Fermentations of Nitrogenous Organic Compounds
H . A . B A R K E R
I. Introduction 151 A. Early Studies 152 II. Fermentations of Single Nitrogenous Compounds 152
A. Amino Acids 152 B. Heterocyclic Compounds 181
III. Fermentations of Pairs of Amino Acids (Stickland Reaction) 195
A. Oxidations 196 B. Reductions 199 IV. Conclusions 201
References
I. Introduction
Living organisms commonly contain from 1 to 1 0 % nitrogen on a dry weight basis. Most of this nitrogen is present in proteins and other poly- meric compounds, although m a n y monomeric compounds, such as urea, uric acid, creatine, glutamine, and asparagine, also occur in m a n y organisms in lesser amounts. T h e majority of these organic nitrogenous compounds are at an oxidation level between carbohydrates and fats and are poten- tially useful as sources of carbon, nitrogen, and energy for both aerobic and anaerobic microorganisms.
M a n y monomeric compounds can be used directly as fermentation sub- strates b y anaerobic microorganisms. T h e more a b u n d a n t polymeric com- pounds, on the contrary, m u s t be hydrolyzed or otherwise degraded into their component monomers before they can be fermented. This depoly- merization is catalyzed b y a variety of mostly hydrolytic enzymes t h a t a t t a c k more or less specific linkages in proteins, polypeptides, nucleic acids, nucleotides, and related compounds. Such enzymes, commonly present in microorganisms, will not be discussed in this chapter. Nevertheless t h e de- polymerizing enzymes are of major importance in t h e microbial decomposi- tion of organic materials in n a t u r e because t h e y usually catalyze the rate- limiting step in t h e over-all process. Once t h e component amino acids, purines, pyrimidines, and related compounds are liberated, t h e y are fre- quently decomposed rapidly b y mixed microbial populations.
This chapter is devoted t o a systematic consideration of t h e decomposi- tion of amino acids, purines, pyrimidines, and a few other nitrogenous com- pounds b y bacteria under anaerobic conditions without t h e intervention of inorganic oxidizing agents such as sulfate, nitrate, or carbonate. This ex-
151
eludes such processes as sulfate reduction, denitrification, and the methane fermentation. T h e processes discussed are mostly fermentations in which either a single organic nitrogenous compound, or a pair of such compounds, or products of the decomposition of one or more such compounds undergo coupled oxidation-reduction reactions. Generally such fermentations, like fermentations of carbohydrates, provide t h e energy and metabolic inter
mediates required for growth of the organism in an appropriate medium.
However, some anaerobic processes t h a t will be discussed have been studied only with cell suspensions and are not yet known t o be capable of supporting growth. A few reactions are mentioned which are known not to support growth b u t deserve attention for some other reason. Emphasis is placed upon t h e over-all chemical transformations of bacterial fermentations and on the metabolic pathways b y which these transformations occur.
A. EARLY STUDIES
M u c h of the early chemical work on the anaerobic decomposition of nitrog
enous compounds b y "putrefactive" bacteria was confused b y t h e use of either mixed cultures or very complex substrates like meat extract, or both.
Observations m a d e on such cultures demonstrated t h a t bacteria can live b y the anaerobic decomposition of amino acids with the production of am
monia, carbon dioxide, hydrogen sulfide, fatty acids, and a variety of other more or less volatile substances, often having unpleasant odors. However, these studies did little to establish the specific substrates decomposed or the chemical reactions catalyzed b y individual species. Information of this type was probably first obtained b y Naviasky,1 who inoculated a medium con
taining asparagine as the main energy source with a pure culture of Bacillus proteus vulgaris and demonstrated t h e formation of carbon dioxide, am
monia, acetate, and succinate. Shortly thereafter, similar observations were m a d e b y Brasch2 on the fermentation of glutamate b y Bacillus (Clostridium) putrificus and b y Liebert3 on t h e fermentation of uric acid b y Bacillus (Clos
tridium) acidiurici. These and other observations demonstrated t h a t t h e fermentation of single nitrogenous compounds, like the fermentation of sugars, can support t h e growth of anaerobic bacteria. A quarter of a century later Stickland4 used suspensions of washed cells of Clostridium sporogenes grown on a complex medium, t o demonstrate t h a t this species normally ob
tains energy b y oxidation-reduction reactions between pairs of different amino acids. Subsequently it was shown t h a t each of these types of amine acid fermentation is used b y m a n y species of anaerobic bacteria.5
II. Fermentations of Single Nitrogenous Compounds
A. AMINO ACIDS
About twenty species of anaerobic or facultatively anaerobic bacteria are known t o ferment single amino acids. Table I lists these bacteria and indi-
T A B L E I
AMINO ACIDS FERMENTED BY ANAEROBIC BACTERIA
Species
Alanine | [ Arginine | | Aspartate | Cysteine | Δ-Ami no valerate | Glutamate 1 Glycine | | Histidine 1 | Hydroxyproline | | Isoleucine | Leucine | Lysine | Methionine | | Phenylalanine | Proline | Serine 1 Tryptophan | Threonine | Tyrosine 1 Valine | Stickland reaction |
Clostridium botulinum6' 7 ±
+ +
Clostridium cochlearium*
+
—Clostridium perfringens9 · 10
+ + + + +
Clostridium propionicum11
+
—+
— — — — — — ± — —+
—+
— ± —Clostridium saccharobutyri-
+ + + + +
cum12
Clostridium sporogenes1* > 14 —
+
—+
— — — — —+
—+ +
—+ + +
—+
Clostridium sticklandii1*
+
—+ + + +
Clostridium tetanil*> 17 —
+ +
—+
—+
—Clostridium tetanomorphum1* — —
+ + +
—+ -
— — =fc-
—+
—+
— —Clostridium species19
+
Fusobacterium nucleatum20 — ±
+ +
—+
— —+
— —+
—+ +
— —Diplococcus glycinophilus21
- - - - - + - - -
—- - - -
—Micrococcus aerogenes22 — — — —
+
—+
— — — — — — —+
—+
— — —Micrococcus anaerobius23 — — — — —
+
— — — — — — — — — — — — — Micrococcus variabilis23 — — — — —+
— — — — — — — — — — — — — Micrococcus prevotii22 — — — —+ - + - - - -
—- - +
—+ -
—-
Micrococcus activus22 — — — —
+
—+
— — — — — — —+
—+
— — —Micrococcus asaccharolyticus22 — —
-
—+
—+ - - -
— — — —+ - + -
—-
Coccus L C2 4
+ + +
Escherichia coli25
+ +
Key: + , fermented rapidly; =fc, fermented slowly; — not, fermented.
cates t h e amino acids t h a t are attacked. T h e ability t o catalyze a n oxida
tion-reduction reaction between pairs of different amino acids (Stickland reaction) is also indicated.
