The Halogen Chemistry of the Actinides
K . W . B A G N A L L *
Atomic Energy Research Establishment, Chemistry Division, Harwell, England
1. Introduction . . 2. T h e Trivalent Actinides
A . General Chemistry B . Trifluorides C. Trichlorides D . Tribromides E . Tri-iodides F . Mixed Halides G. Oxyhalides
3. T h e Tetravalent Actinides A . General Chemistry B . Tetrafluorides C. Tetrachlorides D . Tetrabromides E . Tetraiodides F . Mixed H a l i d e s G. H a l o Complexes H . Oxyhalides
4. T h e P e n t a v a l e n t Actinides A . General Chemistry B . Pentafluorides C. I n t e r m e d i a t e Fluorides D . Pentachlorides . . E . P e n t a b r o m i d e s . . F . P e n t a i o d i d e s G. Mixed halides H . H a l o Complexes . . I. Oxyhalides
5. T h e H e x a v a l e n t Actinides A . General Chemistry B . Hexafluorides C. U r a n i u m Hexachloride D . Oxyhalides
References
304 306 306 307 309 311 313 314 314 315 315 316 319 326 327 328 329 335 337 337 338 339 340 342 343 343 343 347 351 351 352 358 359 367
* Present address: D e p a r t m e n t of Chemistry, U n i v e r s i t y of Manchester, E n g l a n d . 3 0 3
304 κ. w. B A G N A L L
E l e m e n t A c T h P a U N p P u A m
A t o m i c N o . 89 9 0 9 1 9 2 9 3 9 4 9 5
3 3 3 3 3
4 4 4 4 4 4
5 5 5 5 5
6 6 6 6
a T h e m o s t s t a b l e s t a t e i n a q u e o u s s o l u t i o n i s u n d e r l i n e d .
e x t e n d spatially i n t o t h e o u t e r valence regions of t h e a t o m a n d are m o r e accessible for b o n d i n g , which m a y involve 5/-, 6d', 7s- a n d 7^-orbitals.
As a result, t h e a c t i n i d e s form a wide v a r i e t y of complex species, i n c o n t r a s t t o t h e l a n t h a n i d e s in which t h e b o n d i n g is largely ionic. T h e heavier actinides a r e p r e d o m i n a n t l y t e r v a l e n t i n solution a n d i n t h e solid s t a t e , b u t n o definite c o m p o u n d s h a v e been recorded for t h e elements from einsteinium (99) t o l a w r e n c i u m which a r e a t p r e s e n t only available i n e x t r e m e l y m i n u t e q u a n t i t i e s .
T h e elements from u r a n i u m t o a m e r i c i u m a r e best r e g a r d e d as a n inner t r a n s i t i o n series, w i t h t h e c h e m i s t r y of t h e h e x a v a l e n t elements characterized b y t h e u n i q u e l y stable o x y g e n a t e d ions MO b o t h in solution a n d in solid c o m p o u n d s a n d , w i t h t h e e x c e p t i o n of americium, b y volatile hexafluorides MF^, t h e c h e m i s t r y of w h i c h shows some similarity t o t h a t of t h e g r o u p V I d-transition e l e m e n t hexafluorides WFg a n d MoFg. UClg, t h e only o t h e r k n o w n actinide h e x a h a l i d e , also resembles i t s t u n g s t e n analogue t o some e x t e n t . I n t h e p e n t a v a l e n t s t a t e t h e M Oi o n p r e d o m i n a t e s i n a q u e o u s m e d i a , i n c o n t r a s t t o t h e b e h a v i o u r of p r o t a c t i n i u m ( V ) , w i t h which hydrolysis a n d polymeriza
tion t o oxygen-bridged species occurs readily in t h e absence of com- plexing anions, a b e h a v i o u r r a t h e r similar t o t h a t of n i o b i u m a n d t a n t a l u m , a n d t h e r e is n o satisfactory evidence for t h e p r o t a c t i n y l ion, PaOg"^. H o w e v e r , t h e halide complexes of p e n t a v a l e n t p r o t a c t i n i u m a n d u r a n i u m resemble t h o s e formed b y n i o b i u m a n d t a n t a l u m e x c e p t
1. Introduction
T h e classification of t h e h e a v y elements from a c t i n i u m (89) t o lawrencium (103) as a s e c o n d / - t r a n s i t i o n series, t h e actinides, originally suggested b y Seaborg, is n o w well established. T h e earlier m e m b e r s of t h e g r o u p , u p t o a m e r i c i u m (95) exist i n a greater v a r i e t y of valency s t a t e s (Table I ) t h a n d o t h e l a n t h a n i d e s , largely because t h e 5/-electrons h a v e relatively lower b i n d i n g energies, a n d a r e less effectively shielded b y t h e o u t e r electrons, t h a n a r e t h e 4/-electrons. T h e 4/-electrons a r e n o t accessible for b o n d i n g i n t h e l a n t h a n i d e s , whereas t h e 5/-orbitals
TABLE I . O x i d a t i o n s t a t e s o f t h e U g h t e r a c t i n i d e s *
T H E H A L O G E N C H E M I S T R Y O F T H E A C T I N I D E S 305
t h a t t h e former c a n increase t h e i r a p p a r e n t c o o r d i n a t i o n n u m b e r t o 8 in b o t h fluoro a n d chloro complexes w h e r e a s t h e l a t t e r e x h i b i t t h e higher coordination only in t h e fluoro complexes, t h e chloro species being restricted t o 6-coordination.
I n t h e q u a d r i v a l e n t s t a t e t h e t h o r i u m halocomplexes show m a n y resemblances t o t h e i r u r a n i u m analogues, b u t t h e r e are some m a r k e d differences in t h e complexing b e h a v i o u r of t h e t e t r a h a l i d e s w i t h o x y g e n donor ligands. T h e c o m m o n e s t coordination n u m b e r s are 8 a n d 6, b u t 7 a n d 9 are also k n o w n . T h e t e r v a l e n t actinides b e h a v e in m u c h t h e s a m e w a y as t h e l a n t h a n i d e s , b u t w i t h m o r e evidence of complexing in a q u e o u s halogen acids a t high halide ion c o n c e n t r a t i o n s . I n view of t h e s e factors, t h e c h e m i s t r y of t h e halides h a s b e e n t r e a t e d in four sections, b y v a l e n c y s t a t e s r a t h e r t h a n e l e m e n t b y element.
T h e available d a t a on t h e complexing b e h a v i o u r of t h e a c t i n i d e halides indicates t h a t in all v a l e n c y s t a t e s t h e actinides c a n b e r e g a r d e d as n e a r l y p u r e C h a t t - A h r l a n d A - t y p e ions, fluoro complexing in e v e r y case being m u c h stronger t h a n w i t h t h e m o r e polarizable h e a v i e r halogens; as far as is k n o w n , o x y g e n donors, such as iV^iV^-dialkylamides, s u b s t i t u t e d p h o s p h i n e oxides a n d d i m e t h y l s u l p h o x i d e , form m o r e stable complexes t h a n simple n i t r o g e n donors. Studies of such com
plexes h a v e been largely restricted t o t h e lighter actinides a n d little s t r u c t u r a l i n f o r m a t i o n is available.
All of t h e actinides are r a d i o a c t i v e t o a g r e a t e r or lesser degree a n d m a n y of t h e m are therefore e x t r e m e l y t o x i c ; because of t h i s , w o r k w i t h a n y of t h e m , o t h e r t h a n t h o r i u m a n d u r a n i u m , m u s t be carried o u t u n d e r v e r y carefully controlled conditions in glove-boxes or similar enclosures. T h e t e c h n i q u e s u s e d for macroscale ( p r o t a c t i n i u m , n e p t u n i u m , p l u t o n i u m a n d americium) a n d microscale (actinium, c u r i u m (96) a n d later actinides) studies of t h e s e elements h a v e b e e n a d e q u a t e l y described in t h e reviews of t h e c h e m i s t r y of t h e actinides ( K a t z a n d Seaborg, 1957) a n d of t h e i r halides ( K a t z a n d Sheft, 1960), which also include e x t r e m e l y useful compilations of physical a n d t h e r m o d y n a m i c d a t a , which are n o t discussed in t h i s review. R a n d a n d K u b a s c h e w s k i (1963) h a v e also p u b l i s h e d a critical compilation of t h e t h e r m o c h e m i c a l properties of u r a n i u m c o m p o u n d s a n d o t h e r detailed reviews of t h e c h e m i s t r y of t h e g r o u p (Haissinsky, 1962) a n d of t h e fluorides in p a r t i c u l a r (Hodge, 1961 ; T a n a n a e v ei αί., 1961 ), as well a s c o m p r e h e n s i v e reviews of t h e earlier w o r k on t h e halides of t h o r i u m ( K a t z i n , 1954), u r a n i u m ( K a t z a n d R a b i n o w i t c h , 1951), n e p t u n i u m ( C u n n i n g h a m a n d H i n d m a n , 1954) a n d p l u t o n i u m ( C u n n i n g h a m , 1954) are also available.
