrométertől függően n = 1-20) legintenzívebb iont választják ki, azt fragmentálják és megtörténik az MS/MS spektrumok felvétele [50]. A cél, hogy minél több információ kerüljön begyűjtésre, ezért az n értékét megfelelően kell megválasztani. Ha nagyon megnövelik az n értékét, elméletileg több MS/MS spektrumot lehet felvenni, de ez több időt fog igénybe venni, megnő az egy peptid analízisére fordított ciklusidő. Tekintve, hogy az on- line kapcsolt kromatográfból folyamatosan érkeznek a tömegspektrométerbe a peptidek, érdemes az n értékét úgy megválasztani, hogy az összhangban legyen a kromatográfia során elválasztott peptidek elúciójával. A módszer jól automatizálható és elterjedten használt, viszont a hátránya, hogy csak a legintenzívebb ionokról van így információ. Emiatt bizonyos szennyezők, mint pl. a keratin, vagy a mintában levő abundáns fehérjék pl. szérum esetében az albumin, nagyobb arányban jelennek meg a találatok között, megnehezítve a fontosabb, de kisebb mennyiségben jelen levő fehérjék detektálását. Ugyanakkor a reprodukálhatóság is viszonylag alacsony, és a hiányzó adatpontok miatt információveszteséggel is számolni kell [51].
Ezzel szemben az információ független adatgyűjtés (Data Independent Acquisition – DIA) esetében a teljes mérendő tömegtartományt felosztják kisebb részekre (10-20 m/z) és az így definiált tömeg ablakokban levő összes prekurzor iont fragmentálják, függetlenül attól, hogy milyen az intenzitása, ill. milyen a prekurzor ion m/z értéke [52,53]. A folyamatot többször megismételve a teljes tömegtartományon, és az így begyűjtött adatokat
felhasználva, a későbbiek során a fehérjéket azonosítani és kvantitálni lehet. Az információfüggő adatgyűjtéssel szemben, ahol pontosan ismert, hogy melyik MS/MS spektrum melyik prekurzor ionhoz tartozik, az információ független adatgyűjtés során ez nem ennyire egyértelmű [54]. A megfelelő eredmények érdekében először egy könyvtárat célszerű építeni a vizsgálni kívánt mintatípus többszöri információfüggő analízisével. Tipikusan 100 m/z tömegtartományonként felveszik a 16 legintenzívebb prekurzor ionról az MS/MS spektrumokat, és ezeket használják fel a könyvtár építéséhez [52,55].
Tekintve, hogy a DDA és DIA adatgyűjtési módok esetében nincs előzetes információ arra vonatkozóan, hogy milyen fehérjék vannak a mintában, és az ott levő lehető legtöbb fehérjét azonosítják, ezeket a módokat ún. shotgun adatgyűjtési módoknak nevezzük. Ezzel szemben a célzott módszerek mindegyike azon alapul, hogy a mintában egy, vagy több adott fehérjére vonatkozó információt gyűjtenek [56].
A célzott tömegspektrometria alkalmazása során az ún. MRM/SRM (Multiple/Selected Reaction Monitoring) adatgyűjtési módot használják az esetek jelentős részében. Az MRM/SRM módszerek specifikusan képesek detektálni adott molekulákat, és információ nyerhető az illető anyag mennyiségére vonatkozóan is [57]. A módszer elsősorban hármas kvadrupól analizátorokat tartalmazó tömegspektrométerek sajátossága: azon alapul, hogy az első kvadrupólt (Q1) úgy állítják be, hogy csak a meghatározott molekulára jellemző m/z értékkel rendelkező ionokat engedje át, amelyek majd az ütközési cellában fragmentálódnak. A harmadik kvadrupól (Q3) beállításait úgy módosítják, hogy a keletkezett fragmensek közül csak az előre meghatározott fragmenseket engedje át. Tehát csak akkor lesz detektálható jel, ha mindkét, az adott molekulára jellemző feltétel egyidejűleg teljesül.