T h e bacteria presently known t o ferment amino acids fall mainly into two groups, t h e anaerobic spore-formers (Clostridium species) a n d t h e an
aerobic cocci (Micrococcus a n d Diplococcus species). Only one nonsporulat- ing, obligately anaerobic, rod-shaped bacterium is listed, namely, Fuso
bacterium nucleatum. Probably other species of this general morphological and physiological group (family Bacteriodaceae) also will be found t o fer
m e n t amino acids. Although most of t h e bacteria t h a t ferment nitrogenous compounds are obligately anaerobic, one facultative species, Escherichia coli, is probably able t o ferment certain amino acids.
All of t h e amino acids listed in Table I , with t h e exception of proline, hydroxyproline, a n d isoleucine, are known t o b e fermented b y a t least one
bacterial species. T h e three exceptional amino acids are readily decomposed by Clostridia t h a t catalyze t h e Stickland reaction (see Table I ) . Undoubt
edly in time bacteria will be found t h a t ferment these amino acids.
1. A L A N I N E
A fermentation of alanine has so far been observed only with Clostridium propionicum,11 which also ferments β-alanine,2 6 serine, threonine, lactate, pyruvate, and acrylate. W i t h all these substrates, except threonine, t h e products are acetate, propionate, and carbon dioxide; ammonia is also formed from t h e amino acids. T h e fermentation of alanine is represented by equation (1).
3CH3CHNH2COOH + 2 H20 - * 3NH3 + 2CH3CH2COOH + CH3COOH + C 02 (1)
T h e non-nitrogenous products of t h e alanine fermentation are t h e same as those formed b y t h e propionic acid bacteria2 7 (genus Propionihacterium) or Micrococcus lactilyticus2* from lactate, except t h a t C. propionicum does not produce succinate. Despite t h e similarity in products, there is substan
tial evidence t h a t t h e chemistry of t h e two processes differs significantly.
With both propionic acid bacteria2 9 and Μ. lactilyticus,30 succinate h a s been shown t o be a precursor of propionate. I n extracts of Μ. lactilyticus, suc
cinate is first converted to succinyl-CoA and then decarboxylated t o pro-
HOOCCH2CH2COSC0A C 02 + CH3CH2COSC0A (2)
pionyl-CoA, t h e immediate precursor of propionate. C. propionicum, on the contrary, neither forms nor decarboxylates succinate3 1 or succinyl- CoA. Additional evidence against t h e participation of a symmetrical com
pound like succinate is the absence of randomization of t h e a- and β-carbon atoms of lactate during its conversion to propionate.3 2 Also, C. propionicum, unlike t h e propionic acid bacteria, is unable to incorporate carbon dioxide carbon into propionate.3 1 T h e formation of succinate from a C3 substrate is known to involve carbon dioxide.
An indication of t h e p a t h of propionate formation in C. propionicum was provided b y t h e observation t h a t acrylate is fermented b y cell suspensions as rapidly as alanine or lactate.1 1 This suggested t h e possibility t h a t t h e lat
ter substrates are converted t o acrylate which is then reduced t o propionate according t o t h e scheme shown on page 155.
This reaction sequence is similar t o t h a t involved in t h e synthesis and oxi
dation of butyric acid b y Clostridium kluyveri31 except t h a t in t h e latter organism t h e coenzyme A derivatives of t h e acids are t h e actual reactants.
T h e occurrence of such reactions in t h e reverse direction was investigated by S t a d t m a n3 4 by studying t h e oxidation of propionate with extracts and dried cell preparations of C. propionicum. Dried cells catalyzed t h e oxida-
C H3C H N H2C O O H C H3C H O H C O O H Alanine Lactate
\ - N H » /
\ / C H2= C H C O O H
Acrylate
+ 2 H
C H3C H2C O O H Propionate
tion of propionate, lactate, pyruvate, or alanine to acetate and carbon di
oxide using oxygen as an electron acceptor. T h e oxidation of propionate was found t o be absolutely dependent upon the addition of catalytic amounts of acetyl phosphate. Since enzymic reactions are known b y which propionyl- CoA can be synthesized from propionate, acetyl phosphate, and coenzyme A, it seemed probable t h a t acetyl phosphate was required for the synthesis of propionyl-CoA. This conclusion was supported b y the observation t h a t cell-free extracts, unlike dried cell preparations, were unable to oxidize pro
pionate even in the presence of acetyl phosphate; however, propionyl-CoA was oxidized readily. Apparently the enzymic system responsible for the synthesis of propionyl-CoA was inactive in extracts.
T h e immediate oxidation product of propionyl-CoA has not been directly identified because of its instability in the enzymic system. However, indirect evidence for the formation of acrylyl-CoA was obtained. Synthetic acrylyl- CoA was shown to react rapidly with ammonium ion t o form β-alanyl-CoA [equation (3)] in the presence of an extract of cells of C. propionicum grown on a l a n i n e .2 6 , 3 5 This reaction is reversible, b u t the equilibrium strongly
C H2: C H C O S C o A + N H3 ^ C H2N H2C H2C O S C o A (3) favors the formation of β-alanyl-CoA. When C1 4-labeled propionate was
oxidized in the presence of ammonium ion and catalytic amounts of acetyl phosphate and coenzyme A, C1 4-labeled 0-alanine was formed. Since the enzyme preparation catalyzed reaction (3), t h e accumulation of 0-alanine provided presumptive evidence for the intermediate formation of acrylyl- CoA and 0-alanyl-CoA. T h e latter compound could lose t h e CoA moi
e t y b y hydrolysis or a transfer reaction. T h e role of β-alanyl-CoA in t h e metabolism of alanine b y C. propionicum is still completely obscure.
T h e reduction of acrylyl thioester to t h e propionyl derivative is also cata
lyzed b y extracts of C. propionicum under certain circumstances. This t y p e of reaction has been observed with acrylyl pantetheine, using a dye, re
duced safranine, as reducing agent.2 6 All the available evidence indicates
t h a t acrylyl-CoA is probably an intermediate in propionate formation b y C. propionicum.
All a t t e m p t s to demonstrate an enzymic conversion of alanine to acrylyl- CoA have been unsuccessful. There is no evidence for the formation of alanyl-CoA from alanine or for an interconversion of alanyl-CoA and acrylyl-CoA.
T h e reversible amination of acrylyl-CoA to 0-alanyl-CoA under the in
fluence of the enzyme acrylyl coenzyme A aminase suggests t h a t β-alanyl- CoA m a y be an intermediate in the fermentation of 0-alanine in accordance with the following sequence:
β-alanine C o A > jS-alanyl-CoA ,τΝΗ>, acrylyl-CoA *±2H * propionyl-CoA This suggestion is strengthened by the observation t h a t the aminase is about 100 times more active in cells grown on β-alanine t h a n in cells grown on α-alanine.2 6 However, the initial reaction in this sequence, the conver
sion of 0-alanine to 0-alanyl-CoA, has not been demonstrated.
