K ÉMIAI EGYENSÚLYI ÉS KINETIKAI KÖLCSÖNHATÁSOK LEÍRÁSA , ALKALMAZÁSA AZ
IONCSERE - KROMATOGRÁFIÁBAN
Doktori (PhD) értekezés
Készítette
Horváth Krisztián
okleveles környezetmérnök
Témavezet˝o
Dr. Hajós Péter
egyetemi docens
Készült a Pannon Egyetem
„Anyagtudományok és -technológiák” Doktori Iskolája keretében
Pannon Egyetem Mérnöki Kar
Analitikai Kémia Intézeti Tanszék
2007
III
harmadik oldal
Kivonat
A szerz˝o a dolgozat els˝o felében retenciós modellt vezetett le szerves és szervetlen anionok latex-agglomerált pellikuláris állófázisokon történ˝o viselkedésének leírására, Langmuir-tí- pusú egyensúlyok alkalmazásával. Az állófázis felépítésére jellemz˝o állandót,w súlyténye- z˝ot vezetett be, mely az állófázis töltésegyensúlyban lev˝o funkciós csoportjainak teljes ion- cserekapacitáshoz viszonyított arányát mutatja meg. Kísérleti adatbázis alapján, iterációs úton meghatározta a levezetett retenciós modell egyensúlyi és rendszer paramétereit. Vizs- gálta awsúlytényez˝o hatását anionok retenciós profiljára. Statisztikai módszerekkel igazol- ta a levezetett retenciós modell használhatóságát.
A dolgozat második felében egyszeresen negatív töltés˝u anionok retenciós viselkedését vizsgálta n-decil-2.2.2 kriptand molekulát tartalmazó állófázison, NaOH és KOH eluens al- kalmazásával. Retenciós egyenletet vezetett le egynegatív töltés˝u anionok vizsgált állófázi- son történ˝o viselkedésének leírására, figyelembe véve a kriptand molekula protonálódását, alkálifém-kriptát komplex kialakulását, az anionok és az eluens ionok között kialakuló ion- cserét. Iterációs úton meghatározta a retenciós modellben szerepl˝o egyensúlyi paramétere- ket. Az ionpár-képz˝odési állandók értékének nagyságrendjét vizsgálva megállapította, hogy a K+-kriptát funkciós csoportok er˝os, a Na+-kriptát funkciós csoportok gyenge, a proton- ált kriptand molekulák nagyon gyenge anioncserél˝onek tekinthet˝ok. Vizsgálta az elválasztó oszlop ioncsere-kapacitási viszonyait. A levezetett retenciós modellt statisztikailag értékelte.
A dolgozat harmadik felében a kromatográfia sztochasztikus elméletének alkalmazható- ságát vizsgálta az anion-kromatográfiás vízanalitikai elválasztások területén. A sztochasz- tikus elmélet felhasználásával, a momentumok módszerének segítségével meghatározta az egyes mintaionok olyan molekuláris retenciós sajátságait, mint az adszorpciós-deszorpciós lépések száma (n) és az egyes ion fajták állófázison történ˝o átlagos tartózkodási ideje egy- egy megköt˝odés alkalmával (τs). Megállapította, hogy az egyes ionok lépésszáma csekély eltéréseket mutat, a retenciós viselkedésükben mutatkozó különbségek f˝oként a megköt˝od- ve eltöltött id˝ok különbségéb˝ol ered. Elméleti megfontolások alapján kapcsolatot teremtett az ionkromatográfiában használatos többszörös eluens/minta retenciós modellel, így nem csak a kromatográfiás csúcs helyét, hanem a csúcs alakját is becsülni képes módszert dolgo- zott ki analitikai elválasztások céljára.
V
Abstract
In the first part of the thesis a retention model was developed in order to describe retention behavior of inorganic and organic anions on latex-agglomerated pellicular ion exchangers.
The model based on Langmiur-type equilibria. As a new parameter the fractional electros- tatic coefficient,w, of the ion exchange capacity was introduced referring to the ratio of the anion-exchange functional groups are in charge balance with the cation-exchange layer be- hind the latex particles. The system parameters, and the ion-pair formation coefficients for analyte and eluent anions were determined by nonlinear iterative calculation. The effect of w on the retention profile of anions was investigated. The predictive power of the model was verified by statistical methods.
In the second part of the work, the retention behavior of inorganic anions was investiga- ted on a cryptand-based anion exchanger using NaOH and KOH-based eluents. Considering the complexation and protonation equilibria a theoretical retention model was developed for describing the retention behavior of monovalent anions. The parameters of the model were iterated. By investigating the magnitude of the ion-pair formation coefficients it was concluded that the K+-cryptate and Na+-cryptate complexes behaved as strong and weak functional groups. The ion-exchange capacity of the column was investigated as a functi- on of eluent concentration. The validity of the derived retention model was confirmed by statistical evaluation.