Az ilyen, ún. MRM/SRM átmenetek (Q1 és Q3 kvadrupólban beállított m/z érték) alkalmazása egyrészt biztosítja a specificitást, másrészt a kromatográfiás csúcs alatti terület (AUC) arányos a tömegspektrométerbe juttatott molekula mennyiségével. A módszer specificitását, valamint a kvantitálást, ismert mennyiségű, szintetikus, stabil izotóppal jelzett (SIL) peptidek hozzáadásával lehet ellenőrizni. Bár a tömegkülönbség miatt a SIL peptidek MRM/SRM átmenetei értelemszerűen különböznek a vizsgálandó peptidétől, a mintában levő vizsgálandó (endogén) peptid és a szintetikus SIL peptid azonos fiziko-kémiai tulajdonságokkal rendelkezik. Az endogén és a SIL peptidek egyszerre eluálódnak, hasonló módon fragmentálódnak, így a szintetikus, SIL peptidek belső standardként használhatók [57].
A módszer óriási előnye, hogy egyszerre több fehérjére jellemző MRM/SRM átmenetek is beilleszthetők egyetlen módszerfájlba, ezáltal több fehérje mennyiségi analízise is elvégezhető egyetlen mintából [57,58].
A kvadrupól és Orbitrap analizátorokat együtt tartalmazó tandem tömegspektrométerek esetében a fenti hármas kvadrupól segítségével megvalósított MRM/SRM-hez hasonló, de nagyobb áteresztőképességű célzott vizsgálat végezhető [59]. Itt is szükséges a vizsgálandó fehérje ismerete, és a fehérje-specifikus egyedi peptid(ek)nek, az ún. proteotipikus peptid(ek)nek megfelelő m/z értékeket itt is ki kell választani, viszont itt egyszerre több prekurzort is kiválasztható, és az Orbitrap analizátorban a kiválasztott prekurzorok mindegyikének fragmenseit rögzíteni lehet. Így gyakorlatilag MS/MS spektrum lesz az eredmény, ami kiválóan alkalmazható a peptidek azonosítására. A PRM (Parallel Reaction Monitoring) során nem szükséges az átmeneteket megtervezni, ugyanis az összes fragmens rögzítésre kerül, és az adatelemzés során lehet az MRM/SRM átmeneteket kigyűjteni és kvantitatív analízisre felhasználni [59,60].
2.2.5. Fehérjék kvantitálása
A fehérjék tömegspektrometriás kvantitálása történhet shotgun vagy célzott módszerek alkalmazásával [56]. A shotgun adatgyűjtési módok jelölést vagy jelölés nélküli módszereket egyaránt használhatnak [58].
A kémiai jelölés során bármilyen biológiai mintát lehet kémiai reakció révén jelölni, de a jelölés hatékonysága sosem 100%-os. Az iTRAQ (izobár jelölés abszolút és relatív kvantitáláshoz – isobaric tag for relative and absolute quantitation) [61] vagy TMT (tandem mass tag) [62] során olyan SIL jelölő reagenst alkalmaznak, amely elsősorban a fehérjék N- terminálisához, illetve a lizin oldalláncok amin csoportjához kapcsolódik. A jelölő reagens tartalmaz egy jelölő csoportot, egy kiegyenlítő csoportot - amely tömegét úgy választják meg, hogy a jelölő csoporttal együtt azonos tömeget biztosít (izobár) mindegyik jelölő reagensnek -, valamint egy reaktív csoportot, amely a mintában levő fehérjék amino csoportjához való kapcsolódást biztosítja. A különböző izobár jelölő reagensekkel jelölt mintákat összekeverik, és egy mintaként vizsgálják. A tömegspektrometriás analízis során az MS spektrumban a peptidekre jellemző csúcsok azonos m/z értéknél detektálhatók, de a fragmentáció során a jelölő csoportra jellemző fragmensek is megjelennek. Az iTRAQ jelölők esetén az utóbbi fragmensek a 113-121 Da, míg a TMT jelölők esetén a 126-134 Da tartományba esnek. Ezen fragmenseket külön-külön detektálni lehet az MS/MS felvételeken, és a csúcsok alatti
területek minden esetben arányosak az adott jelölő csoport koncentrációjával, vagyis a jelölt peptid, és az annak megfelelő fehérje mennyiségével [61,62].