A reversible hydration of an acrylyl thioester t o a lactoyl thioester [re-
C H 2= C H C O - C o A + H20 ^ CH3CHOHCOC0A (4) action (4)] could not be detected in extracts of C. propionicum. This reac
tion in the forward direction has so far been observed only with extracts of a Pseudomonas species which was grown aerobically on propionate.2 6 2. δ-AMINO VALERATE
An unnamed Clostridium, isolated from sewage sludge after a preliminary anaerobic enrichment in a medium containing S-aminovalerate as the main energy source has been shown1 9 to convert this substrate to ammonia, valerate, propionate, and acetate according to the following equation:
2H2NCH2CH2CH2*CH2COOH + 2 H20 - *
(5) 2NH8 + CH3CH2CH2*CH2COOH + CH3CH2COOH + *CH3COOH I n a fermentation of 2-C1 4-5-aminovalerate, the C1 4 was found only in t h e α-carbon of valerate and in the methyl carbon of acetate. These results sug
gest t h a t the fermentation involves a reductive deamination and a 0-oxida- tion of valerate or some intermediate C5 compound to propionate and ace
t a t e . N o enzymic studies have been reported. T h e substrate specificity of t h e organism is rather high since several compounds related to δ-amino- valerate, including ^-alanine, 7-aminobutyrate, norvaline, ornithine, and e-aminocaproic acid, cannot support its growth.
3. ARGININE, CITRULLINE, AND ORNITHINE
N o fermentations of these compounds have been reported. Arginine can serve as a hydrogen acceptor in the Stickland reaction for Clostridium sporo-
genes, probably after an initial conversion t o ornithine, since three moles of ammonia are formed per mole of arginine and ornithine serves as a hydrogen acceptor.3 6 Clostridium sticklandii is able t o use arginine, citrulline, or orni- thine as a n oxidant.1 5
T h e conversion of arginine to ornithine has been demonstrated with C.
perfringens,27 Streptococcus faecalis™ S. lactis*9 and S. haemolyticus.40 At least two enzymic steps are involved. Cell-free extracts of these organisms convert arginine to citrulline under appropriate conditions according t o the equation:
N H N H
I I
C = N H C = 0
I I
NH N H
I + H20 —• I + N H3 (6)
(CH2)3 (CH2)3
C H N HI I 2 C H N H2
I I
COOH COOH Arginine Citrulline
W i t h extracts of S. faecalis or S. lactis a large accumulation of citrulline re- quires t h e absence of orthophosphate. T h e enzyme responsible for the de- amination is called arginine desiminase.4 1 T h e cleavage of t h e ureido group of citrulline has been demonstrated with extracts of S. faecalis?**42 and S.
lactis?9 and with cell suspensions of C. perfringens.27 W i t h the former organ- isms, t h e reaction requires the presence of orthophosphate and a phosphate acceptor such as adenosine-5'-phosphate or adenosine diphosphate. T h e reaction probably proceeds according to equation (7), although a stoichio-
citrulline + Pi + ADP -» ornithine + C 02 + N H8 + ATP (7) metric yield of adenosine triphosphate (ATP) has not been observed be-
cause of the presence of A T P a s e in the enzyme preparations.4 3 T h e enzyme system catalyzing reaction (7) has been called "citrullinase" or "citrulline ureidase." However, it is now known t h a t two enzymic steps are involved in t h e reaction,3 9 one being a phosphorolysis of citrulline [reaction (8)] t o ornithine and carbamylphosphate, the other being the transfer of the phos- phoryl group from carbamylphosphate to adenosine diphosphate (ADP) t o form A T P [reaction (9)]. Carbamylphosphate has been shown to serve
L-citrulline + H P 04" ^ L-ornithine + N H2C O O P 03" (8) NH2COOPOr + ADP i=± N H3 + C 02 + ATP (9) b o t h as a carbamyl donor in citrulline synthesis [the reverse of reaction
(8)] and as a phosphate donor to A D P [reaction (9)] in t h e presence of ex-
tracts of S. faecalis.44 T h e enzymes catalyzing the reactions have been sepa
rated a n d partially purified.4 5 T h e equilibrium in reaction (8) is far t o t h e left. Therefore t h e decomposition of citrulline is dependent upon t h e re
moval of carbamylphosphate b y reaction (9). T h e equilibrium in the latter reaction is far in t h e direction of A T P formation.
T h e multienzyme system responsible for t h e conversion of arginine t o ornithine, carbon dioxide, a n d ammonia was formerly called "arginine di
hydrolase" in the mistaken belief t h a t t h e reaction was caused b y a single enzyme.
Although a fermentation of ornithine h a s n o t been described, reactions are known b y which a n extensive decomposition of ornithine can occur in a system containing a suitable reducing agent and a mixture of two Clostridia.
An organism catalyzing t h e Stickland reaction can reduce ornithine t o δ-aminovalerate which can be fermented b y a n unnamed Clostridium1 9 (see Section I I , A, 2 on δ-aminovalerate).
4. ASPARTATE AND ASPARAGINE
Asparagine was probably t h e first amino acid shown t o be decomposed anaerobically b y a pure bacterial culture. I n 1908 Naviasky1 found t h a t Bacillus proteus vulgaris can ferment asparagine with the formation of sev
eral products among which succinate, acetate, carbon dioxide, and ammonia were positively identified. Since this early work, relatively few studies on the fermentation of asparagine or aspartate have been carried out.
T h e products of aspartate fermentation b y Clostridium tetani (Table I I ) are ammonia, carbon dioxide, volatile acids (acetate a n d b u t y r a t e ) , h y droxy acids (lactate a n d malate), a n d alcohol (ethanol a n d possibly buta- nol). Although succinate was n o t mentioned as a product of aspartate fer
mentation, it is probably formed because succinate is a major product (yield about 50 %) of the fermentation of malate b y C. tetani.17 Clostridium
TABLE II
PRODUCTS OF L-ASPARTATE FERMENTATION BY Clostridium tetani16
Product Yielda
Ammonia (100) Carbon dioxide 105
Acetate 186
Lactate 55c
Ethanol 30
β The yield of product is expressed in moles per 100 moles of substrate decomposed.
Cell suspension experiment.
b Probably includes some butyrate.
e Probably includes some malate.
saccharobutyricum was reported4 6 to form acetic and butyric acids from aspartate in a molar ratio of 2 t o 1.
T h e available d a t a are insufficient to permit firm conclusions concerning t h e p a t h of aspartate decomposition. M a n y facultative bacteria form t h e enzyme aspartase,4 7 which deaminates aspartate according to equation (10).