T h e properties of u r a n i u m hexafluoride (De W i t t , 1960) a n d p l u t o n i u m hexafluoride (Steindler, 1963) h a v e also b e e n reviewed.
306 κ . w. BAGNALL
2. The Trivalent Actinides A. General chemistry
T h e stable o x i d a t i o n s t a t e for a c t i n i u m , a m e r i c i u m a n d t h e heavier actinide elements is + 3 , b o t h in solution a n d in solid c o m p o u n d s . T h e r e is n o evidence for t h i s o x i d a t i o n s t a t e for t h o r i u m or p r o t a c t i n i u m in a q u e o u s solution, or for t h e l a t t e r in solid c o m p o u n d s , a n d one would e x p e c t such species t o reduce w a t e r . A l t h o u g h t h e r e h a v e been a n u m b e r of r e p o r t e d p r e p a r a t i o n s of solid bipositive or tripositive t h o r i u m halides, b y w a y of r e d u c t i o n of t h e t e t r a h a l i d e w i t h t h o r i u m or alu
m i n i u m a t high t e m p e r a t u r e s , t h e results are often conflicting a n d it is o n l y r e c e n t l y t h a t t h e lower v a l e n c y t h o r i u m iodides h a v e become well established. I n a q u e o u s solution u r a n i u m ( I I I ) oxidizes readily, b u t n e p t u n i u m ( I I I ) a n d p l u t o n i u m ( I I I ) are a good deal m o r e stable in t h i s respect a n d t h e i r complexing b e h a v i o u r is easier t o investigate. M a n y of t h e d a t a on t h e halide complexing of t h e a c t i n i d e s ( I I I ) i n a q u e o u s solution are derived from work on m e t h o d s of s e p a r a t i n g t h e actinides from one a n o t h e r or from flssion p r o d u c t s . Such complexing in a q u e o u s solution is generally r a t h e r weak, as w i t h t h e l a n t h a n i d e s ( I I I ) , associa
t i o n w i t h halide ions being m a i n l y t h r o u g h electrostatic interactions.
I t seems t h a t halide ions can only displace t h e h y d r a t i o n w a t e r from t h e a c t i n i d e ( I I I ) ions in v e r y c o n c e n t r a t e d halide solutions; such stability c o n s t a n t d a t a as are k n o w n indicate t h a t t h e stabilities of t h e acti- n i d e ( I I I ) halo complexes a r e c o m p a r a b l e t o those of t h e l a n t h a n i d e s , a l t h o u g h s o m e w h a t m o r e stable t h a n t h e l a t t e r where /-electrons are involved in t h e b o n d i n g . S p e c t r o p h o t o m e t r i c evidence for t h e UCla"'" ion h a s b e e n o b t a i n e d (Shiloh a n d Marcus, 1962) a n d similar studies show t h a t n e p t u n i u m ( I I I ) , p l u t o n i u m ( I I I ) a n d a m e r i c i u m ( I I I ) (Shiloh a n d Marcus, 1962,1964,1966) form t h e ions M C P + a n d MCI2+. A m e r i c i u m ( I I I ) species of t h e s e t y p e s h a v e also been identified b y ion e x c h a n g e (Grenthe, 1962; P e p p a r d et aL, 1962), while e x t r a c t i o n of a m e r i c i u m ( I I I ) from solutions of high chloride c o n c e n t r a t i o n indicates t h e formation of anionic complexes such as AmCl4- or AmClg^- (Marcus et aL, 1964), as also i n d i c a t e d b y cation exchange studies ( D i a m o n d et aL, 1954;
Choppin a n d C h e t h a m - S t r o d e , 1960; Choppin a n d Dinius, 1962). T h e r e is some evidence t h a t t h e complex CmCP+ is m o r e stable t h a n P u C P + or AmCP+ ( W a r d a n d Welch, 1956) a n d t h e r e is q u a l i t a t i v e evidence showing t h a t t h e higher actinides t e n d t o form chloro complexes even m o r e readily (Choppin a n d C h e t h a m - S t r o d e , 1960; Choppin a n d Dinius, 1962; I s a a c et aL, 1960). B r o m i d e ion complexes m o r e w e a k l y t h a n chloride ion (Shiloh a n d Marcus, 1962, 1964) a n d t h e r e is n o evidence for iodo complexes. Anionic complexes of a n u m b e r of actinide trihalides
T H E H A L O G E N C H E M I S T R Y O F T H E A C T I N I D E S 307
«0 Co ( g c m- 3 )
A c F s W h i t e H e x a g o n a l , PQJmmc - 4 - 1 7 7-53 7-88
Dtj, ( L a F g t y p e ) U F 3 V i o l e t - r e d H e x a g o n a l , P6.Jmmc -
o r b l a c k D U ( L a F g t y p e ) 4 - 1 4 6 7 - 3 4 8 8-95
N p F 3 P u r p l e o r H e x a g o n a l , P6Jmmc -
b l a c k Dtn ( L a F g t y p e ) 4 - 1 0 8 7 - 2 7 3 9 - 1 2
P u F g P u r p l e H e x a g o n a l , PQ^jmrnc -
Dtn ( L a F g t y p e ) 4 - 0 8 7 7 - 2 4 0 9 - 3 2 A m F g ^ P i n k H e x a g o n a l , PQ^/mmc -
Dtn ( L a F g t y p e ) 4 - 0 6 7 7 - 2 2 5 9 - 5 3 CmFgto W h i t e H e x a g o n a l , P6^/mmc -
Dtn ( L a F g t y p e ) 4 - 0 4 1 7 - 1 7 9 9 - 7 0 a C o r r e c t e d d a t a c o l l e c t e d i n T a b l e I V o f t h e r e v i e w b y K a t z a n d S h e f t ( 1 9 6 0 ) u n l e s s s t a t e d o t h e r w i s e .
to A s p r e y et al. ( 1 9 6 5 a ) .
h a v e been identified in fused salt m e d i a a n d t e m p e r a t u r e - c o m p o s i t i o n d i a g r a m s for p l u t o n i u m trifluoride a n d trichloride i n alkali a n d alkaline e a r t h fluoride or chloride p h a s e s h a v e been s u m m a r i z e d (Leary, 1962),
T h e electronic configurations of t h e lighter actinides a r e still b y n o m e a n s certain, since t h e 6d- a n d 5/-electron energies a r e v e r y similar even a t u r a n i u m ; t h u s spectral a n d m a g n e t i c studies of t h e ion c a n b e i n t e r p r e t e d o n t h e basis of a S/^Bii^-configuration (Dawson, 1 9 5 1 ; J e z o w s k a - T r z e b i a t o w s k a , 1963) whereas t h e m a g n e t i c b e h a v i o u r of UCI3 in LaClg a p p r o a c h e s t h a t of N d( E t S 0 4 ) 3 .9 H 2 O a n d is consistent w i t h a 5/3-configuration ( H a n d l e r a n d H u t c h i s o n , 1956). 5/^-configurations for t h e higher actinides a r e , however, well established, for e x a m p l e b y m a g n e t i c studies (Dawson et al., 1951).
B. Trifluorides
T h e k n o w n actinide trifiuorides h a v e t h e LaFg t y p e s t r u c t u r e (Table I I ) , in w h i c h t h e central m e t a l a t o m lies o n a twofold axis a n d h a s 9 fluorine a t o m s a t n e a r l y equal distances (Zalkin et al., 1966). T h e y a r e insoluble in w a t e r a n d h y d r a t e d salts a r e p r e c i p i t a t e d from a q u e o u s solutions of t h e t e r v a l e n t actinides o n a d d i t i o n of hydrofluoric acid or a soluble fluoride. T h e a n h y d r o u s c o m p o u n d s a r e o b t a i n e d b y d r y i n g a slurry of t h e p r e c i p i t a t e d fluoride in c o n c e n t r a t e d hydrofluoric acid (CmFg a t 200°; F e a y , 1954) or h e a t i n g t h e p r e c i p i t a t e d fluoride in h y d r o g e n fluoride a t 400° (Asprey et al., 1965a), a n d b y h e a t i n g t h e
TABLE I I . C r y s t a l l o g r a p h i c d a t a f o r t h e a c t i n i d e t r i f l u o r i d e s ^
C o l o u r S y m m e t r y a n d L a t t i c e p a r a m e t e r s (A) C a l c u l a t e d
s p a c e g r o u p d e n s i t y
308 κ. w. BAGNALL
sesquioxides in hydrogen fluoride (AcFg at 700°, Fried et al., 1950, and AmFg at 650°, Fried, 1951; Westrum and Eyring, 1951). The more readily oxidized trifluorides (UFg, NpFg, PuFg) are usually prepared under reducing conditions; thus UFg is conveniently made b y the reduction of the tetrafluoride in the absence of water with hydrogen at
1000° (Spencer-Palmer, 1944), with aluminium in a vacuum at 900°, aluminium monofluoride subliming from the reaction vessel (Runnalls,
1953):
U F 4 + Al U F 3 + A l F t
and with finely divided uranium in an argon atmosphere at 1050°
(Warf, 1949):
3 U F 4 -f U ^ 4 U F 3
or with magnesium at 560° (reduction to uranium occurs at 600°;
Schwarz and Vaughan, 1953). A n oxyfluoride, and at high tempera
tures, an oxide, is formed if water vapour is present. Uranium trifluoride begins to disproportionate above 1000°; it precipitates silver from aqueous silver perchlorate and is oxidized to uranyl fluoride b y boiling water. Its magnetic properties have also been recorded (Nguyen-Nghi et al, 1964).