A jelölés nélküli kvantitálás (label-free quantification) teljes mértékben tömegspektrometriás módszer, amely nem alkalmaz semmiféle jelölő anyagot. A vizsgálat során bekövetkező MS/MS események számát, vagy a prekurzor ion intenzitását használják a kvantitáláshoz. Ugyanis minél több MS/MS készül egy fehérjéről, vagy minél nagyobb a kiválasztott prekurzor ion intenzitása, annál nagyobb koncentrációban van jelen a fehérje a mintában. Mind a jelölés nélküli, mind a kémiai jelölést alkalmazó módszer jól használható biológiai minták fehérjéinek azonosítására és relatív kvantitálására [58,63].
A proteomikai feladatok megoldására a komplex biológiai kérdések megválaszolásánál egyre szélesebb körben elterjedt a célzott proteomika alkalmazása. A kémiai jelölést alkalmazó vagy a jelölés nélküli kvantitálási módszerekkel ellentétben, a célzott módszerek során szükség van a vizsgálni kívánt fehérje ismeretére. Viszont azáltal, hogy pontos információval rendelkezünk a vizsgálandó fehérjéről, specifikus és szenzitív MRM/SRM vagy PRM módszert lehet tervezni. A célzott módszerek során az egyes átmenetekre kapott jelekből a kapcsolt kromatográfiás elválasztás (online LC) során kialakuló csúcs, pontosabban a csúcs alatti terület arányos a fehérje mennyiségével [57,59,60,64].
A kvantitálási módszerek mindegyikével meghatározható a fehérjék relatív mennyisége, viszont csak a célzott módszerek segítségével nyílik lehetőség az abszolút kvantitálásra. Az ismert mennyiségű SIL peptidek mint „belső standardok” alkalmazásával végzett abszolút kvantitálás segítségével pontosan meg lehet állapítani a vizsgált fehérje/peptid koncentrációját vagy anyagmennyiségét az adott mintában [57].
2.2.6. Adatgondozás, adatelemzés
A proteomikai módszerekkel generált minőségi vagy mennyiségi információkat az esetek igen jelentős részében adatbázisokban tárolják. A megfelelő paraméterek, sok esetben a MIAPE (Minimum Information About a Proteomic Experiment) irányelveknek (http://www.psidev.info/miape) megfelelő információk megadásával a nyers és elemzett adatokat fel lehet tölteni adatbázisokba. Ilyen adatbázisok például az EMBL-EBI (https://www.ebi.ac.uk/) által gondozott Pride [65], a Peptide Atlas [66], a célzott adatokra specializálódott Panorama [67,68], stb. Ezek az adatok szabadon hozzáférhetők és a ProteomeXchange-n keresztül letölthetők. A ProteomeXchange integrálja az egyes adatbázisokat, így egy egységes felületet biztosít az adatok feltöltéséhez és letöltéséhez [69].
Jelenleg 10848 adathalmazt tartalmaz, amelyből 4489 humán
(http://www.proteomexchange.org/, 2020.04.03.). A proteomikai kutatóközönség számára nagyon fontos a megbízható és szabadon hozzáférhető adatok generálása, emiatt a terület vezető folyóirataiban a publikálás feltétele az adatok feltöltése a MIAPE kritériumoknak megfelelően a szabadon hozzáférhető adatbázisba, lehetőség szerint a ProteomeXchange felületen keresztül.
Az omikai módszerekkel begyűjtött nagy adatmennyiség egyik legátfogóbb és legjobb vizualizációs lehetőséget biztosító megjelenítése a hálózatok alkalmazása [70,71]. A hálózatok rajzolásánál a rendelkezésre álló kísérleti vagy predikciós adatok felhasználásával ténylegesen igazolt, vagy becsült fehérje-fehérje interakciókat használják fel. A hálózatrajzoló szoftverek jelentős része csak a bináris információt, a kapcsolat meglétét vagy hiányát alkalmazza, de az egyre nagyobb mértékben rendelkezésre álló mennyiségi adatokat csak kismértékben használják [72,73].