In the third part of the dissertation the applicability of the stochastic theory was in- vestigated in the field of anion chromatography. The stochastic parameters of the eluted anions, such as the residence time of the molecule on the surface of the stationary phase (τs) during one adsorption event, and the average number of adsorption steps (n) were cal- culated on the basis of a retention database of organic and inorganic anions. It was shown that the retention of the analytes, and the selectivity of the separation was primarily due to the variation ofτs, the number of sorption steps did not affect it significantly. The stochastic theory and the multiple species eluent/analyte retention model were integrated on theore- tical considerations. By simultaneous application of the two theories full chromatograms (retention time and peak shape) of a sample mixture can be predicted.
VII
Riassunto
Nella prima parte della tesi, e stato sviluppato un modello di ritenzione per la descrizione del comportamento cromatografico di anioni inorganici ed organici su scambiatori ionici pellicolari lattice-agglomerati. Il modello e basato su equilibri di tipo Langmuir. Come nu- ovo parametro e stato introdotto il coefficiente elettrostatico frazionario,wdella capacita di scambio ionico, che si riferisce al rapporto dei gruppi funzionali di scambio ionico che sono in bilancio di carica con lo strato di scambio cationico dietro le particelle di lattice. I para- metri del sistema, le costanti di formazione di coppia ionica e di selettivita di scambio ionico tra l’analita e gli ioni eluente, sono stati determinati, mediante calcolo iterativo non lineare, impiegando un database sperimentale. E stato studiato l’effetto di w sul profilo di ritenzione degli anioni e il potere predittivo del modello e stato supportato da metodi statistici.
In una seconda parte del lavoro e stato studiato il comportamento cromatografico di anioni inorganici su una colonna di scambio anionico contenente un macrociclo criptan- do, utilizzando eluenti contenenti NaOH o KOH. Tenendo in considerazione gli equilibri di complessazione e di protonazione coinvolti nel sistema, sono stati sviluppati un modello di ritenzione e un’equazione che descrive l’effetto simultaneo del tipo e della concentrazione dello ione eluente sul fattore di ritenzionek. Mediante l’utilizzo di un database di dati speri- mentali, e stato possibile iterare i parametri del modello. Dal confronto dei valori dei coeffi- cienti di formazione delle coppie ioniche, si e potuto concludere che i complessi K+-criptati agiscono come gruppi funzionali di scambio ionico forti, mentre i complessi Na+-criptati e il criptando protonato si comportano, rispettivamente, come gruppi funzionali deboli e debolissimi. La capacita di scambio ionico della colonna e stata valutata sulla base della co- noscenza delle costanti di formazione di coppia e di protonazione. La validita del modello di ritenzione sviluppato e stata supportata da una valutazione statistica.