COOH H O O C — C H
I II
H2N C H H C — C O O H
I ^ N H 3 + (10)
CH2
I
COOH
L-Aspartate Fumarate However, t h e presence of this enzyme has not been demonstrated in a n y obligately anaerobic bacterium.
Clostridium welchii has been shown4 8 * 4 9 to catalyze a decarboxylation of C O O H C H2C H N H2C O O H — C 02 + CH3CHNH2COOH (11) L-aspartate to L-alanine [reaction (11)]. This decarboxylation is greatly accelerated by p y r u v a t e and a variety of other α-keto acids and is inhibited by carbonyl reagents. T h e stimulation b y p y r u v a t e has been interpreted to mean t h a t the reaction involves a transamination between aspartate and p y r u v a t e to give alanine and oxalacetate [equation (12)] followed by de
carboxylation of the latter c o m p o u n d4 9 [equation (13)]. This interpretation
aspartate + pyruvate —• oxalacetate -f- alanine (12)
oxalacetate —* pyruvate -f C 02 (13)
is incorrect, because Meister et al.AB have shown t h a t C1 4-labeled p y r u v a t e is not incorporated into alanine as the above reactions predict. T h e mech
anism b y which keto acids stimulate aspartate decarboxylation has not been determined.
C O O H C H2C H N H2C O O H -> C O O H C H2C H2N H2 + C 02 (14) A very slow decarboxylation of L-aspartate to β-alanine [reaction (14)]
is also catalyzed by some b a c t e r i a5 0 b u t is not known t o occur in Clostridia.
T h e reaction sequence involved in t h e reductive conversion of L-aspartate to threonine5 1 · 5 2 should also be considered as a possible p a t h of aspartate fermentation. If this reaction sequence does occur in anaerobic bacteria, t h e decomposition of aspartate m u s t be coupled with suitable oxidative re
actions.
A t present there is no evidence t h a t a n y of t h e above reactions of L- aspartate is directly involved in its fermentation b y anaerobic bacteria.
5 . CYSTEINE, HOMOCYSTEINE, AND M E T H I O N I N E
Some microorganisms and animal tissues have long been known t o con
tain an enzyme, cysteine desulfhydrase, t h a t converts cysteine to pyruvate, ammonia, and hydrogen sulfide according to the following reaction.5 3 This
HSCH2CHNH2COOH + H20 -> H2S + N H3 + CH3COCOOH (15) enzyme has been found in Proteus vulgaris,5* P. morganii,bb Escherichia coli,56 Propionihacterium pentosaceum,51 and Bacillus subtilis.bS T h e E. coli and B. subtilis enzymes a t t a c k only L-cysteine, whereas extracts of P.
pentosaceum act on D and L isomers a t the same rate. These differences m a y indicate the existence of two stereospecific desulfhydrases or of a cysteine racemase.
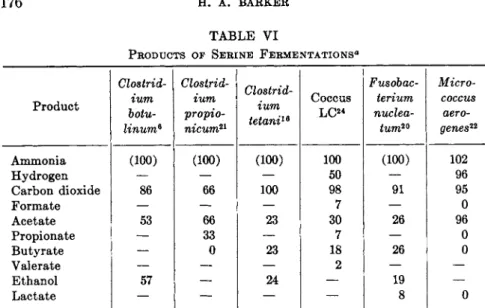
T h e fermentation of cysteine with formation of ammonia and hydrogen sulfide and other products has been shown with an obligately anaerobic coccus (Table I I I ) , b u t a stoichiometric formation of t h e products of t h e cysteine desulfhydrase reaction has not been demonstrated with a n y an
aerobic species. Formation of pyruvate from cysteine b y rumen coccus L C2 4 is probable because the products of fermentations of p y r u v a t e and cysteine b y this organism are very similar except for ammonia and hydrogen sulfide.
Quantitative d a t a on t h e products of cysteine fermentation b y other ana
erobic bacteria are not available.
On the basis of the above evidence it is probable t h a t fermentations of cysteine generally involve conversion of the substrate t o p y r u v a t e b y t h e cysteine sulfhydrase reaction, followed b y a fermentation of p y r u v a t e ac
cording to t h e enzymic constitution of t h e organism.
DL-Homocysteine is decomposed b y cell-free extracts of Proteus morganii more rapidly t h a n DL-cysteine, DL-serine, or DL-threonine. T h e products of
TABLE III
PRODUCTS OF L-CYSTEINE FERMENTATION BY Coccus L C2 4
Product Yield8
Ammonia 100
Hydrogen 8
Hydrogen sulfide
+
Carbon dioxide 103
Formate 14
Acetate 26
Propionate 2
Butyrate 31
Valerate 5
β The figures give the yield of product in moles per 100 moles of cysteine decom
posed.
homocysteine decomposition are ammonia, hydrogen sulfide, and a-keto- HSCH2CH2CHNH2COOH + H20 -> H2S + N H3 + CH2CH2COCOOH (16) b u t y r a t e [reaction (16)]. T h e enzyme catalyzing this reaction apparently requires pyridoxal phosphate as a cofactor.5 9 T h e enzymes attacking cysteine and homocysteine appear to be different. N o information appears to be available concerning t h e anaerobic decomposition of α-ketobutyrate b y P . morganii.
Methionine is attacked slowly b y several anaerobic bacteria (Table I ) . Chemical and enzymic studies of methionine decomposition have been carried out only with Clostridium sporogenes}4 Extracts of this organism were found t o convert L-methionine t o α-ketobutyrate, ammonia, and m e t h - CHaS—CH2CH2CHNH2COOH + H20 — CH3SH + N H3 + CH3CH2COCOOH (17) ylmercaptan [reaction (17)]. T h e enzyme system responsible for this reac
tion has been called b o t h methionine demercapto-deaminase1 4 and dethio- m e t h y l a s e .6 0 I n C. sporogenes, the enzyme system does not act upon D- methionine, although in some bacteria t h e dethiomethylase is accompanied b y a methionine racemase which permits the utilization of b o t h isomers.
T h e partially purified dethiomethylase has been shown to require pyridoxal phosphate as a coenzyme.
T h e further transformations of α-ketobutyrate and t h e final products of methionine fermentation by C. sporogenes have not been determined.
6. GLUTAMATE
G l u t a m a t e is fermented with great facility b y Clostridium tetanomor-
phum1*' 6 1'6 2; indeed it appears t o be t h e preferred substrate for this spe
cies. T h e rate of glutamate decomposition is similar t o t h a t of glucose de
composition b y other organisms. For example, when a suitable medium containing 0.1 Μ glutamate is inoculated with 1 volume per cent of an ac
tive culture of C. tetanomorphum, t h e substrate is completely decomposed within 18 t o 24 hours a t 37°C.