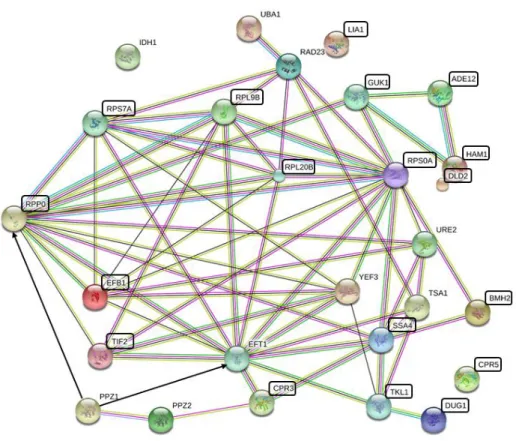
A hálózatok rajzolásához egyik leggyakrabban alkalmazott adatbázis és szoftverkörnyezet a String, amely szabadon hozzáférhető és felhasználóbarát módon, folyamatosan frissülő adatbázissal biztosítja a könnyen értelmezhető eredményeket [74,75].
A hálózatok elemzésekor fontos a hálózati csomópontok, jelen esetben a fehérjék, illetve a csomópontok közötti élek, esetünkben a fehérje-fehérje interakciók ismerete. Ezen információkat felhasználva a hálózat változására jellemző paraméterek számíthatók, illetve a hálózati paraméterek vizsgálatával a hálózatok változásai számszerűsíthetők. A String adatbázis a hálózat megjelenítésén kívül alkalmas a funkcionális elemzés elvégzésére is: a vizsgált hálózatban a feldúsult, a véletlentől statisztikailag szignifikáns módon gyakrabban előforduló funkciójú fehérjéknek megfelelő génontológiai (Gene Ontology – GO) funkciókat kilistázza, és a funkcióknak megfelelően a fehérjéket színezi.
Jelenleg az adatbázis - viszonylag bonyolult matematikai módszereket alkalmazva - használható a mennyiségi adatok elemzésére is, de vizsgálataink idején még nem állt rendelkezésre hasonló funkció [76].
2.3. A proteomikai módszerek alkalmazási területei
A proteomikai technikákat leggyakrabban biológiai jelenségek hátterében húzódó molekuláris mechanizmusok megértésére, fehérjék szerepének, illetve betegségek patomechanizmusának vizsgálatára és biomarkerek azonosítására használják az orvostudománnyal kapcsolatos területeken. Tekintve, hogy az adatintenzív módszerek alkalmazása jelentős mennyiségű adatot hoz létre, ezek megfelelő elemzésével az adatokban
rejlő biológiai információ kinyerhető. Ezért gyakran alkalmazzák a proteomikai technikákat szignalizációs útvonalak analízisére, szekretált fehérjék vizsgálatára, különböző állapotok összehasonlítására azzal a céllal, hogy a jelenségek mögött meghúzódó, biológiai szempontból releváns információ nyilvánvalóvá váljon [33,77–80]. Változatos alkalmazási lehetőségeit tekintve a proteomika a fehérjékkel kapcsolatos kutatások egyik nagyon gyakori eszköze. Jól mutatja ezt a tényt a FEBS (Federation of the European Biochemical Societies) (https://2019.febscongress.org/program) a HUPO (Human Proteome Organization) (http://hupo-2019.p.asnevents.com.au/), valamint az EuPA (European Proteomics Association) (https://www.eupa2019.org/program/scientific-program/) által szervezett éves konferenciák tematikája. A proteomika jól használható a baktériumok mennyiségi analízisétől kezdve (metaproteomika) [81,82] a genomikai és proteomikai információkat ötvöző proteogenomikán [83] át a fehérjék sejten belüli lokalizációjának és előfordulási gyakoriságának vizsgálatáig [84,85] a biológia és elméleti orvostudományok majdnem minden területén.
2.3.1. Fehérjék szerepének vizsgálata
A proteomikai technikákat gyakran alkalmazzák fehérjék szerepének vizsgálatára. Az ilyen jellegű módszerek általában ötvözik a hipotézis-vezérelt és adatintenzív megközelítéseket. Egy adott fehérje pontos funkciójának felderítésére még mindig a legjobb módszer - ha kivitelezhető - a fehérjét kódoló gén eltávolítása a vizsgált biológiai rendszerből (élőlény, szerv, sejtvonal stb.), és a kieső, illetve megváltozott funkciók tanulmányozása [86].