Nella terza parte della tesi, e stata studiata l’applicabilita della teoria stocastica della cro- matografia sui processi di scambio ionico. In particolare, i picchi cromatografici sono stati descritti da funzioni gaussiane esponenziali modificate i cui parametri sono stati determi- nati mediante fitting del picco. I parametri stocastici degli anioni eluiti, come il tempo di residenza della molecola nella superficie della fase stazionaria (τs) durante un evento di ad- sorbimento e il numero medio di stadi di adsorbimento (n), sono stati calcolati utilizzando
IX
un database di dati cromatografici per anioni organici ed inorganici. E stato dimostrato che il meccanismo di ritenzione e la selettivita della separazione sono dovuti principalmente al- la variazione di s, mentre il numero di stadi di adsorbimento non li influenza in maniera sig- nificativa. La teoria stocastica e il modello di ritenzione per specie multiple eluente/analita sono stati integrati da considerazioni teoriche. Mediante la contemporanea applicazione delle due teorie, possono essere descritti a priori cromatogrammi completi (tempo di riten- zione e forma del picco) di miscele di campioni.
Tartalomjegyzék
Bevezetés 1
1. Irodalmi összefoglaló 3
1.1. Folyadékkromatográfia . . . 3
1.1.1. A folyadékkromatográfiás elválasztások folyamata . . . 3
1.1.2. A folyadékkromatográfia eszközei . . . 4
1.1.3. Kromatográfiás alapfogalmak . . . 5
1.1.4. A kromatográfia sztochasztikus elmélete . . . 9
1.2. Az ionkromatográfia . . . 12
1.2.1. Az ionkromatográfia típusai . . . 12
1.2.2. Az ionkromatográfia eszközei . . . 13
1.2.3. Az ionkromatográfia el˝onyei . . . 15
1.2.4. Elválasztási és detektálási rendszer választása . . . 17
1.3. Az ionkromatográfia alkalmazása a környezeti analízisben . . . 17
1.4. Anioncsere-kromatográfia . . . 22
1.4.1. Állófázisok . . . 22
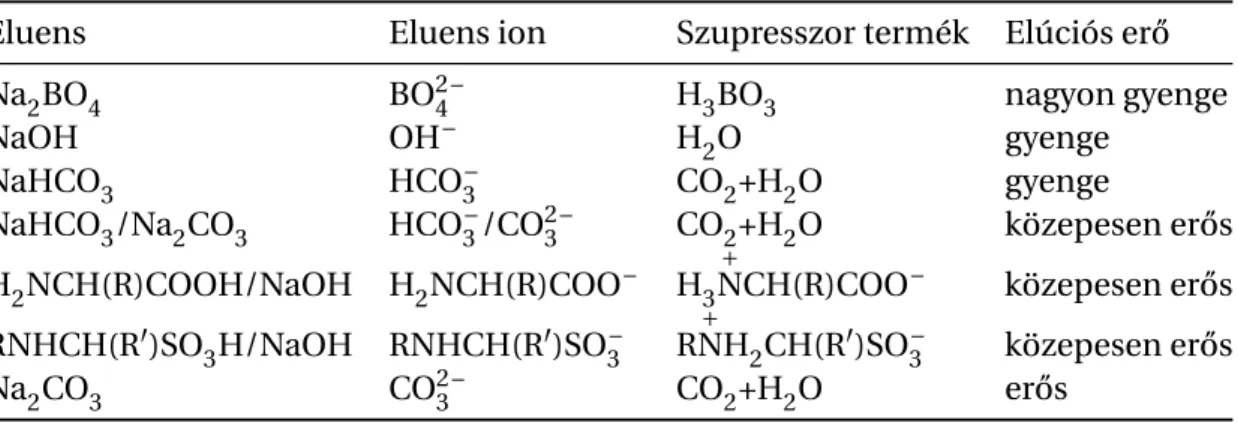
1.4.2. A leggyakrabban használatos anionkromatográfiás eluensek . . . 25
1.4.3. Anionkromarográfiás retenciót leíró elméletek . . . 28
2. Kísérleti rész 37 2.1. Felhasznált eszközök, anyagok . . . 37
2.1.1. Alifás karbonsavak és szervetlen anionok retenciós tulajdonságainak vizsgálata latex alapú anioncserél˝on . . . 37
2.1.2. Szervetlen anionok retenciós viselkedésének tanulmányozása makro- ciklikus állófázison . . . 37
2.1.3. Szerves és szervetlen anionok vizsgálata nagykapacitású oszlopon . . . . 38
2.2. Ioncsere-kapacitás meghatározása . . . 39
2.3. Retenciós modell egyensúlyi paramétereinek meghatározása iterációs úton . . 40