T h e main products of glutamate fermentation are acetate, b u t y r a t e , a m monia, carbon dioxide, and hydrogen (Table I V ) . T h e yields of ammonia a n d carbon dioxide are essentially constant a t one mole per mole of gluta
m a t e under a variety of conditions. T h e yields of t h e other three products are somewhat dependent on t h e p H of t h e m e d i u m .6 1 When t h e p H is in
creased from 7 to 8 the yield of hydrogen is about doubled and t h e molar ratio of acetate t o b u t y r a t e is increased from 2.9 to 3.8.
T h e p a t h of glutamate decomposition in C. tetanomorphum is of consider
able interest because it is completely different from t h e well-known p a t h of glutamate metabolism via α-ketoglutarate and t h e tricarboxylic acid cycle.
TABLE IV
PRODUCTS OF GLUTAMATE FERMENTATION4*
Product
Clostridium tetanomorphumi
glutamate
Clostridium tetani17 glutamate
Micrococcus aerogenes22 glutamate Ammonia
Carbon dioxide
(100) 91
0 5 116 41 0
(100) 94
103 98 0 5 101 45 0 Formate
Hydrogen Acetate Butyrate
Lactate trace
90 45
α The figures give the yield in moles per 100 moles of substrate decomposed. The data were obtained with cell suspensions.
T h e first indication of an unusual p a t h of glutamate breakdown was pro
vided by t h e low yield of carbon dioxide. A conversion of glutamate to ace
t a t e and carbon dioxide via the tricarboxylic cycle would produce 3 moles of carbon dioxide per mole of glutamate instead of t h e observed 1 mole. A significant reutilization of carbon dioxide was excluded b y an experiment with C1 4-labeled carbon dioxide.
Further information concerning the fate of the individual carbon atoms of glutamate was obtained by tracer experiments.6 1»6 3 ·6 4 Samples of glutamate labeled with C1 4 in positions 1, 2, 4, or 5 were fermented b y washed cells, and the products were isolated and degraded to locate the isotope. T h e re
sults of these experiments are summarized diagrammatically in Fig. 1.
Glutamate carbon atoms 1 and 2 are converted largely to acetate, with only a small conversion to b u t y r a t e . Carbon atoms 3 and 4 are converted mainly to b u t y r a t e in such a way t h a t carbon atom 4 appears in the carboxyl and presumably the 0-carbon atoms of b u t y r a t e , whereas carbon a t o m 3 prob
ably occupies the a- and 7-positions of b u t y r a t e . Carbon a t o m 5 is con
verted exclusively to carbon dioxide. These results indicate the formation of two different C2 units during glutamate fermentation. One unit, derived from glutamate carbon atoms 1 and 2, is converted t o free acetate which is not readily activated b y this organism for conversion to b u t y r a t e . T h e sec-
6COOH -* eC 02
FIG. 1. Fermentation of C1 4-glutamate by Clostridium tetanomorphum.
ond C2 unit, derived from glutamate carbon a t o m s 3 and 4, m u s t be an ac
tivated acetyl group, since it is preferentially converted t o b u t y r a t e . T h e results of the tracer experiments are consistent with t h e idea t h a t pyruvate, derived from glutamate carbon a t o m s 3, 4, and 5, is an intermediate in t h e fermentation.
F u r t h e r analysis of t h e chemical reactions in t h e glutamate fermentation was done with enzyme preparations.6 5 Crude, particle-free extracts of C.
tetanomorphum convert glutamate anaerobically to ammonia, acetate, car
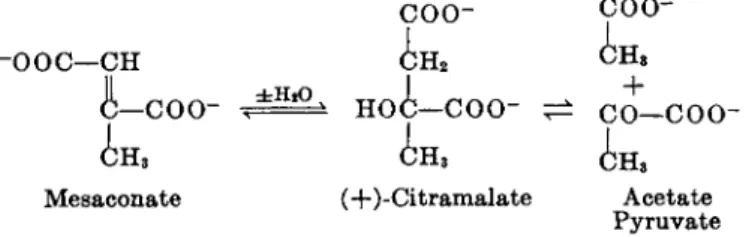
bon dioxide, hydrogen, and several minor products including p y r u v a t e and mesaconate, b u t do not form b u t y r a t e under the same conditions. T h e identification of mesaconate (methylfumarate) b y W a c h s m a n6 5 was a major contribution because it pointed t h e way t o t h e further elucidation of t h e role of branched chain dicarboxylic acids in glutamate breakdown. W h e n mesaconate was added as a substrate, it was rapidly decomposed b y cell-free extracts to carbon dioxide, hydrogen, p y r u v a t e , and acetate. B o t h t h e rate of mesaconate formation from glutamate, and t h e rate of its decomposition are more t h a n adequate to justify t h e conclusion t h a t mesaconate is an in
termediate in glutamate fermentation.
During t h e decomposition of mesaconate b y crude extracts a n additional product sometimes accumulated in appreciable amounts. This compound, which was detectable either b y paper chromatography or b y partition chromatography on a silica gel column, h a d t h e properties of a dicarboxylic acid containing one or more hydroxyl groups. N o t enough of t h e acid was a t first available to permit its identification b y chemical methods, b u t a consideration of t h e structure of mesaconate and t h e possibility of the en
zymatic hydration of its double bond, suggested t h a t t h e dicarboxylic acid might be either a- or β-methylmalic acid. T h e former compound, also known as citramalic acid, seemed to be a more likely intermediate because a simple aldol t y p e cleavage would yield p y r u v a t e and acetate, known products of mesaconate decomposition in this system. DL-Citramalic acid was synthe
sized and found t o be decomposed rapidly to pyruvate and acetate b y cell- free extracts.
Figure 2 shows the reactions involved in the conversion of mesaconate to p y r u v a t e and acetate b y C. tetanomorphum. T h e reversible hydration of the
c o o - c o o -
-OOC—CH
+
c o — c o o -
Mesaconate (+)-Citramalate Acetate Pyruvate FIG. 2. Conversion of mesaconate to acetate and pyruvate.
double bond of mesaconate t o form ( + ) - c i t r a m a l a t e is catalyzed by an en
zyme referred to as mesaconase. This enzyme has not been studied in de
tail, b u t it appears t o be distinct from fumarase and aconitase. T h e activity of mesaconase is dependent upon the presence of ferrous ion and cys
teine. T h e enzyme is strongly inhibited by chelating agents such as α,α'- dipyridyl or o-phenanthroline, which combine with ferrous ion. This in
hibition provides a convenient method for blocking the p a t h of glutamate fermentation a t the mesaconate level. T h e equilibrium of the mesaconase reaction favors the formation of citramalate ( Ke q. £^ 8).