Az adatintenzív proteomikai módszerek alkalmazásával átfogó képet kaphatunk a fehérjék minőségi és mennyiségi változásait illetően a kontroll és génkiütött biológiai mintákban, ezeket az információkat pedig felhasználhatjuk a megváltozott biológiai funkciók feltérképezésére [87]. Ugyanakkor a poszt-transzlációs módosítások vizsgálatával olyan fontos szabályozó folyamatokról nyerhetünk információt, mint a foszforiláció-defoszforiláció [88,89]. A foszforilációt a kinázok, míg a defoszforilációt a protein foszfatázok katalizálják.
Számos kutatási eredmény bizonyítja a kinázok szerepét a jelátviteli folyamatokban, de összefüggésbe hozták több patofiziológiás állapot kialakulásával is, mint például a daganatok kialakulása [90,91]. Egyre több bizonyítékot találtak arra vonatkozóan, hogy a foszfatázok nem csak kikapcsolói a kinázok által okozott változásoknak, hanem fontos részt vállalnak a szignalizációs folyamatok szabályozásában is [92]. A Ser/Thr foszfatázok a Ser/Thr kinázok által a Ser, ill. Thr aminosavakra helyezett foszfát csoportokat távolítják el. A Ser/Thr
specifikus PPP foszfoprotein foszfatázok egyik jelentős alcsoportját képezik az 1 típusú foszfatázok, melyek legismertebb képviselője a PP1 [92].
A PPP foszfatáz család egy új, gombákra jellemző tagja a PPZ1 foszfatáz [93], amelyet elsőként a Saccharomyces cerevisiae modell organizmusban jellemeztek [92,94].
Ortológjait számos gomba fajban megtalálták, mint a Schisosaccharomyces pombe, Neurospora crassa, Aspergillus nidulans és Candida albicans [95–98]. A deléciós mutáns C.
albicans törzsek vizsgálatával úgy találták, hogy a CaPPZ1 gén által kódolt CaPpz1 enzimnek szerepe van a kation homeosztázis szabályozásában, a sejtfal bioszintézisében, az oxidatív stresszre adott válasz kialakításában, a morfogenezisben, valamint módosíthatja a patogén virulenciáját [97,99–101]. Annak ellenére, hogy a deléciós kísérletekkel viszonylag sok információt sikerült gyűjteni a foszfatáz funkciójáról, fiziológiás szubsztrátja és a pontos hatásmechanizmusa még megismerésre vár. A proteomikai technikák alkalmazása kiváló lehetőséget biztosít a CaPpz1 szerepének tanulmányozására proteomikai és foszfoproteomikai szinten egyaránt.
2.3.2. Betegségek patomechanizmusának vizsgálata
A fent említett alkalmazási területek mellett a proteomikát leggyakrabban betegségek patomechanizmusának felderítésére, molekuláris szintű vizsgálatára alkalmazzák [79]. A betegektől és a megfelelő kontrolloktól származó humán mintákat, vagy az egyes állatfajokban létrehozott betegségmodellekből származó mintákat összehasonlítják, és adatintenzív technikákkal minden elérhető, fehérjéről származó információt begyűjtenek. Ez után bonyolult adatelemzési módokat használva az adatokat elemzik és értelmezik.
Munkám során az adatintenzív proteomikai módszereket a proliferatív vitreoretinopátia (PVR) patomechanizmusának tanulmányozására használtam. A PVR-ről viszonylag keveset tudunk, sajnos etiológiája nem ismert pontosan. Multifaktoriális betegségről van szó, amely a rhegmatogén retinaleválás egyik komplikációja. A rhegmatogén retinaleválást korrigáló műtétek után az esetek 8-25 %-ában kifejlődhet [102,103]. A rendelkezésre álló adatok alapján úgy tűnik, hogy a PVR tulajdonképpen a retina fiziológiás javítási mechanizmusának kóros folyamata, amely nemkívánatos, sok esetben súlyos látásromlással járó állapotokat okoz. A szérum eredetű vagy üvegtesti faktorok hatására egy epidermális-mesenchimális átalakulás figyelhető meg. A retina pigment epitél (RPE) sejtek és gliasejtek migrációját a RPE sejtek agresszív proliferációja követi és kialakul az epiretinális membrán, amely a betegség egyik vezető tünete [104,105]. A membrán ez után összehúzódik, közben extracelluláris kollagén termelődik, és ezen hatások eredményeként a