2.4. Kromatográfiás csúcsok momentumainak meghatározása görbeillesztéssel . . 41
XI
3. Eredmények 45
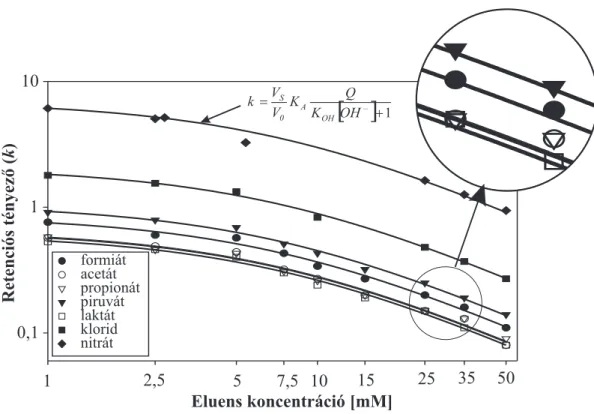
3.1. Anionok elválasztásának mechanizmusa latex-alapú állófázisokon . . . 45
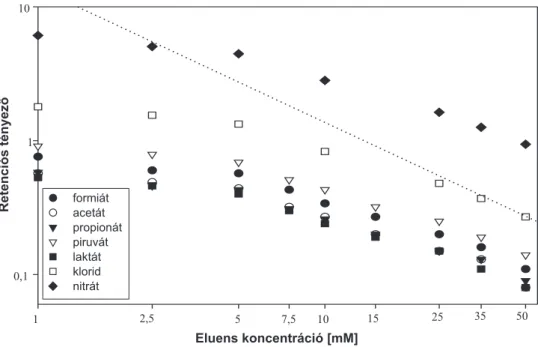
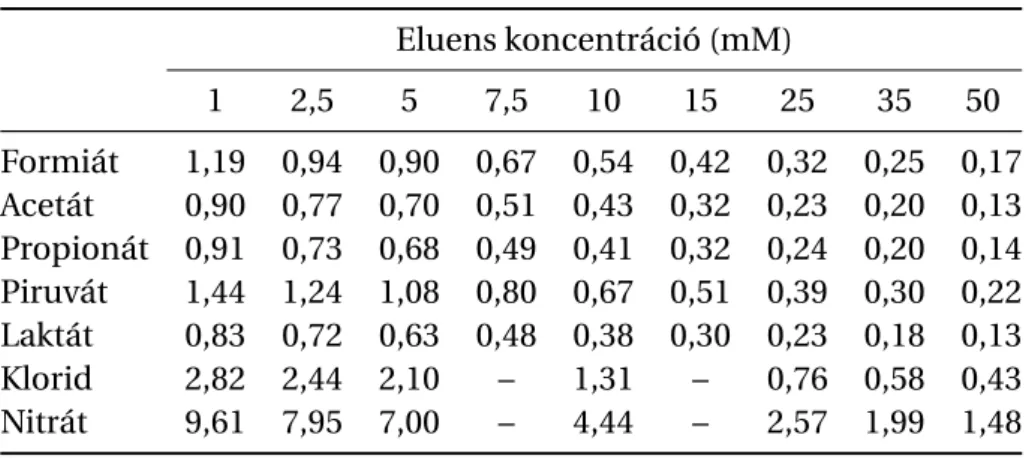
3.1.1. Alifás karbonsavak és szervetlen anionok retenciós viselkedése latex- alapú állófázisokon . . . 45
3.1.2. Eluens- és mintaionok kémiai egyensúlyai latex-alapú állófázisokon . . 49
3.1.3. Elválasztó oszlop ioncsere-kapacitása . . . 51
3.1.4. Mintaionok retenciós tényez˝oje . . . 53
3.1.5. Eluens és mintaionok egyensúlyi állandójának meghatározása . . . 53
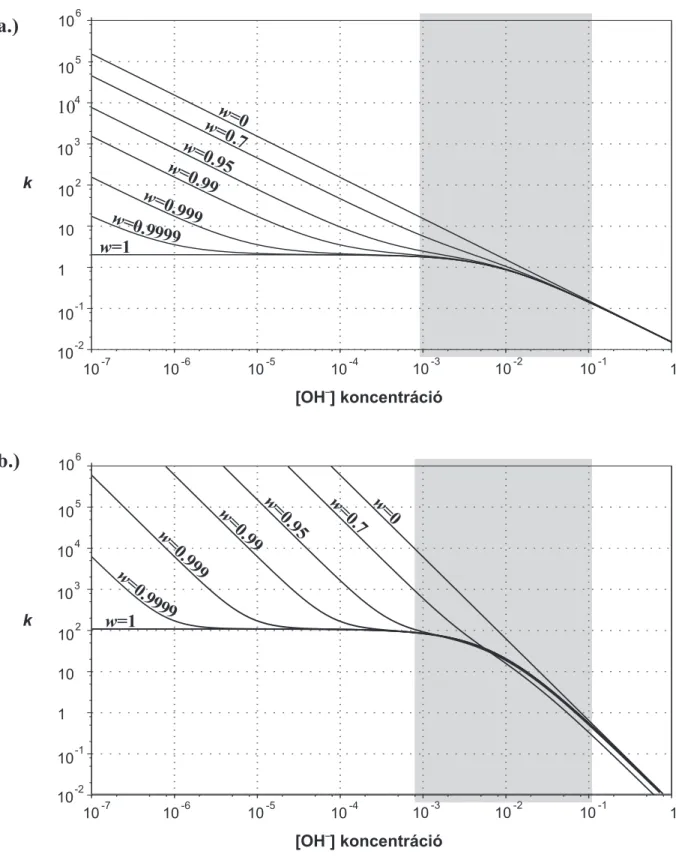
3.1.6. Awsúlytényez˝o hatása anionok retenciós profiljára . . . 55
3.1.7. A retenciós modell statisztikai értékelése . . . 56
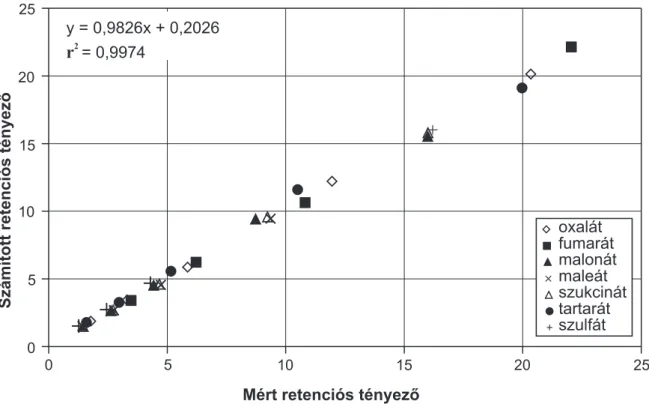
3.2. Makrociklikus egyensúlyok az anionkromatográfiában . . . 60
3.2.1. Anionok retenciós viselkedése makrociklikus állófázison . . . 60
3.2.2. Elválasztás során lejátszódó komplex- és ioncsere-egyensúlyok . . . 63
3.2.3. Egyensúlyi állandók meghatározása . . . 66
3.2.4. A levezetett retenciós modell statisztikai értékelése . . . 69
3.3. Anionok retenciós viselkedésének integrált leírása . . . 73
3.3.1. Sztochasztikus paraméterek meghatározása . . . 73
3.3.2. A megköt˝odések gyakoriságának és az ioncserekapacitás kihasználtsá- gának meghatározása . . . 77
3.3.3. A többkomponens˝u eluens/minta retenciós modell kapcsolata a szto- chasztikus elmélettel . . . 80
3.3.4. A vízanalitikában fontos ionok kromatogramjának becslése a többkom- ponens˝u eluens/minta és a sztochasztikus modell egyesítésével . . . 81
4. Összefoglalás 89 Irodalomjegyzék 94 A szerz˝o tudományos munkássága i 4.1. Publikációk . . . i
4.2. Konferencia el˝oadások . . . ii
4.3. Az eredmények hasznosítása . . . v
4.4. Elismerések . . . vi
Tézispontok vii
Theses xi
Bevezetés
A XX. század második felében az elválasztási módszerek többsége ugrásszer˝u fejl˝odésen ment keresztül. Az elválasztások sebessége, hatékonysága, megbízhatósága és felbontása egyaránt javult. A kromatográfia történetében az els˝o jelent˝os mérföldk˝o a megoszlásos- és papírkromatográfia megjelenése volt az 1940-es években, ezt a különféle gélkromatográfi- ás módszerek térhódítása követte a hatvanas években. Dönt˝oen fontos lépés volt a szerves vegyületek analízisében a gázkromatográfia megjelenése. A legjelent˝osebb áttörést azon- ban a – Horváth Csaba nevéhez f˝uz˝od˝o – nagyteljesítmény˝u folyadékkromatográfia (HPLC:
high performance liquid chromatography) kifejlesztése jelentette a hetvenes években. Ez- zel sokkal gyorsabban, pontosabban, reprodukálhatóbban és szelektívebben lehetett igen bonyolult elválasztási feladatokat megoldani.