T h e conversion of ( + ) - c i t r a m a l a t e to pyruvate and acetate is catalyzed by the enzyme citramalase. This enzyme is highly active b u t very un
stable, having a half-life in extracts of only a few hours a t 0°C. W h e n ex
tracts are incubated a t 37°C. for an hour, they completely lose t h e ability to decompose citramalate. Since most of t h e other enzymes involved in glutamate fermentation are more stable, this provides a method for ob
taining an accumulation of citramalate during t h e decomposition of other substrates.
Citramalase shows a high degree of substrate specificity. ( + ) - C i t r a m a l a t e is the only substrate so far found t o be decomposed by the enzyme a t an appreciable rate. T h e equilibrium of the reaction strongly favors the de
composition of citramalate
particularly when t h e substrate concentration is low. T h e only cofactor known to be required for the citramalase reaction is magnesium ion.
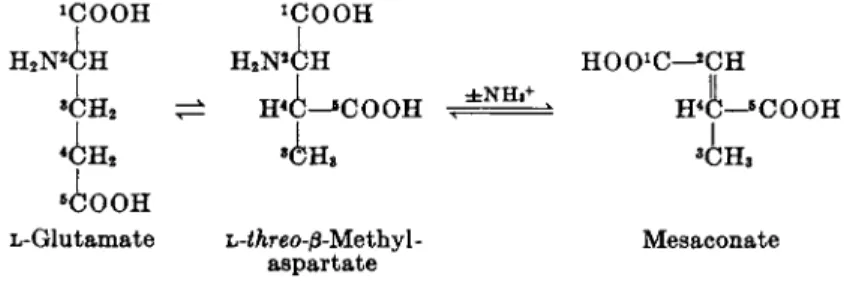
T h e conversion of glutamate to mesaconate and ammonia involves a t least two reactions, shown in Fig. 3. T h e first reaction results in a rear
rangement of the carbon chain of L-glutamate with formation of L-threo-β- methylaspartate, a branched-chain C6 dicarboxylic amino acid. T h e second reaction is a deamination of β-methylaspartate to mesaconate.
T h e formation of β-methylaspartate in the C. tetanomorphum system was [pyruvate] [acetate]
[citramalate] « 7
Κ)ΟΟΗ ^ O O H
8C O O H
L-Glutamate e L·-threo-β-Methy\- Mesac
aspartate
FIG. 3. Conversion of glutamate to mesaconate.
Mesaconate
detected while studying the effect of charcoal treatment on the ability of cell-free extracts to interconvert glutamate, mesaconate, and ammonia. Un
treated extracts catalyze the reaction in both directions. After charcoal treatment, the decomposition of glutamate was largely suppressed, whereas the formation of amino acid from mesaconate and ammonia proceeded at an undiminished rate. This result indicated that an amino acid other than glutamate had been formed by the charcoal-treated extract. This was con
firmed by the observation that the amino acid was not decarboxylated by the specific L-glutamic decarboxylase of
E. coli.Later, a procedure was de
veloped for isolating several grams of the crystalline product of the enzymic reaction.
66The compound was shown to be an L-jS-methylaspartate, prob
ably the
h-threoisomer.
Following the isolation of 0-methylaspartate, the enzyme 0-methylaspar- tase that catalyzes the reversible conversion of the amino acid to mesac
onate and ammonia was purified about 45-fold.
67The enzyme was found to have a high substrate specificity, L-^reo-0-methylaspartate being the only amino acid decomposed at a rapid rate.
The equilibrium constant of the 0-methylaspartase reaction
_ [mesaconate] [ N H4 +]
e q' [j8-methylaspartate]
is 0.24 at pH 7.9 and 25°C. The equilibrium strongly favors the formation of mesaconate and ammonia at relatively low substrate concentrations (<0.01 M). Therefore the enzyme can be used as a convenient reagent for the quantitative estimation of L-//ire0-0-methylaspartate, using a spectro- photometric method based upon the ultraviolet light absorption of mesaco
nate at 240 πΐμ. At high substrate concentrations, the equilibrium favors the synthesis of the amino acid. Consequently the enzyme can also be used for the preparation of L-<Areo-/3-methylaspartate.
Another property of the enzyme that proved to be useful in studying the role of β-methylaspartate in glutamate fermentation is its sensitivity to inhibition by calcium ion. Relatively low concentrations of calcium ion, of the order of 0.01 M, cause a high degree of inhibition by competing with the essential magnesium ion. Addition of calcium ion to crude extracts can be used to inhibit β-methylaspartase specifically and cause the accumula
tion of L-#ireo-0-methylaspartate under appropriate conditions.
With the aid of the above information the role of L-£fereo-j3-methylas- partate as an intermediate between glutamate and mesaconate has been established. The formation of 0-methylaspartate from glutamate was dem
onstrated by decomposing glutamate with a crude extract in the presence of
0.015 Μ calcium ion and showing that β-methylaspartate accumulated to
a concentration 30 times that permitted by the 0-methylaspartase equi-
librium. I n the absence of calcium ion, t h e conversion of /3-methylaspartate t o mesaconate and ammonia is extremely rapid in relation to t h e rate of glutamate breakdown. Extracts usually contain sufficient β-methylaspar- tase to decompose 350 Mmoles of L-J/ireo-/3-methylaspartate per minute per milliliter at 37°C. Most extracts decompose glutamate at less t h a n one- t e n t h of t h a t rate.
T h e equilibrium constant for t h e reaction
L-glutamate ^ L-^reo-j8-methylaspartate (18) expressed by the equation
j£ [0-methylaspartate]
C Q' [L-glutamate]
is approximately 0.13 at p H 8. Since the equilibrium constant for t h e β- methylaspartase reaction is 0.24, t h e equilibrium constant for t h e over-all conversion of L-glutamate to mesaconate and ammonium ion [reaction (19)]
L-glutamate *-> mesaconate + N H4 (19) is 0.032. This corresponds to a standard free energy change (AF) a t p H 7.9
of 2.0 kilocalories. Despite this somewhat unfavorable standard free energy change, the reaction is pulled in the direction of glutamate decomposition b y t h e formation of two products and by the subsequent conversion of mesaconate to pyruvate and acetate via citramalate.
T h e rearrangement of the straight C6 chain of glutamate t o t h e branched chain of β-methylaspartate is a reaction of considerable interest because no similar reaction has been observed in other enzymic systems. T h e chemical mechanism of this rearrangement is not known, b u t the origin of t h e indi
vidual carbon atoms of β-methylaspartate has been established b y tracer experiments. Reference to Fig. 4 will facilitate interpretation of t h e results.