The neptunium and plutonium compounds are prepared b y the action of an equimolar mixture of hydrogen and hydrogen fluoride on the dioxides, NpFg at 500° (Fried and Davidson, 1948) and PuFg at 600° (Florin, 1949). The latter is also obtained b y heating almost any plutonium compound in hydrogen-hydrogen fluoride; thus pluto- nium(III) oxalate reacts at 550-660° (Reavis et αί.,1959). I t is also formed when hydrated plutonium tetrafluoride ( P u F 4 . 2-5ll20) is heated in a vacuum, apparently b y w a y of hydrolysis to the dioxide which then reacts with the tetrafluoride (Dawson et al., 1954b):
3PUF4 + PuOg ^ 4 PuFg + O2
and b y heating plutonium (III) or (IV) oxalate in dichlorodifluoro- methane (Freon 12) at 400-450° (Burger and Roake, 1952, 1961).
Phase diagrams describing the fluorocomplexes formed in fused salt media have been summarized in some detail (Leary, 1962). A few tetra- fluoro complexes have been prepared b y solid state reactions; thus the sodium plutonium (III) and americium (III) salts, N a M F 4 , have been made b y heating sodium fluoride with the actinide trifluoride, or sodium carbonate or fluoride with the actinide sesquioxide or dioxide in a stream of hydrogen fluoride either alone (MgOg) or m i x e d with hydrogen (MO2) at 450-650°. They are hexagonal, isostructural with NaLaF^ (Keller and Schmutz, 1964).
THE HALOGEN CHEMISTRY OF THE ACTINIDES 309
s p a c e g r o u p
Go Co
D e n s i t y (gcm~^)
A c C l g W h i t e H e x a g o n a l , C6Jm --Cln 7-62 4 - 5 5 4 - 8 1
UC13 R e d e H e x a g o n a l , G6Jm--Gin 7-442 4 - 3 2 0 5-51
N p C l g G r e e n ^ H e x a g o n a l , C6Jm--GQU 7 - 4 0 5 4 - 2 7 3 5-58
P u C l a E m e r a l d G r e e n H e x a g o n a l , CG^Im --Ce% 7 - 3 8 0 4 - 2 3 8 5-70
AmCl3i> P i n k H e x a g o n a l , CGJm--Gl, 7 - 3 9 0 4 - 2 3 4 5-78
C m C l g ^ W h i t e H e x a g o n a l , GQ^jm--Gin 7-368 4 - 2 2 8 5-81
a C o r r e c t e d d a t a c o l l e c t e d i n T a b l e I V o f t h e r e v i e w b y K a t z a n d S h e f t (1960) u n l e s s s t a t e d o t h e r w i s e .
^ A s p r e y et al. ( 1 9 6 5 a ) .
c A l s o d e s c r i b e d a s o l i v e g r e e n ( J o h n s o n et al., 1 9 5 8 ) .
^ F r e q u e n t l y d e s c r i b e d a s w h i t e , p r o b a b l y d u e t o a fine s t a t e o f s u b d i v i s i o n o f t h e c o m p o u n d .
b y the action of hydrogen chloride on t h e hydride at 250° (Johnson et al., 1958); reduction of the tetrachloride with hydrogen at 550-650°
under pressure or with metals such as zinc at 450-480° (Young, 1958) or with hydrogen iodide at 300-350° have also been used. I t dispro
portionates at 840° (Shchukarev et al., 1956b) and is a strong reducing agent, being oxidized b y water. Chlorine at 250° oxidizes it to the tetra
chloride, while bromine or iodine react t o form uranium (IV) mixed halides; uranyl chloride and uranium tetrachloride are formed when it is heated in air. I t is insoluble in acetone, carbon tetrachloride, chloroform.
B y analogy with europium, one would expect americium to form stable dihalides, but t h e only evidence for these t o date is a report of the reduction of americium(III) in a calcium fluoride matrix b y its own radiation or b y electrolysis at 600°, the presence of Am'^+ being shown by its electron spin resonance spectrum (ground state ^Srj/^ as against '^FQ for Am^+). Reoxidation occurs on heating at about 500° (Edelstein et al, 1966).
C. Trichlorides
The properties of the known actinide trichlorides are somewhat similar t o those of the lanthanide compounds; some crystallographic data are given in Table I I I . Actinium trichloride, a white solid, is pre
pared b y heating the hydroxide with ammonium chloride at 250° in a vacuum (Farr et al., 1953) or b y heating t h e hydroxide or oxalate in carbon tetrachloride at higher temperatures (Fried et al., 1950); it sublimes in a vacuum at 960°. The uranium compound is best prepared
TABLE I I I . C r y s t a l l o g r a p h i c d a t a f o r t h e a c t i n i d e t r i c h l o r i d e s *
C o l o u r S y m m e t r y a n d L a t t i c e p a r a m e t e r s (A) C a l c u l a t e d
310 κ . w. BAGNALL
pyridine a n d n o n p o l a r solvents a n d r e a c t s w i t h gaseous a m m o n i a a t 450-500° t o form t h e amidochlorides UNHgCla a n d U(NH2)2C1. A t higher t e m p e r a t u r e s (^^800°) these c o m p o u n d s decompose t o form t h e imidochloride a n d t h e nitride U N 1. 7 3_ 1. 7 5 (Berthold a n d K n e c h t , 1965a). T h e imidochloride h a s t h e s a m e s t r u c t u r e as t h e oxychloride (Berthold, 1966).
N e p t u n i u m trichloride is p r e p a r e d b y chlorination of t h e dioxide w i t h a m i x t u r e of h y d r o g e n a n d c a r b o n t e t r a c h l o r i d e a t 350-400° or b y r e d u c t i o n of t h e t e t r a c h l o r i d e w i t h h y d r o g e n a t 450° (Fried a n d D a v i d s o n , 1948) or a m m o n i a a t 350-400° (Sheft a n d F r i e d , 1953) a n d it is also formed in t h e p r e p a r a t i o n of n e p t u n i u m t e t r a c h l o r i d e b y chlor
ination of t h e dioxide w i t h hexachloropropene, p r o b a b l y as a result of r e d u c t i o n of t h e t e t r a c h l o r i d e b y carbonaceous m a t e r i a l (Bagnall a n d Laidler, 1966). T h e p l u t o n i u m c o m p o u n d is m a d e on t h e milligram scale b y reaction of t h e dioxide w i t h carbonyl chloride a t 500-850° (Ras- m u s s e n a n d H o p k i n s , 1961; B o r e h a m et al,, 1960); reaction is appreci
able even a t 350-400° (Tolley, 1953). I t is also formed b y t h e a c t i o n of carbon t e t r a c h l o r i d e on t h e dioxide a t 450-500° ( F o m i n et al,, 1958a) b u t for g r a m scale p r e p a r a t i o n s t h e action of carbonyl chloride on p l u t o n i u m ( I I I ) c a r b o n a t e a t 500-550° or of h y d r o g e n chloride on p l u t o n i u m ( I I I ) or p l u t o n i u m ( I V ) o x a l a t e a t 140-500° is m o r e effective (Boreham et al,, 1960). R e a c t i o n of a m i x t u r e of h y d r o g e n a n d h y d r o g e n chloride w i t h oxalates (Garner, 1959) or of h e x a c h l o r o p r o p e n e w i t h p l u t o n i u m ( I I I ) o x a l a t e a t 180-190° (Christensen a n d Mullens, 1952;
H a r d e r et al,, 1958) a n d of h y d r o g e n chloride w i t h p l u t o n i u m h y d r i d e ( A b r a h a m et al., 1949; R e a v i s et al,, 1959) or direct p r e p a r a t i o n from t h e elements ( A b r a h a m et al,, 1949) are also satisfactory.
T h e p l u t o n i u m c o m p o u n d can also be m a d e b y h e a t i n g t h e dioxide m i x e d w i t h carbon, s u l p h u r or p h o s p h o r u s in chlorine, b y t h e action of s u l p h u r dichloride a n d chlorine on t h e dioxide a t 800°, b y t h e action of p h o s p h o r u s pentachloride on t h e dioxide or b y liquid p h a s e chlorination of p l u t o n i u m peroxide w i t h s u l p h u r monochloride a n d chlorine a t 280°
(Davidson a n d K a t z , 1958). T h e r m a l decomposition of d i p y r i d i n i u m h e x a c h l o r o p l u t o n a t e ( I V ) in a v a c u u m a t 390-600° or in argon a t 220-470° does n o t yield a p u r e p r o d u c t ( H a r d e r et al,, 1958), a n d t h e action of h y d r o g e n chloride on p l u t o n i u m dioxide a t 200-1000° yields a m i x t u r e of trichloride a n d oxychloride (Davidson a n d K a t z , 1960). T h e green a n h y d r o u s c o m p o u n d is hygroscopic, forming t h e blue hexa- h y d r a t e , which is i s o m o r p h o u s w i t h t h e n e o d y m i u m c o m p o u n d (David
son a n d K a t z , 1960).