A HPLC – és ezzel együtt az ion- és az ionpár-kromatográfia – a legjelent˝osebb módszerré n˝otte ki magát az elválasztásos analízisen belül. Mind a vízanalitika, a bioanalitika, a gyógy- szergyártás, mind a környezetvédelmi analízis területén kiemelt fontossággal bír. Jelenleg valószín˝uleg több elválasztást végeznek HPLC módszerekkel, mint az összes többi elválasz- táson alapuló módszerrel együttvéve.
A folyadék fázisú analitikai elválasztások tervezése komplikált feladat, mivel egyidej˝uleg több tényez˝o, az eluensoldat, az elválasztóoszlop és a mintakomponensek kémiai szerkeze- tét, fizikai-kémiai tulajdonságait, továbbá a detektálhatóság paramétereit is figyelembe kell venni. Többféle matematikai algoritmus létezik az ionkromatográfiás elválasztási folyama- tok leírására. A dolgozatban bemutatott munka retenciós modellek fejlesztését mutatja be különböz˝o típusú állófázisokon a nagyhatékonyságú ionkromatográfia területén, valamint az ionkromatográfiás elválasztások integrált leírását adja.
1
1. Irodalmi összefoglaló
A dolgozat els˝o részében a kromatográfiás elválasztások elvét, m˝uködését mutatom be a módszer jobb megértése céljából. Részletesen ismertetem a folyadékkromatográfia a szto- chasztikus retenciós elméletét. Emellett rövid áttekint˝o ismertetést adok az ionkromatográ- fia jelent˝oségér˝ol, felhasználásáról, fontosabb elméleti modelljeir˝ol.
1.1. Folyadékkromatográfia
A kromatográfia különböz˝o kémiai tulajdonságú komponensek fizikai elválasztásának olyan módszere, melynek során az elválasztandó komponensek megoszlanak két fázis között. Az egyik fázist, mely egyhelyben áll, álló fázisnak, míg a másik fázist, mely egy jól definiált irány- ban mozog, mozgó fázisnak nevezzük [1]. Ha a mozgó fázis folyadék, folyadékkromatográ- fiáról (LC) beszélünk.
1.1.1. A folyadékkromatográfiás elválasztások folyamata
A folyadékkromatográfiás elválasztások a különböz˝o típusú mintakomponensek és a kroma- tográfiás állófázis között fellép˝o kölcsönhatás eltér˝o erején alapulnak [2, 3]. Két különböz˝o kémiai tulajdonságú komponens elválasztása a következ˝oképpen történik: tegyük fel, hogy az egyik komponens (A) hasonló módon köt˝odik meg az állófázison, mint az eluens, míg a másik komponens (B) jóval er˝osebben. Ha a két komponenst tartalmazó elegy kis mennyi- ségét az oszlopra injektáljuk, akkor azAtípusú molekulák, mivel az eluenshez hasonló mér- ték˝u visszatartást szenvednek, ugyanakkora sebességgel haladnak el˝ore az oszlopban, mint az eluens molekulák. ABmolekulák ellenben, köszönhet˝oen az állófázissal szemben mu- tatott er˝osebb kölcsönhatásuknak, több id˝ot töltenek az állófázison, azaz az eluens és azA molekuláknál kisebb sebességgel mozognak el˝ore. Ha megfelel˝oen nagy a két komponens megköt˝odésének er˝ossége közti különbség, akkor a két komponens eltér˝o id˝oben hagyja el az oszlopot, így módunkban áll két különböz˝o komponensként detektálni ˝oket (1.1. ábra).
Elválasztás során általában meglehet˝osen keskeny sávokat injektálunk (5–20µl), azon- ban a folyamat lezajlása során a komponensek sávjai kiszélesednek, köszönhet˝oen a lassú
3
Detektorjel
Idő
1.1. ábra:Elválasztás mechanizmusa a komatográfiában
adszorpciós kinetikának, a longitudinális- és örvénydiffúziónak, valamint egyéb tényez˝ok- nek [2, 3]. Ezek a folyamatok együttesen okozzák a kromatográfiás csúcs sávszélesedését.