T h e numbering of the carbon atoms in compounds of Fig. 4 is based upon t h e previously mentioned tracer experiments, in which glutamate variously labeled with C1 4 was fermented by intact cells. T h e results, considered in relation to the known and relatively simple reactions from β-methylaspar- t a t e t o t h e final products, indicate t h a t the methyl group of β-methylas- p a r t a t e originates from carbon atom 3 of glutamate and is attached to car
bon atom 4 of glutamate. This conclusion was confirmed b y an experiment in which glutamate-4-C1 4 was converted to mesaconate, which was degraded chemically to establish the position of the isotope.6 8 T h e results proved t h a t t h e carbon atom adjacent t o t h e methyl group was derived exclusively from glutamate carbon a t o m 4. Since β-methylaspartate and mesaconate have identical carbon skeletons, t h e same conclusion applies t o 0-methylaspar- t a t e .
K S O O H
2^ Η Ν Η2
3C H2
4<^Η2
6i( ) O H
Glutamate 0-Methylaspartate
*ΟΟΟΗ
2C H N H2
3CH3—4H
6C O O H
± N H t
H O C M C —2C H C H3—* h —6C O O H
Mesaconate
3C H3—4C H2—3C H2—4C O O H Butyrate
5C 02 + 3C H3—4C O "
Acetyl
- 2 H
^ O O H ΪΟ Ο Ο Η
2(^H3 2^ Η2
+ ^ ± 3C H3—4i —6C O O H Ο
Η
3C H3—4C O —5C O O H Acetate
Pyruvate Citramalate FIG. 4. Path of glutamate fermentation.
From a knowledge of the correspondence of carbon atoms in glutamate and β-methylaspartate, the nature of the reaction involved in the intercon
version of the straight- and branched-chain carbon structures is apparent.
A cleavage m u s t occur between carbon atoms 2 and 3 of glutamate. T h e C2 group, consisting of glutamate carbon atoms 1 and 2, is then transferred, directly or indirectly, back to t h e C3 moiety so as to form a new bond be
tween carbon atoms 2 and 4. This leaves carbon atom 3 on the side in the methyl group. There is no evidence t h a t t h e C 3 chain consisting of carbon atoms 3, 4, and 5 is broken during the C2 transfer reaction.
T h e effect of charcoal t r e a t m e n t of crude extracts on the decomposition of glutamate has already been mentioned. This effect was shown to be caused b y the removal of an essential coenzyme. T h e coenzyme has recently been isolated from C. tetanomorphum and shown to be a yellow-orange com
pound which is rapidly inactivated b y exposure to visible light.6 9 I t is a derivative of pseudovitamin Bi 2 containing an additional molecule of ade
nine. T h e precise function of t h e coenzyme in the interconversion of gluta
m a t e and 0-methylaspartate has not been determined.
T h e decomposition of pyruvate and the formation of b u t y r a t e have not been studied extensively in the C. tetanomorphum system. However, the formation of hydrogen, carbon dioxide, and acetate from pyruvate under certain conditions suggests the functioning of a pyruvate-decomposing sys
tem similar to t h a t found in Clostridium butyricum 7 0 ·7 1 shown in reaction CH3COCOOH + HSCoA -» CH3COSCoA + C 02 + H2 (20)
(20). T h e further conversion of the acetyl group derived from p y r u v a t e t o b u t y r a t e m a y follow the p a t h used b y Clostridium kluyveri?1
So far the chemical reactions of glutamate fermentation have been in
tensively studied only in C. tetanomorphum. Several other species, includ
ing Micrococcus aerogenes, C. saccharobutyricum, C. tetani, C. botulinum, and Fusobacterium nucleatum, are known to ferment glutamate readily (Ta
ble I ) . Presumptive evidence for t h e mesaconate p a t h of glutamate decom
position is provided b y t h e observation t h a t these species, like C. tetano
morphum, form approximately one mole of carbon dioxide per mole of glutamate fermented. However, cells or cell extracts of Μ. aerogenes, grown on glutamate, were found not to decompose β-methylaspartate, mesaconate or DL-citramalate.2 2 This apparently eliminates these compounds from con
sideration as intermediates, b u t does not exclude participation of their co
enzyme A derivatives. Itaconyl-CoA and possibly mesaconyl-CoA are in
volved in t h e decomposition of itaconic acid b y r a t liver mitochondria.7 2
7. GLYCINE
T h e only anaerobic bacteria known to carry out a true fermentation of glycine are Diplococcus glycinophilus,21 Micrococcus anaerobius, and M.
variabilis.23 T h e Diplococcus and Micrococcus species m a y be more closely related t h a n t h e different generic names imply. All three organisms have a similar morphology and they form almost the same products.
T h e fermentation of glycine is described approximately b y equation (21).
4CH2NH2COOH + 2 H20 -> 4 N H3 + 2 C 02 + 3CH3COOH (21) I n addition t o the products shown, hydrogen gas is formed b y D. glycinoph
ilus in significant b u t variable amounts. T h e variability is determined b y the fact t h a t the hydrogen-producing reaction is readily reversible. H y d r o gen is evolved only when t h e partial pressure of t h e gas in t h e medium is low. At a pressure of about 0.3 atmosphere equilibrium is reached; a t higher pressures hydrogen is consumed. Because of these relations and t h e low solubility of hydrogen in water, a considerable evolution of hydrogen is observed only when t h e medium is shaken and the gas phase above t h e culture is large. T h e formation of hydrogen results in a n increased yield of carbon dioxide and a decreased yield of acetic acid. T h e two Micrococcus species do not form hydrogen from glycine.
Inspection of equation (21) suggests t h a t t h e fermentation of glycine consists of a dismutation in which the complete oxidation of one mole of glycine is coupled with the reduction of three moles of glycine to acetate and ammonia. Tracer experiments in which C1 4-labeled glycine was fermented by cell suspensions of D. glycinophilus have shown t h a t this interpretation is incorrect.7 3 W i t h glycine-l-C1 4 as substrate, C1 4 was found mainly in car-
bon dioxide b u t also in lesser amounts in b o t h carbons of acetate. T h e spe- cific activity of the carbon dioxide was almost the same as t h a t of the car- boxyl carbon of the substrate. W i t h glycine-2-C1 4 as substrate, both carbon atoms of acetate were strongly labeled whereas carbon dioxide was very weakly labeled. W i t h C 02- C1 4, b o t h carbons of acetate were again labeled.
These results show t h a t t h e carbon dioxide is derived mainly from t h e carboxyl carbon of glycine, whereas b o t h carbons of acetate are derived partly from t h e methylene carbon of glycine and partly from carbon dioxide.
T h e d a t a exclude both a complete oxidation of glycine t o carbon dioxide and a significant direct reduction of glycine to acetate. T h e y demonstrate t h a t t h e formation of acetate involves a condensation of the methylene carbon atoms of two molecules of glycine or a derivative thereof.