T h e r m a l analysis of t h e PUCI3-KCI p h a s e d i a g r a m indicates t h e existence of KaPuClg (m.p. 685°) a n d , possibly, KaPuClg (Benz et al.,
T H E H A L O G E N C H E M I S T R Y O F T H E A C T I N I D E S 311
1959); KaUClg and K2UCI5 have been identified in the UCI3-KCI system (Kraus, 1943) but there is no evidence of compound formation in the PuCla-LiCl and PuCla-NaCl systems (Bjorklund et al, 1959). Similarly, there is evidence for Rb3(Cs3)PuCl6, Rb(Cs)Pu2Cl7 and RbgPuCls in the PuCl3-RbCl and PUCI3-CSCI fused salt systems (Benz and Douglass, 1961b) and for the formation of complexes of the type M3PUCI9 (M = Sr, Ba) under similar conditions, but there is no evidence of compound formation with magnesium or calcium chlorides (Johnson, K.W.R.
et al, 1961). The caesium chloro complex, CS3PUCI6.2H2O, has been isolated from 6 N hydrochloric acid solution of plutonium (III) in the presence of an excess of caesium chloride; it melts in air at 100° with oxidation of the plutonium to the dioxide (Stevens, 1965). Americium- (III) behaves in a more complex manner than plutonium(III), the hydrated complex CsAmCl4.4H20 being obtained from l l M hydro
chloric acid; in the presence of sodium chloride, however, the hexa
chloro complex, CsgNaAmClg, is obtained (Bagnall et al, 1967c). The anhydrous triphenylphosphonium salt (Ph3PH)3AmCl6, crystallizes from alcoholic hydrochloric acid, and the visible spectrum of its solution shows that the AmClg^" group is octahedral (J. L. Ryan, personal communication).
Americium trichloride is prepared by the reaction of the dioxide with carbon tetrachloride at 800° to 900° (Fried, 1951 ; Hall and Markin,1957) or with hydrogen chloride (Broido and C u n n i n g h a m , 1950). It sublimes in a vacuum at 850°. Both americium and curium trichlorides, however, are most easily made by evaporating to dryness a hydrochloric acid solution of the trivalent element containing ammonium chloride and subliming the last from the residue (Asprey et al, 1965a). The califor
nium compound has been made on the submicrogram scale by the action of hydrogen chloride on the sesquioxide at 450° (Cunningham, 1961).
D. Tribromides
Most of the actinide tribromides (Table IV) have been made by heating the oxide at moderate temperatures with aluminium bromide, formed in situ from the elements; thus AcBr3 is formed at 750° ( F r i e d et al, 1950), N p B r g from the dioxide at 3 5 0 - 4 0 0 ° in the presence of excess aluminium to reduce the tetrabromide which is formed (Fried and Davidson, 1948) and A m B r 3 from the dioxide at 500° ( F r i e d , 1951).
Americium and curium tribromides are, however, most easily prepared by heating the trichlorides with ammonium bromide at 4 0 0 - 4 5 0 ° in hydrogen (Asprey et al, 1965a).
Thorium tribromide is said to be formed by reduction of the tetra
bromide by hydrogen at 360° in the absence of moisture (Shchukarev
312 κ . w. BAGNALL
A c B r g W h i t e H e x a g o n a l C6Jm — C i » 8-06 4 - 6 8 5 - 8 5
U B r g R e d H e x a g o n a l C6Jm —Glj^ 7-942 — 4:'U0 6-53
a - N p B r g G r e e n H e x a g o n a l CQJm—C^j^ 7-917 — 4 - 3 8 2 6 - 6 2
j g - N p B r g G r e e n O r t h o r h o m b i c Cmcm — DU 4 - 1 1 1 2 - 6 5 9 - 1 5 6 - 6 2 P u B r g G r e e n O r t h o r h o m b i c Cmcm — DU 4 - 0 9 1 2 - 6 2 9 - 1 3 6 - 6 9 A m B r g t o W h i t e O r t h o r h o m b i c Cmcm —DU 4 - 0 6 4 1 2 - 6 6 9 - 1 4 4 6-79 CmBrgto W h i t e O r t h o r h o m b i c Cmcm — D\l 4 - 0 4 8 1 2 - 6 6 9 - 1 2 4 6-87
* C o r r e c t e d d a t a c o l l e c t e d i n T a b l e I V o f t h e r e v i e w b y K a t z a n d S h e f t (1960) u n l e s s s t a t e d o t h e r w i s e .
t> A s p r e y et al. ( 1 9 6 5 a ) .
et al., 1956a) but this claim, hke others for thorium trichloride and tri
bromide, is very doubtful. Uranium tribromide is made b y the action of hydrogen bromide on t h e hydride at about 300° (Spedding et al., 1958) and, less satisfactorily, b y hydrogen reduction of t h e tetrabromide at 600-700° and b y reaction of stoicheiometric quantities of the elements a t about 570° (Eastman et al., 1958a). I t disproportionates above 900° and is more hygroscopic than t h e trichloride; it dissolves in water with t h e evolution of hydrogen and with dry ammonia gas forms t h e ammine U B r g. e N H g . Uranium tribromide is insoluble in non-polar solvents and dissolves in, or reacts with, polar solvents and cannot be recovered from its solutions unchanged (Spedding et al., 1958). Uranium is soluble in t h e molten tribromide b u t lower halides have n o t been isolated (Eastman et al., 1958b; Corbett et al., 1963). I t reacts with glass or quartz at high temperatures, forming the tetrabromide, dioxide and disilicide, t h e last presumably being formed b y reaction of silica with uranium metal liberated in t h e disproportionation. Plutonium tribro
mide is best made from t h e elements at 300° (Davidson et al., 1949) or b y t h e action of hydrogen bromide on the hydride at 600° (Reavis et al., 1 9 5 9 ) or on plutonium(IV) oxalate hexahydrate at 500° (Fomin et al., 1958b). The reaction of plutonium dioxide with hydrogen bromide, or with a mixture of carbon monoxide and bromine, is never quantitative even at temperatures above 800°. I t is also reported t o be formed b y reaction of plutonium dioxide with bromine and sulphur bromides (Davidson and Katz, 1 9 5 8 ) and b y evaporating a solution of plutonium- (IV) hydroxide in 5M hydrobromic acid t o dryness in a stream of hydrogen bromide and subsequent heating in hydrogen bromide at 300°
or with ammonium bromide at 350° and 10~^ m m (Davidson and H y d e ,
TABLE I V . C r y s t a l l o g r a p h i c d a t a f o r t h e a c t i n i d e t r i b r o m i d e s *
C o l o u r S y m m e t r y a n d s p a c e g r o u p L a t t i c e p a r a m e t e r s (A) C a l c u l a t e d d e n s i t y
«0 60 C O ( g c m- 3 )
T H E H A L O G E N C H E M I S T R Y O F T H E A C T I N I D E S 313
U I 3 B l a c k O r t h o r h o m b i c , Cmcm- 4 - 3 2 1 4 - 0 1 1 0 - 0 1 6 - 7 6 N p l a B r o w n O r t h o r h o m b i c , Cmcm - 4 - 3 0 1 4 - 0 3 9 - 9 5 6-82 P u l 3 ^ B r i g h t
g r e e n
O r t h o r h o m b i c , Cmcm-'DU 4 - 3 3 1 3 - 9 5 9 - 9 6 6-92
A m l s ^ . c Y e l l o w H e x a g o n a l , Β'^-ΟΙ^ 7-42 — 2 0 - 5 5 6 - 0 4
Cml3c W h i t e H e x a g o n a l , 7?--σ|^ 7-44 — 2 0 - 4 6-37 a C o r r e c t e d d a t a c o l l e c t e d i n T a b l e I V o f t h e r e v i e w b y K a t z a n d S h e f t ( 1 9 6 0 ) u n l e s s o t h e r w i s e s t a t e d .
i> A s p r e y et al. ( 1 9 6 4 ) . c A s p r e y et al. ( 1 9 6 5 a ) .
formed b y r e a c t i o n of t h e h y d r i d e w i t h m e t h y l iodide a t 275-300°
(Ayres, 1944) or w i t h iodine v a p o u r (Corbett et al., 1963), b y r e d u c t i o n of t h e t e t r a i o d i d e in h y d r o g e n ( K a t z a n d R a b i n o w i t c h , 1951, p . 538) a n d from stoicheiometric q u a n t i t i e s of t h e e l e m e n t s a t 700-750°
( P o p o v a n d Senin, 1957) or a t 525° a t low pressure (Gregory, 1944), a l t h o u g h r e a c t i o n of iodine v a p o u r w i t h t h e massive m e t a l is e x t r e m e l y slow (Corbett et al., 1963). U r a n i u m tri-iodide m e l t s a t 766-5° ( P o p o v a n d Senin, 1957) a n d a t t a c k s glass a t 800° (Ayres, 1944). T h e p l u t o n i u m c o m p o u n d is formed b y h e a t i n g t h e m e t a l in h y d r o g e n iodide a t 450°
( H a g e m a n n et al., 1949) or w i t h m e r c u r i c iodide a t 500° i n a sealed t u b e (Asprey et al., 1964).