Általánosságban elmondható, hogy minél nagyobb egy komponens visszatartása az oszlop- ban, annál szélesebb zónában hagyja el az oszlopot.
1.1.2. A folyadékkromatográfia eszközei
A folyadékkromatográfiás rendszerek állandó tartozékai a szivattyú, a mintabeviteli rend- szer, az elválasztó oszlop, a detektor és az adatgy˝ujt˝o/feldolgozó egység (1.2. ábra) [2].
A rendszer lelke a kromatográfiás oszlop, ahol a tulajdonképpeni elválasztás zajlik. Mi- vel az állófázis mikrométer nagyságú részecskékb˝ol áll, nagy nyomású pumpa szükséges a mozgófázis áramoltatására a rendszerben. A kromatográfiás elválasztás folyamata a minta- elegy rendszerbe történ˝o bejuttatásával kezd˝odik. Az elválasztás az id˝o alatt történik meg, amíg a mintakomponensek az oszlopban tartózkodnak. Az oszlopot elhagyva az egyes min- tamolekulák, -ionok a detektorba jutnak, ami azok koncentrációjával arányos elektromos
1.1. Folyadékkromatográfia 5
Os lopz Detektor Interfész
Adatfeldolgozó
Injektáló szelep Pumpa Mozgó fázis
Hurok
1.2. ábra:A folyadékkromatográfia eszközei
jelet továbbít az adatgy˝ujt˝o egység felé. A detektor által szolgáltatott jelet az id˝o fügvényé- ben ábrázolva kapjuk az ún. kromatogramot (1.1. ábra alsó része), mely minta összetételére vonatkozó min˝oségi és mennyiségi információkat egyaránt tartalmazza.
1.1.3. Kromatográfiás alapfogalmak
A dolgozatban többször el˝oforduló, a kromatográfiában általánosságban használt alapfogal- makat igyekszem tisztázni ebben a fejezetben.
Retenciós id˝o, -térfogat, -tényez˝o
Egy komponens visszatartásának mértéke jellemz˝o az adott vegyületre, így annak min˝oségi azonosítására is szolgálhat. A legegyszer˝ubb módszer a visszatartás (retenció) számszer˝usí- tésére az injektálás kezdete és az adott komponens detektorban történ˝o megjelenése közti id˝o mérése. Utóbbit célszer˝uen a detektor maximális válaszjeléhez szokás társítani. Ezt a mér˝oszámot a kromatográfiában retenciós id˝onek (tR) hívjuk [1]. A retenciós id˝o arányos a komponens visszatartásával és fordítottan arányos az eluens térfogatáramával.
A retenciós id˝o és az eluens térfogatáramának szorzata a retenciós térfogat (VR), ami a retenciós id˝onél egy sokkal inkább általános, retenciót jellemz˝o mér˝oszám, ugyanis nem függ az eluens térfogatáramától, csak a komponens visszatartásától és az elválasztó oszlop geometriai méreteit˝ol, tulajdonságaitól. A retenciós térfogat azt a mozgófázis térfogatot je- lenti, amely ahhoz szükséges, hogy az adott komponenst az oszlopról eluáljuk. A retenciós térfogat két részb˝ol tev˝odik össze:
1. Holttérfogat: az az eluens térfogat, amely az id˝o alatt áramlik keresztül az oszlopon, amíg a mintakomponens a mozgófázisban tartózkodik. A holttérfogat megegyezik az elválasz- tó oszlop térfogatának azon részével, melyet a mozgófázis tölt ki (V0), értéke ennélfogva
minden mintakomponensre nézve állandó.
2. Korrigált retenciós térfogat: a mozgófázis azon térfogata, mely az id˝o alatt áramlik keresz- tül az oszlopon, amíg a mintakomponens az állófázison tartózkodik. Értéke a különböz˝o mintamolekulák esetén más és más.
A leguniverzálisabb retenciót jellemz˝o mér˝oszám a retenciós tényez˝o (k). A retenciós tényez˝o a komponens álló- és mozgófázisbeli mennyiségének hányadosa [1]:
k = ns
nm, (1.1)
aholnsa vizsgált komponens állófázison,nma mozgófázisban lev˝o móljainak száma.
Az 1.1 egyenlet átrendezésével a következ˝o egyenletet írhatjuk fel:
k+1= ns nm+nm
nm =ns+nm
nm . (1.2)
Legyen a mozgófázis áramlási sebessége az oszlopbanu(cm/sec), a vizsgáltAminta ré- szecskéinek (molekula, ion stb.) átlagos vándorlási sebessége pediguA, melynek értéke attól függ, hogy azAionok mekkoraRhányada van a mozgófázisban, és mekkora a mozgófázis uáramlási sebessége, azaz:
uA=uR (1.3)
és
R= nm
ns+nm = 1
1+k. (1.4)
Ha az Aionok mozgófázisban lev˝o hányada zérus (R= 0), akkoruA is zérus, az ionok nem jutnak el˝ore az oszlopban. Ha mindenAion az eluensben van (R= 1), akkoruA=u, az ionok ugyanakkora sebességgel áramlanak, mint a mozgófázis, nincs visszatartásuk az oszlopban.