F u r t h e r elucidation of t h e actual p a t h of t h e glycine fermentation had t o await the development of concepts concerning t h e interconversions of gly- cine and serine in other organisms and the role of tetrahydrofolic acid in this process.7 4 On the basis of such concepts and b y the application of re- cently developed spectrophotometric techniques for detecting various de- rivatives of tetrahydrofolic acid ( T H F A ) , Sagers and Gunsalus7 5 obtained evidence t h a t crude cell-free extracts of D. glycinophilus catalyze the fol- lowing reactions, which m a y account for the conversion of glycine to pyru- v a t e .
Reaction (22) was not observed directly, b u t the formation of methylene- T H F A , a cyclic compound containing t h e Ci unit at the oxidation level of formaldehyde, was deduced from the fact t h a t C1 4-formaldehyde was formed on acidification of the reaction mixture, presumably b y chemical decompo- sition of the enzymic product. Furthermore, when t h e reaction was carried out in t h e presence of substrate a m o u n t s of triphosphopyridine nucleotide ( T P N ) , a more oxidized product, m e t h e n y l - T H F A , containing t h e Ci unit a t t h e oxidation level of formate, was found to accumulate. T h e immediate product of glycine oxidation would be expected t o be glyoxylic acid, which is known to be formed from glycine b y a Pseudomonas and an Achromobacter species. However, this compound has not yet been identified as an inter- mediate in t h e D. glycinophilus fermentation.
Reaction (23) represents t h e transfer of T H F A - b o u n d formaldehyde t o glycine with formation of serine. T h e occurrence of this reaction was de- duced from observations on t h e reverse reaction. T h e serine dehydrase re- action [reaction (24)] was observed directly. I n this connection it m a y be
C H2N H2C O O H + THFA ~2 H > (—CH2—THFA) + C 02 + N H3 (22) ( — C H2— T H F A ) + C H2N H2C O O H - > C H2O H C H N H2C O O H + THFA (23)
C H2O H C H N H2C O O H C H3C O C O O H + N H3 (24)
noted t h a t serine is decomposed m u c h more slowly t h a n glycine b y cell suspensions of D. glycinophilus probably because of a permeability barrier.
T h e above reactions result in the conversion of glycine-2-C1 4 to pyruvate- 2 , 3 - C1 4. P y r u v a t e is slowly decomposed by intact cells. Evidence for t h e oxidation of pyruvate to acetate and carbon dioxide by D. glycinophilus has not yet been presented. If this oxidation does occur, t h e p a t h of forma
tion of a c e t a t e - l , 2 - C1 4 and unlabeled carbon dioxide from glycine-2-C1 4 is clearly indicated, though not yet conclusively established.
T h e p a t h of glycine oxidation in D. glycinophilus appears to be identical with t h a t in the purine-fermenting Clostridia7 5 b u t quite different from t h a t in Pseudomonas species.7 6 Glycine is metabolized in an entirely different manner by organisms using the Stickland reaction.
T h e previously mentioned tracer experiments with intact cells demon
strated t h a t the partial oxidation of glycine is coupled with a reduction of carbon dioxide to acetic acid. T h e chemistry of this type of carbon dioxide reduction has not been elucidated. Carbon dioxide could be incorporated into the methyl carbon of acetate via formate, formyl-THFA, hydroxy- m e t h y l - T H F A , serine, and pyruvate. However, this p a t h does not account for the preferential incorporation of carbon dioxide carbon into t h e carboxyl group of acetate b y D . glycinophilus73 and Butyribacterium rettgeri77 W i t h the latter organism evidence has been obtained which indicates t h a t pyru
v a t e probably is not an intermediate in acetate synthesis.7 8 For example, when glucose was fermented in the presence of C1 402 under conditions per
mitting the accumulation of both pyruvate and acetate, it was found t h a t the specific activity of the methyl carbon atom of acetate was much higher t h a n t h a t of pyruvate. T h e chemistry of acetate synthesis from carbon di
oxide still needs extensive study.
D. glycinophilus decomposes several glycine-containing peptides with subsequent fermentation of the glycine moiety.2 1 T h e peptides decomposed most rapidly are hippuric acid, p-aminohippuric acid, DL-leucylglycine, DL- alanylglycine, diglycine, acetylglycine and hippurylglycine. T h e nonglycine moieties are not attacked. T h e peptidases responsible for the decomposition of peptides in this organism have not been studied.
8. HISTIDINE
This amino acid is fermented b y Clostridium, Fusobacterium, and Micro
coccus species (Table I ) . T h e fermentation products of three species are given in Table V. T h e p a t h s of histidine degradation b y these organisms appear to be similar to those used b y aerobic systems such as mammalian liver,7 9 Pseudomonasfluorescens,*0 and Aerobacter aerogenes*1'82 Most of t h e basic studies of t h e p a t h of histidine breakdown were done with aerobic organisms.
Clostridium tetano- Clostridium Micrococcus morphum63'83»84 tetani11 aerogenes22
Ammonia ~ 2 0 0 293 290
Carbon dioxide 63 68 103
Formamide 82 — 0
Formate 0 — 5
Hydrogen 16 0 0
Acetate 93 211 191
Butyrate 33 31 5
Lactate 0 — 35
α The figures give the yield of products in moles per 100 moles of substrate de
composed. The data were obtained with cell suspensions.
T h e first step in histidine breakdown by all of the above species is a de- amination to urocanate which can be readily detected and identified b y its C H — Ν C H — Ν
CH CH
C — N H
I
C H2
I
C H N H2
I
COOH Histidine
- N H i C — N H
I
CH CH
I
COOH Urocanate
+2H*o
COOH
I
C H N H — C H = N H
I
CH2 (25)
CH2
COOH
Formiminoglutamate characteristic ultraviolet absorption spectrum. This deamination, cata
lyzed b y t h e enzyme histidase, appears t o be irreversible. Formation of urocanate from histidine has been shown with C. tetanomorphum™ A.
aerogenes?1 and Micrococcus aerogenes.22 These bacteria, or enzymic prepara
tions derived from them, also degrade urocanate to the same products formed from histidine. T h e rate of urocanate breakdown under appropriate conditions is adequate to justify the assumption it is on the main p a t h of histidine decomposition.
Urocanate is further degraded t o L-a-formiminoglutamate b y an enzyme system called urocanase. Formiminoglutamate has been isolated as a prod
uct of urocanate decomposition in relatively crude enzyme preparations derived from mammalian liver7 9 and P. fluorescens*0 and its properties have been carefully compared with those of the synthetic compound. T h e evi
dence for the accumulation of formiminoglutamate in extracts of C. tetano- TABLE V
PRODUCTS OF HISTIDINE FERMENTATION*
Product