T h e earlier, a n d conflicting, r e p o r t s of t h e p r e p a r a t i o n of t h o r i u m tri-iodide a n d di-iodide h a v e n o w been resolved b y r e c e n t w o r k w h i c h shows t h a t a b l a c k tri-iodide, which m a y c o n t a i n t h e Th^+ ion, is
1958). V e r y little is k n o w n a b o u t t h e s e c o m p o u n d s , or a b o u t t h e cor
responding iodides, a p a r t from p r e p a r a t i v e details, crystallographic d a t a (Tables I V a n d V) a n d m a g n e t i c d a t a for UBrg a n d UI3 (Dawson, 1951).
E. Tri-iodides
T h e actinide tri-iodides a r e usually m a d e b y r e a c t i o n of a l u m i n i u m iodide or a m m o n i u m iodide w i t h t h e oxide; Aclg, a l t h o u g h n o t definitely identified, a p p e a r s t o b e formed a t 500-700° (Pried et al., 1950), N p l g from t h e dioxide a n d a l u m i n i u m iodide a t 350-400° (Fried a n d D a v i d s o n , 1948) a n d A m l g similarly a t 500° (Fried, 1951) or b y h e a t i n g t h e t r i chloride w i t h a m m o n i u m iodide a t 400° in h y d r o g e n , a m e t h o d w h i c h is equally applicable t o t h e c u r i u m c o m p o u n d . A m e r i c i u m tri-iodide is n o t r e d u c e d b y h y d r o g e n a t high t e m p e r a t u r e s (Asprey et al., 1965a).
Crystallographic d a t a a r e s u m m a r i z e d i n T a b l e V . U r a n i u m tri-iodide is
TABLE V . C r y s t a l l o g r a p h i c d a t a f o r t h e a c t i n i d e t r i - i o d i d e s *
C o l o u r S y m m e t r y a n d s p a c e g r o u p L a t t i c e p a r a m e t e r s (A) C a l c u l a t e d d e n s i t y
«0 Κ Co (gcm-3)
314 κ . w. B A G N A L L
formed b y t h e r e d u c t i o n of t h e t e t r a i o d i d e w i t h t h o r i u m m e t a l i n t a n t a l u m or p l a t i n u m vessels. I t is n o t , however, isomorphous w i t h t h e u r a n i u m c o m p o u n d . T w o forms of t h e di-iodide h a v e also been identified, likewise m a d e b y r e d u c t i o n of t h e t e t r a i o d i d e w i t h t h o r i u m m e t a l ; t h e dull black α form is o b t a i n e d a t 600°, b u t t h e r e a c t i o n is n e v e r complete, a n d t h e golden β form is o b t a i n e d a t 800°. T h e χ to β t r a n s f o r m a t i o n occurs sluggishly a t 600-700°; b o t h a r e of hexagonal s y m m e t r y . All t h r e e c o m p o u n d s d i s p r o p o r t i o n a t e a t higher t e m p e r a t u r e s a n d decom
pose w a t e r vigorously w i t h t h e evolution of h y d r o g e n (Scaife a n d W y l i e , 1964).
F. Mixed halides
M a n y u r a n i u m ( I I I ) m i x e d halides h a v e been recorded, p r e p a r e d b y t h e r m a l decomposition or h y d r o g e n r e d u c t i o n of t h e u r a n i u m ( I V ) m i x e d halides, a n d b y fusing t h e stoicheiometric q u a n t i t i e s of t h e tri
halides; a detailed a c c o u n t of t h e s e is given b y Gregory (1958).
G. Oxyhalides
M a n y of t h e actinide trihalides a r e c o n v e r t e d t o oxyhalides b y v a p o u r p h a s e hydrolysis, t h e p r o d u c t s usually being identified b y X - r a y crystallography (Table V I ) ; m o s t of t h e m a r e k n o w n t o b e insoluble in
TABLE V I . C r y s t a l l o g r a p h i c d a t a f o r t h e a c t i n i d e ( I I I ) o x y h a l i d e s
C o l o u r S y m m e t r y a n d s p a c e L a t t i c e p a r a m e t e r s ( A ) C a l c u l a t e d g r o u p o r s t r u c t u r e t y p e
« 0 bo Co
d e n s i t y ( g c m- 3 )
A c O F a W h i t e C u b i c , C a F a 5-94 8-28
P u O F i > M e t a l l i c T e t r a g o n a l , P b F C l 4 - 0 5 — 5-72 9 - 7 0 ( P 4 / n m m - Z ) i f t )
A c O C l a W h i t e T e t r a g o n a l , P b F C l 4 - 2 5 — 7-08 7-23
{Pélnmm-Dlj,)
U O C l c R e d T e t r a g o n a l , P b F C l 4 - 0 0 — 6 - 8 5 8-78
P u O C l d G r e e n o r T e t r a g o n a l , P b F C l 4 - 0 1 — 6-79 8-8
b l u e - g r e e n {P4:lnmm-Dl^)
A m O C l e W h i t e T e t r a g o n a l , P b F C l 4 - 0 0 — 6-78 8-96
( P 4 / n m m - D Î , )
A c O B r a W h i t e T e t r a g o n a l , P b F C l 4 - 2 8 — 7-41 7-9
{P4:lnmm-Dlj,)
P u O B r d D e e p G r e e n T e t r a g o n a l , P b F C l 4 - 0 2 — 7-57 9-1
{P4:lnmm — Dlj,)
P u O I d B r i g h t G r e e n T e t r a g o n a l , P b F C l 4 - 0 4 — 9-17 8-5 {P4:lnmm-Dlj,)
a F r i e d et al. ( 1 9 5 0 ) .
^ Z a c h a r i a s e n ( 1 9 5 1 ) .
c S h c h u k a r e v a n d E f i m o v ( 1 9 5 7 ) .
d Z a c h a r i a s e n ( 1 9 4 9 a ) .
e T e m p l e t o n a n d D a u b e n ( 1 9 5 3 ) .
THE HALOGEN CHEMISTRY OF THE ACTINIDES 315
w a t e r , b u t soluble in dilute acids. T h e a c t i n i u m c o m p o u n d s are m a d e by- hydrolysis w i t h a m m o n i a a n d w a t e r v a p o u r , A c O F a n d AcOCl being o b t a i n e d a t 900-1000°, A c O B r a n d A c O I a t 500°. A c O F , in c o n t r a s t t o L a O F , c a n n o t be m a d e b y h e a t i n g t h e trifluoride in air (Fried et at.,
1950). T h e only recorded u r a n i u m ( I I I ) oxyhalide is t h e red chloride;
this r e m a i n s in t h e residue, m i x e d w i t h u r a n i u m dioxide, left w h e n u r a n i u m trichloride is sublimed; it is s e p a r a t e d from t h e oxide b y dé
c a n t a t i o n in w a t e r , t o which it is q u i t e inert, in c o n t r a s t t o t h e u r a n i u m trihalides ( S h c h u k a r e v a n d Efimov, 1957).
N e p t u n i u m ( I I I ) oxyhalides are n o t k n o w n , p r e s u m a b l y because t h e y h a v e n o t been sought, b u t all four p l u t o n i u m ( I I I ) c o m p o u n d s h a v e b e e n described. T h e oxyfluoride was observed w h e n t h e trifluoride was m e l t e d in argon, p r e s u m a b l y a result of t h e presence of traces of w a t e r (Robinson, 1944); t h e chloride h a s been m a d e b y h e a t i n g t h e h y d r a t e d trichloride in a sealed t u b e a t 400° ( A b r a h a m et al., 1949) a n d b y h y d r o lysis of t h e trichloride w i t h a m i x t u r e of h y d r o g e n , w a t e r v a p o u r a n d h y d r o g e n chloride a t 400-520° (Davidson a n d K a t z , 1960). T h e b r o m i d e is o b t a i n e d b y hydrolysis of t h e t r i b r o m i d e a t 400° ( D a \ â d s o n et al.,
1949) or b y h e a t i n g t h e dioxide in moist h y d r o g e n b r o m i d e a t 750°
(Sheft a n d D a v i d s o n , 1949), a n d t h e iodide b y h e a t i n g dried p l u t o - n i u m ( I V ) h y d r o x i d e w i t h h y d r o g e n a n d h y d r o g e n iodide a t 750° (Hage
m a n n et al. 1949).
Americium ( I I I ) oxychloride is o b t a i n e d b y v a p o u r p h a s e hydrolysis of t h e trichloride or b y h e a t i n g t h e sesquioxide in a m i x t u r e of h y d r o g e n chloride a n d w a t e r v a p o u r a t 500° ( K o c h a n d C u n n i n g h a m , 1954), a reaction which, a t 450°, h a s been used t o p r e p a r e t h e californium c o m p o u n d (Cunningham, 1961).