Az (1.2) és az (1.4) összefüggésekb˝ol következik, hogy:
uA= u
1+k. (1.5)
AzuA, az oszlopL hossza és az elválasztás id˝oszükséglete között kapcsolat teremthet˝o, ha figyelembe vesszük, hogy az id˝o az út és a sebesség hányadosa. Esetünkben azAkom- ponensnektRid˝ore van szüksége ahhoz, hogy azL hosszúságú oszlopon áthaladjon. AzA
1.1. Folyadékkromatográfia 7
sáv áthaladási ideje ezek alapján:
tR = L
uA. (1.6)
Ehhez hasonlóan az oldószer vagy más vissza nem tartott komponens
t0= L
u (1.7)
id˝o alatt halad át az oszlopon. Ha az (1.6) és (1.7) összefüggésekb˝ol kiejtjük L-t, akkor a következ˝o egyenlethez jutunk:
tR = u t0
uA, (1.8)
amib˝ol, az (1.5) egyenletet figyelembe véve adódik, hogy:
tR=t0(1+k) . (1.9)
Az (1.9) összefüggés átrendezésével akszámítását lehet˝ové tev˝o egyenlethez jutunk:
k = tR−t0
t0 . (1.10)
Látható, hogy a retenciós tényez˝o a vissza nem tartott és a vizsgált komponens csúcsa közti távolság és az injektálás, valamint a vissza nem tartott komponens csúcsa közötti távol- ság hányadosával egyenl˝o. Adott oszlop, eluens, elválasztási h˝omérséklet ésXkomponens esetén, kis mintamennyiséget alkalmazvakállandó, így az adott mintakomponens min˝osé- gi azonosítását lehet˝ové teszi.
Megoszlási hányados
Az el˝oz˝o fejezetben láthattuk, hogy az elválasztóoszlopban a komponensek vándorlási se- bessége azok álló- és mozgófázis közötti egyensúlyi megoszlásától függ:
µdx dv
¶
A
= 1
DA, (1.11)
ahol dxaz állófázis infinitezimálisan kis mennyisége, amelyen a megoszló anyag dv térfo- gatú fluid fázis bejuttatásának hatására el˝orehalad. DA a megoszlási hányados. Az elvá-
lasztandó komponensek oldódnak a mozgófázisban, ill. megköt˝odnek az állófázison. Ezt a folyamatot a megoszlási hányadossal jellemezhetjük [1]:
DA= (A)
[A], (1.12)
ahol (A) a minta állófázisbeli, [A] pedig a mozgófázisbeli koncentrációja. DA értéke a két fázis kémiai tulajdonságaitól függ, így általában egyt˝ol különböz˝o értékeket vesz fel. DA nagy, ha nagy a komponens affinitása az állófázishoz, és kicsi, ha az inkább a mozgó fázisban tartózkodik.
HaDAállandó az elúció folyamán, akkor aVNnettó retenciós térfogat az alábbiak szerint számítható kiVStérfogatú állófázist tartalmazó oszlop esetén:
VN=VSDA. (1.13)
ÁllandóDAesetén tehátVNtérfogatú fluid fázis szükséges ahhoz, hogy a minta aVStérfo- gatú állófázist tartalmazó oszlopon áthaladjon.
A megoszlási hányados és az el˝oz˝o fejezetben ismertetett retenciós tényez˝o (DA) között az (1.1) és az (1.12) összefüggések alapján a következ˝o kapcsolat állítható fel:
k = VS(A) V0[A] = VS
V0DA. (1.14)
Elválasztás hatékonysága
Az elválasztás hatékonysága a komponens retenciójától és kromatográfiás csúcsának szé- lességét˝ol függ. Egy mintakomponens retenciója a molekula és az állófázis közötti kölcsön- hatás er˝osségére, valamint az állófázis fajlagos felületére utal. A komponens sávjának szé- lesedése ellenben alapvet˝oen kinetika által szabályzott, és az állófázis részecskéinek átmé- r˝ojét˝ol, porozitásától, pórus méretét˝ol és az oszlop mérett˝ol függ. A kromatográfiás oszlop hatékonyságára jellemz˝o, igen fontos mér˝oszám az elméleti tányérszám, mely a kromato- gráfia tányérelméletéb˝ol eredeztethet˝o [4, 5]. Értéke a következ˝o összefüggés segítségével számítható ki [6]:
N = tR2
σ2, (1.15)
aholtRésσa komponens retenciós ideje ill. varianciája.
Minél nagyobb egy szeparációs rendszer hatékonysága, annál keskenyebb csúcsokat
1.1. Folyadékkromatográfia 9
kaphatunk adott retenciós id˝o esetén, azaz annál több komponens kromatográfiás csúcsát tudjuk megkülönböztetni egy adott retenciós id˝o tartományban.
Szelektivitás
A két különböz˝o komponens szelektivitásán azok retenciós tényez˝oinek vagy korrigált reten- ciós id˝oinek hányadosát értjük [2, 3]. A szelektivitás egy kromatográfiás rendszer elválasztási erejét mutatja meg adott komponensekre vonatkozóan. Értéke nagyobb 1-nél, mivel mindig a nagyobb retencióval rendelkez˝o komponens retenciós tényez˝ojét osztjuk a kisebbével.