3. The Tetravalent Actinides A. General chemistry
Tetrafluorides are k n o w n for all t h e actinides from t h o r i u m t o c u r i u m inclusive a n d , b y a n a l o g y w i t h t h e l a n t h a n i d e s , b e r k e l i u m should also form a tetrafluoride; c o m p o u n d s w i t h halogens of higher a t o m i c n u m b e r , however, become increasingly less s t a b l e on passing u p t h e series from t h o r i u m . T h u s t h e simple tetrachlorides a n d t e t r a bromides of p l u t o n i u m a n d of t h e higher actinides are u n k n o w n , a l t h o u g h complexes derived from b o t h p l u t o n i u m t e t r a c h l o r i d e a n d t e t r a b r o m i d e can be p r e p a r e d . P l u t o n i u m trichloride (Benz, 1962) a n d t r i b r o m i d e ( F o m i n et al., 1958b) b e c o m e a p p r e c i a b l y m o r e volatile in t h e presence of t h e a p p r o p r i a t e halogen, which suggests t h a t t h e s e t e t r a h a l i d e s m a y exist in t h e v a p o u r p h a s e u n d e r such conditions.
316 κ . w. B A G N A L L
Tetraiodides of n e p t u n i u m a n d of t h e higher actinides do n o t exist a n d e v e n u r a n i u m t e t r a i o d i d e is relatively u n s t a b l e t o h e a t .
T h e tetrafluorides are all insoluble in w a t e r a n d are n o t appreciably hygroscopic, whereas t h e o t h e r t e t r a h a l i d e s are all v e r y hygroscopic, readily forming h y d r a t e s in moist air. T h e q u a d r i v a l e n t actinides can be r e g a r d e d as nearly p u r e C h a t t - A h r l a n d A - t y p e elements, t h e fluoro complexes being t h e m o s t stable, a n d t h e iodo complexes t h e least stable of t h e halo complexes; in general, oxygen donor ligands a p p e a r t o form complexes w i t h t h e t e t r a h a l i d e s m o r e readily t h a n nitrogen d o n o r ligands a n d simple p h o s p h o r u s a n d s u l p h u r d o n o r ligands do n o t form stable complexes.
T h e ions of p r o t a c t i n i u m a n d t h e higher actinides all h a v e t h e 5/^-configuration, established from a b s o r p t i o n spectroscopy of t h e t e t r a h a l i d e s (J0rgensen, 1959; Pa^+—Fried a n d H i n d m a n , 1954; A x e et al., 1960; — J e z o w s k a - T r z e b i a t o w s k a a n d B u k i e t y n s k a , 1961), m a g n e t i c susceptibility d a t a (U^+—Dawson, 1951; Jezowska-Trzebia
t o w s k a , 1963) a n d p a r a m a g n e t i c resonance a b s o r p t i o n (Pa^+—Axe et al., 1961). T h e general m e t h o d s used for t h e p r e p a r a t i o n of t h o r i u m (Katzin, 1954) a n d u r a n i u m ( K a t z a n d R a b i n o w i t c h , 1951; Gregory, 1958) t e t r a h a l i d e s h a v e been e x h a u s t i v e l y reviewed.
B. Tetrafluorides
T h o r i u m tetrafluoride h a s been m a d e b y t h e reaction of t h e m e t a l with h y d r o g e n fluoride, for e x a m p l e in a sealed t u b e a t 225° (Muetterties a n d Castle, 1961) or b y reaction of fluorine w i t h t h e t e t r a c h l o r i d e or t e t r a b r o m i d e a t r o o m t e m p e r a t u r e (Moissan a n d Martinsen, 1905) a n d b y t h e action of h y d r o g e n fluoride on t h o r i u m h y d r i d e a t 250-350°
(Lipkind a n d N e w t o n , 1952), which is t h e e q u i v a l e n t t o t h e use of t h e finely divided m e t a l because of t h e r m a l decomposition of t h e h y d r i d e , or on t h e t e t r a b r o m i d e a t 350-400° (Chauvenet, 1911). I t is m o r e con
veniently m a d e b y reaction of h y d r o g e n fluoride w i t h t h e low-fired dioxide a t 550° ( N e w t o n et al., 1952) or w i t h a n h y d r o u s t h o r i u m a c e t a t e (Gentile a n d Snyder, 1957) a n d b y d e h y d r a t i o n of t h e tetrafluoride h y d r a t e s (ThF4.2-5H20, ThF4.0-5H2O) a t 250-300°, usually in a v a c u u m ( D ' E y e a n d B o o t h , 1955, 1957; Gagarinskii a n d Mashirev, 1959a). R e a c t i o n of dichlorodifluoromethane (Freon 12) w i t h t h o r i u m dioxide a t 330-400°, or w i t h t h e h y d r a t e d tetrafluoride or a m m o n i u m fluoro complexes a t 350-500°, provides a useful a l t e r n a t i v e r o u t e t o t h e a n h y d r o u s tetrafluoride (Cacciari et al., 1956, 1957).
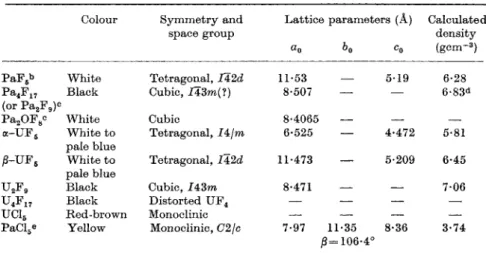
A n h y d r o u s p r o t a c t i n i u m tetrafluoride, a reddish b r o w n solid, is o b t a i n e d b y hydrofluorination of t h e dioxide or h y d r a t e d p e n t o x i d e (Sellers et al., 1954) a n d b y t h e action of a n equimolar m i x t u r e of
T H E H A L O G E N C H E M I S T R Y O F T H E A C T L N I D E S 317
C o l o u r S j r m m e t r y a n d L a t t i c e p a r a m e t e r s (A) C a l c u l a t e d
s p a c e g r o u p d e n s i t y
Κ Co ( g c m- 3 )
T h F ^ W h i t e M o n o c l i n i c , C210—0^2 η 1 3 1 1 1 0 1 8-6 5-71
α 2= 1 2 6 ± 1 °
G r e e n M o n o c l i n i c , C2lc—C^ji 1 2 - 7 3 1 0 - 7 5 8-43 6 - 7 0 a a = 1 2 6 ° 2 0 '
N p F , G r e e n M o n o c l i n i c , C2lc—G^ji 1 2 - 7 0 1 0 - 6 4 8-41 6-8 α 2- 1 2 6 ° 1 0 '
P U F 4 B r o w n M o n o c l i n i c , C2lc--G92h M o n o c l i n i c , C2lc--G92h 12-62 1 0 - 5 7 8-28 7-0 α 2= 1 2 6 ° 1 0 '
A m F ^ T a n M o n o c l i n i c , (72/c—(7f ^ 1 2 - 4 9 10-47 8 - 2 0 7-34
α 2= 1 2 6 ° 1 0 '
C m F i G r e e n i s h - M o n o c l i n i c , C2lc—Clf^ 1 2 - 4 5 1 0 - 4 5 8-16 7-49 t a n
M o n o c l i n i c , C2lc—Clf^
α 2= 1 2 6 ° ± 3 0 '
a C o r r e c t e d d a t a c o l l e c t e d i n T a b l e I V o f t h e r e v i e w b y K a t z a n d S h e f t ( 1 9 6 0 ) u n l e s s o t h e r w i s e s t a t e d .
to L a r s o n et al. ( 1 9 6 4 ) .
h y d r o g e n a n d h y d r o g e n fluoride o n t h e p e n t o x i d e a t 500° (Stein, 1964).
I t is slowly h y d r o l y s e d i n moist air a n d is isomorphous w i t h t h e u r a n i u m c o m p o u n d .
U r a n i u m tetrafluoride is m a d e i n m u c h t h e s a m e w a y a s t h e t h o r i u m c o m p o u n d , b y h e a t i n g t h e m e t a l w i t h liquid h y d r o g e n fluoride i n a sealed t u b e a t 250° (Muetterties a n d Castle, 1961), b y t h e action of h y d r o g e n fluoride o n t h e dioxide (e.g. D a w s o n et al,, 1954a), a process used o n t h e industrial scale ( K u h l m a n a n d Swinehart, 1958) a n d b y t h e reaction of t h e dioxide w i t h s u l p h u r tetrafluoride a t 500° (Johnson, C. E . , et al, 1961) or of t h e trioxide w i t h F r e o n 12 a t 400° (Booth et al, 1946;
Cacciari et al, 1956, 1957). I t is also o b t a i n e d b y h e a t i n g u r a n i u m dioxide w i t h a n excess of a m m o n i u m fluoride or bifluoride ( B r a d d o c k a n d Copenhafer, 1943) a reaction i n which t h e p e n t a f l u o r o u r a n a t e ( I V ) , NII4UF5, is flrst formed (Van I m p e , 1954; N e u m a n n et al, 1962). T h i s decomposes t o t h e tetrafluoride a b o v e 320° (Galkin et al, 1961). A similar reaction w i t h u r a n i u m ( I V ) a c e t a t e a t 450° also yields u r a n i u m tetrafluoride (Sahoo a n d P a t n a i k , 1959).
T h e s t r u c t u r e of u r a n i u m tetrafluoride consists of 8 fluorine a t o m s a r r a n g e d a r o u n d t h e u r a n i u m a t o m i n a slightly d i s t o r t e d a n t i p r i s m configuration (Larson et al, 1964), a n 8-coordinate a r r a n g e m e n t w h i c h p r o b a b l y applies t o t h e r e m a i n i n g actinide tetrafluorides, all of which h a v e t h e s a m e crystal s y m m e t r y (Table V I I ) .
T h e p r e p a r a t i o n of t h e tetrafluorides of t h e higher actinides r e q u i r e s progressively stronger oxidizing conditions. N e p t u n i u m tetrafluoride is
TABLE V I I . C r y s t a l l o g r a p h i c d a t a f o r t h e a c t i n i d e t e t r a f l u o r i d e s *
318 κ. w. B A G N A L L
o b t a i n e d b y t h e action of a m i x t u r e of h y d r o g e n fluoride a n d oxygen on t h e trifluoride a t 500° (Fried a n d D a v i d s o n , 1948), conditions u n d e r w h i c h u r a n i u m w o u l d oxidize t o u r a n y l fluoride. T h e p l u t o n i u m com
p o u n d is m a d e b y t h e action of fluorine on t h e trifluoride a t 300°, a n d of a m i x t u r e of h y d r o g e n fluoride a n d oxygen on t h e trifluoride a t 550°
or o n t h e dioxide a t 550-600° (Florin a n d H e a t h , 1944). T h e h y d r a t e d tetrafluoride, o b t a i n e d from a q u e o u s solution, can be d e h y d r a t e d in a m i x t u r e of h y d r o g e n fluoride a n d o x y g e n a t 350° (Meyer a n d Zvolner, 1944). P l u t o n i u m tetrafluoride is also formed b y reaction of t h e dioxide w i t h s u l p h u r tetrafluoride a t 600° ( J o h n s o n , C.E., et aL, 1961) a n d b y h e a t i n g t h e dioxide w i t h a m m o n i u m bifluoride; t h e a m m o n i u m p e n t a - fluoroplutonate(IV) formed in t h i s reaction decomposes a t a b o u t 280°
(Maly et aL, 1961; Tolley, 1954). P l u t o n i u m tetrafluoride a p p e a r s t o d i s p r o p o r t i o n a t e a b o v e 1200°, w i t h t h e a p p e a r a n c e of a m o r e volatile species (Mandleberg a n d Davies, 1961), p r e s u m a b l y t h e hexafluoride formed b y reaction of t h e tetrafluoride w i t h traces of oxygen; t h e o n l y species which sublimes below 1000° is t h e tetrafluoride (Berger a n d G a u m a n n , 1961).
Americium tetrafluoride is o b t a i n e d b y t h e action of fluorine on t h e trifluoride or dioxide a t 500° (Asprey, 1954) a n d t h e c u r i u m c o m p o u n d b y t h e action of fluorine on t h e trifluoride a t 400°, b u t only w i t h t h e longer-lived isotope, curium-244, r a d i a t i o n d a m a g e inhibiting t h i s reaction w i t h t h e short-lived curium-242 (Asprey et aL, 1957). T h e existence of curium tetrafluoride shows clearly t h a t t h e stability of t h e half-filled 5/-shell is m u c h lower t h a n in t h e l a n t h a n i d e case. T h e visible s p e c t r a of t h e s e solid tetrafluorides h a v e b e e n r e c o r d e d (Asprey a n d K e e n a n , 1958).
H y d r a t e d tetrafluorides, M F 4. 2- 5 H 2 0 , of t h o r i u m ( D ' E y e a n d B o o t h , 1955, 1957), u r a n i u m ( K a t z a n d R a b i n o w i t c h , 1951; D a w s o n et aL, 1 9 5 4 a ) a n d p l u t o n i u m (Dawson et aL, 1 9 5 4 b ; D e i c h m a n n a n d T a n a n a e v , 1961) are p r e c i p i t a t e d from a q u e o u s solutions of t h e q u a d r i v a l e n t ele
m e n t s b y hydrofluoric acid; t h e w h i t e c o m p o u n d p r e c i p i t a t e d from p r o t a c t i n i u m ( I V ) solution (Haissinsky a n d Bouissières, 1951) is also p r o b a b l y of this t y p e . A n e p t u n i u m c o m p o u n d of t h i s form should exist, b u t does n o t a p p e a r t o h a v e been recorded. H y d r a t e d a m e r i c i u m a n d curium tetrafluorides c a n n o t be o b t a i n e d from a q u e o u s solutions since b o t h americium(IV) a n d curium(IV) are u n s t a b l e in w a t e r in t h e absence of high c o n c e n t r a t i o n s of fluoride ion.
T h e h y d r a t e s M F 4. 2- 5 H 2 0 lose w a t e r on h e a t i n g , t h e t h o r i u m com
p o u n d yielding t h e h e m i h y d r a t e , ThF4.0-5H2O ( D ' E y e a n d B o o t h , 1955, 1957), a n d t h e u r a n i u m c o m p o u n d yielding UF4.0-4H2O (Gagarinskii a n d Mashirev, 1959b; Gal'chenko et aL, 1960). B o t h can be completely
THE HALOGEN CHEMISTRY OF THE ACTINIDES 319
d e h y d r a t e d b y h e a t i n g , usually in a v a c u u m . T h e p r e c i p i t a t i o n of UP4.2-5H20, w h i c h exists in b o t h cubic a n d monoclinic modifications, from s u l p h a t e solution b y fluoride ion a p p e a r s t o t a k e place b y w a y of a n i n t e r m e d i a t e UFg^^ species ( T a n a n a e v a n d S a v c h e n k o , 1962b) a n d a fluoro-oxalate, UF2(C204).1-5H20, h a s b e e n o b t a i n e d b y h e a t i n g UF4.2-5H20 w i t h s a t u r a t e d oxalic acid a t 100° a n d t h e a n h y d r o u s c o m p o u n d (UF)2(C204)3 b y h e a t i n g UF4.2-5H20 w i t h oxalic acid d i h y d r a t e a t 200° ( T a n a n a e v a n d S a v c h e n k o , 1962a).
Similar b e h a v i o u r h a s b e e n r e p o r t e d for p r o t a c t i n i u m ( I V ) , t h e c o m p o u n d PaF2(S04).2H20 being isolated from a q u e o u s solution (Stein, 1965) a n d for p l u t o n i u m ( I V ) , p a r t i c u l a r l y in s u l p h a t e solution ( D e i c h m a n n a n d T a n a n a e v , 1961). T h e solubility of UF4.2-5H20 in hydrofluoric-perchloric acid m i x t u r e s c a n likewise b e explained b y t h e presence of UF2^+ species (Savage a n d B r o w n e , 1960).
T h e r e are m a n y p u b l i s h e d p r o c e d u r e s for t h e p r e p a r a t i o n of UF4.2-5H20 (see K a t z a n d R a b i n o w i t c h , 1951); a p a r t i c u l a r l y con
v e n i e n t one is b y electrolytic r e d u c t i o n of u r a n y l fluoride solution a t a m e r c u r y c a t h o d e (Nikolaev a n d L u k ' y a n y c h e v , 1961). A h y d r a t e of composition UF4.4/3H2O is also k n o w n (Gagarinskii et al., 1965).
C. Tetrachlorides
(i) Preparation and Properties
R e v i e w s of t h e p r e p a r a t i o n a n d p r o p e r t i e s of t h o r i u m ( F l a h a u t , 1963) a n d u r a n i u m (Oxley, 1962) t e t r a c h l o r i d e s h a v e r e c e n t l y a p p e a r e d ; t h e s e discuss t h e following, a n d o t h e r , p r e p a r a t i v e p r o c e d u r e s in g r e a t e r detail.
T h e t e t r a c h l o r i d e s are c o m m o n l y m a d e b y chlorination of t h e dioxide w i t h c a r b o n t e t r a c h l o r i d e v a p o u r , t h e t h o r i u m c o m p o u n d a t a b o u t 800° (Matignon a n d Delépine, 1901, 1908), p r o t a c t i n i u m t e t r a c h l o r i d e a t 500° (Sellers et al., 1954), u r a n i u m t e t r a c h l o r i d e a t 450° ( K a t z a n d R a b i n o w i t c h , 1951) or 500° (Harrison, 1958) a n d t h e n e p t u n i u m com
p o u n d a t 530° (Fried a n d D a v i d s o n , 1948). M i x t u r e s of t h o r i u m or u r a n i u m oxides a n d carbon r e a c t quite readily w i t h chlorine a t e l e v a t e d t e m p e r a t u r e s , a p r o c e d u r e long u s e d for t h e p r e p a r a t i o n of t h o r i u m (Berzelius, 1829) a n d u r a n i u m (Péligot, 1842b) t e t r a c h l o r i d e s ; t h e reaction proceeds r a t h e r b e t t e r in t h e presence of ferric chloride in m o l t e n K C l - N a C l a t 800° (Gibson et al, 1960). T h e chlorination of u r a n i u m oxides b y c a r b o n t e t r a c h l o r i d e or h e x a c h l o r o p r o p e n e proceeds b y w a y of UOCI3 ( B u d a e v a n d Vol'skii, 1958). M i x t u r e s of c a r b o n t e t r a chloride a n d chloroform h a v e also been used t o chlorinate u r a n i u m dioxide a t 400-500° (Rosenfeld, 1960).
T h o r i u m t e t r a c h l o r i d e is conveniently m a d e from t h e elements a t
320 κ. w. BAGNALL
800° (Fowles a n d Pollard, 1953), b y reaction of t h e carbide w i t h chlorine (Dean a n d Chandler, 1957, w h o h a v e also reviewed t h e p r e p a r a t i v e procedures available for this c o m p o u n d ) or of h y d r o g e n chloride w i t h t h e h y d r i d e a t 250-350° (Lipkind a n d N e w t o n , 1952) or w i t h t h e m e t a l (Kruss a n d Nilson, 1887) a n d b y d e h y d r a t i o n of t h e h y d r a t e w i t h t h i o n y l chloride (Bradley et al., 1954; F r e e m a n a n d S m i t h , 1958), or b y h e a t i n g w i t h pyridine hydrochloride (Didchenko, 1959). I t can be purified b y h e a t i n g w i t h a m m o n i u m chloride, followed b y sublimation t h r o u g h t h o r i u m m e t a l t u r n i n g s (Skaggs a n d P e t e r s o n , 1958).
P u r e t h o r i u m tetrachloride is also said t o be o b t a i n e d b y t h e reaction of carbon tetrachloride w i t h t h o r i u m tetraiodide a t 100-200° ( W a t t a n d Malhotra, 1960). O t h e r m e t h o d s of p r e p a r i n g t h o r i u m tetrachloride include t h e r m a l decomposition of a m m o n i u m p e n t a c h l o r o t h o r a t e a t 500° a n d chlorination of t h o r i u m dioxide w i t h carbonyl chloride a t 650-700° (Chauvenet, 1911).
A more convenient procedure for t h e p r e p a r a t i o n of p r o t a c t i n i u m tetrachloride is b y h y d r o g e n reduction of t h e pentachloride a t 800°
(Elson et al., 1950; Sellers et al., 1954); because of t h e volatility of b o t h t h e s t a r t i n g m a t e r i a l a n d t h e p r o d u c t t h e reaction is best carried o u t in a sealed t u b e a t 400-500° or, m o r e safely, w i t h a l u m i n i u m (Brown a n d J o n e s , 1966c).
U r a n i u m tetrachloride is best m a d e b y reaction of UO3 or UgOg w i t h hexachloropropene ( H e r m a n n a n d Suttle, 1957), b u t reaction of hexa
chloropropene w i t h NpOa.HgO or NpgOg yields a m i x t u r e of n e p t u n i u m tetrachloride a n d trichloride (Bagnall a n d Laidler, 1966). U r a n i u m tetrachloride is also o b t a i n e d b y t h e action of chlorine on t h e trichloride a t 250° or of a m i x t u r e of chlorine a n d helium (1:10) on t h e hydride.
There is t h e r m a l a n d X - r a y evidence for a crystal t r a n s f o r m a t i o n a t 545° ( J o h n s o n et al., 1958). O t h e r m e t h o d s include reaction of t h e tetrafiuoride w i t h a l u m i n i u m or b o r o n trichloride a t 250-500° in a sealed t u b e (Calkins a n d Larsen, 1945; Fried, 1945), reaction of sulphur m o n o chloride w i t h u r a n i u m t r i o x i d e u n d e r reflux ( U h l e m a n n a n d Fischbach, 1963), a n d r e a c t i o n of t h e elements a t a b o u t 650° (Reynolds a n d Wilkinson, 1956).
T h o r i u m a n d u r a n i u m tetrachlorides h a v e a n 8-coordinate s t r u c t u r e w i t h t h e m e t a l - c h l o r i n e b o n d i n g i n t e r m e d i a t e b e t w e e n covalent a n d ionic, four of t h e Th—Cl distances being 2-46 A a n d four 3-11 A; in UCI4 t h e U—Cl distances are 2-41 a n d 3-09 A (Mooney, 1949). P r o t a c t i n i u m a n d n e p t u n i u m tetrachlorides are isostructural w i t h u r a n i u m tetrachloride. Crystallographic d a t a are s u m m a r i z e d in Table V I I I .
A wide v a r i e t y of h y d r a t e d a n d partially hydrolysed species derived from t h o r i u m a n d u r a n i u m tetrachloride h a v e been recorded in t h e early
THE HALOGEN CHEMISTRY OF THE ACTINIDES 321
T h C l ^ W h i t e T e t r a g o n a l , làjamd- 8 - 4 7 3 — 7 - 4 6 8 4 - 6 0 PaCl4 G r e e n i s h - T e t r a g o n a l , I^tjamd--Dl\ 8 - 3 7 7 — 7 - 4 8 2 4 - 7 2
Y e l l o w
UCI4 G r e e n T e t r a g o n a l , 14:jamd- 8 - 2 9 6 — 7 - 4 8 7 4 - 8 7 NPCI4 R e d - b r o w n T e t r a g o n a l , / 4 / a m c Z - 8-29 — 7-46 4 - 9 2
T h B r ^ W h i t e T e t r a g o n a l 8 - 9 6 3 — 7 - 9 4 6 5-69
U B r , B r o w n M o n o c l i n i c , 21c —I— 1 0 - 9 2 8 - 6 9 7-05 5-55 j 3 = 9 3 ° 9 '
N p B r ^ R e d d i s h - M o n o c l i n i c , 21c —1 — b r o w n
Thl^to W h i t e M o n o c l i n i c P 2 i / „ M o n o c l i n i c P 2 i / „ 1 3 - 2 1 6 8 - 0 6 8 7 - 7 6 6 6 - 0 0 )δ = 9 8 - 6 8 °
a C o r r e c t e d d a t a c o l l e c t e d i n T a b l e I V o f t h e r e v i e w b y K a t z a n d S h e f t ( 1 9 6 0 ) u n l e s s o t h e r w i s e s t a t e d .
i> Z a l k i n et al. ( 1 9 6 4 ) .
l i t e r a t u r e ; s u m m a r i e s of t h e p r e p a r a t i o n a n d properties of t h e s e com
p o u n d s a r e given b y F l a h a u t (1963), K a t z a n d R a b i n o w i t c h (1951), K a t z i n (1954) a n d Oxley (1962). T h e u r a n i u m t e t r a c h l o r i d e h y d r a t e s (4-5 t o 9 HgO) h a v e recently been t h e subject of further investigation (Pommier, 1966).
Studies of t h e c o n d u c t i v i t y of u r a n i u m t e t r a c h l o r i d e i n a q u e o u s e t h a n o l indicate t h e presence of complex species such a s UCI3+ a n d UClg^"^ ( R o a c h a n d Amis, 1962) a n d similar species h a v e b e e n s h o w n t o exist in a q u e o u s hydrochloric acid solutions of t h o r i u m ( I V ) (Zebroski et al,, 1951; W a g g o n e r a n d S t o u g h t o n , 1952) a n d p l u t o n i u m ( I V ) (Grenthe a n d N o r e n , 1960). T h e visible s p e c t r a of u r a n i u m t e t r a c h l o r i d e in n o n a q u e o u s solvents h a v e also been recorded a n d discussed (Ewing,
1961; J e z o w s k a - T r z e b i a t o w s k a et al,, 1958).
(ii) Complexes
Triscyclopentadienyl chlorides a r e k n o w n for b o t h t h o r i u m a n d u r a n i u m , t h e former o b t a i n e d b y reaction of p o t a s s i u m cyclopenta- dienide w i t h t h o r i u m t e t r a c h l o r i d e i n e t h e r (Ter H a a r a n d D u b e c k ,
1964), t h e l a t t e r b y reaction of sodium cyclopentadienide w i t h u r a n i u m tetrachloride i n t e t r a h y d r o f u r a n (Reynolds a n d Wilkinson, 1956). B o t h c o m p o u n d s a r e v e r y sensitive t o m o i s t u r e . T h e u r a n i u m c o m p o u n d is monoclinic (P2i/^), w i t h t h e t h r e e cyclopentadiene rings a n d t h e chlo
rine a t o m a r r a n g e d a p p r o x i m a t e l y t e t r a h e d r a l l y a r o u n d t h e u r a n i u m a t o m (Chi-Hsian W o n g et al,, 1965).
TABLE V I I I . C r y s t a l l o g r a p h i c d a t a f o r the a c t i n i d e t e t r a c h l o r i d e s , t e t r a b r o m i d e s a n d t e t r a i o d i d e s *
C o l o u r S y m m e t r y a n d s p a c e g r o u p L a t t i c e p a r a m e t e r s (A) C a l c u l a t e d d e n s i t y
«0 60 Co ( g c m- 3 )