α=kB kA =tB
tA. (1.16)
Ez a paraméter független az oszlop hatékonyságától, értéke csak az elválasztott kompo- nensek természetét˝ol, az eluens fajtájától és összetételét˝ol, valamint az állófázis tulajdonsá- gaitól függ. Ha két komponens szelektivitása 1, akkor az adott kromatográfiás rendszerben nincs mód azok elválasztására, még az oszlop hatékonyságának növelésével sem.
Felbontás
A folyadékkromatográfiás elválasztások, s így az ionkromatográfiás elválasztások célja álta- lában a minta komponenseinek kielégít˝o mérték˝u szétválasztása. E cél eléréséhez a relatív elválasztást, más néven megfelel˝o felbontást kell biztosítanunk. Két szomszédos A és B sáv felbontása definíciószer˝uen a két sáv középpontja közti távolság és az átlagos sávszélesség hányadosával egyenl˝o [1]:
RS=2|tRA−tRB|
(twA+twB), (1.17)
aholtRAéstRBazAésBsávtRértéke,twAéstwBpedig a sávok alaplapi szélessége ugyan- abban az id˝o egységben, mint atRértékek.
Ha RS=1, akkor ez azt jelenti, hogy a két sáv jól elválik egymástól, maximum 2%-ban fednek át. Nagyobb felbontás érték jobb, kisebb pedig kevésbé hatékony elválasztást jelent.
AdottRSesetén az átfedés nagyobb mérték˝uvé válik, ha a két sáv közül az egyik sokkal ki- sebb, mint a másik.
1.1.4. A kromatográfia sztochasztikus elmélete
A kromatográfia az elválasztási módszerek széles spektrumát öleli fel. A szeparáció során gyakran igen komplex fizikai-kémiai folyamatok játszódnak le. A retenciós mechanizmus-
tól (adszorpció, megoszlás, ioncsere) függ˝oen meglehet˝osen sok modell áll rendelkezésre a kromatográfiás csúcsok helyének és alakjának becslésére [7]. A kromatográfia sztochaszti- kus elmélete egy molekuláris szint˝u modell, amely valószín˝uleg a legegyszer˝ubb képet festi a kromatográfiás folyamatról. A kromatográfiás csúcs kialakulását a részecskék véletlensze- r˝u vándorlásával írja le az oszlop mentén, ugyancsak véletlenszer˝uen fellép˝o adszorpciós- deszorpciós folyamattal kombinálva [8].
A sztochasztikus elméletet Giddings és Eyring alkotta meg 1955-ben [9], majd Giddings fejlesztette tovább [10]. A kromatográfiás folyamatok számos sajátosságát megérthetjük a modell segítségével. Közvetlen betekintést nyújt a szeparációs folyamatba, mivel olyan ki- fejezéseket használ, melyek könnyen megérthet˝ok (adszorpciós lépések száma, átlagos ad- szorpciós id˝o stb.), s melyek a sávszélesedésben jelent˝os szerepet játszanak. A szeparáci- ós folyamat alapvet˝o tulajdonságai (momentumok, tányérszám stb.) néhány egyszer˝u szá- mítással megkaphatók a modellb˝ol. Mindemellett vonzó alternatívát kínál a hagyományos makroszkopikus modellekkel szemben, amelyek egy-egy, gyakran analitikusan nem meg- oldható, differenciális tömegmérleget írnak fel minden komponensre a kromatográfiás fo- lyamat kívánt részletesség˝u fizikai-kémiai leírásának érdekében.
A sztochasztikus elmélet a kromatográfiás folyamatot Poisson-folyamatként szemléli.
Amikor egy részecske az elúció során adszorbeálódik, meg˝orzi oszlopon belüli helyzetét egé- szen addig, míg el nem hagyja az állófázist. Deszorpció után folytatja útját a mozgófázissal, amíg újra meg nem köt˝odik az állófázison. A részecske tartózkodási ideje az állófázison (τ∗s) és a mozgófázisban (τ∗m) véletlenszer˝u, exponenciális eloszlású értékek, melyeknek várható- értékeτsésτm. Minden egyes részecske különböz˝o pályán halad, és különböz˝o id˝opontban éri el az oszlop végét. Ez id˝o alatt minden egyes molekula különböz˝o,r számú adszorpci- ós/deszorpciós lépésen esik át. Utóbbi változó Poisson-eloszlású, várható értéken.
P(τ∗s)=
( 0 hat<0
e−τs/t ha 0≤t (1.18)
P(τ∗m)=
( 0 hat<0
e−τm/t ha 0≤t (1.19)
P(r)=e−nnr
r! , (1.20)
aholtaz id˝o változó.
A véletlenszer˝u hatások következtében ugyanabban az id˝opillanatban az oszlopba belé- p˝o két teljesen azonos kémiai tulajdonságú részecske hamarosan eltér˝o helyen található. Az elúciós profil, azaz kromatográfiás csúcs alakját leíró görbe, az egyes részecskék retenciós
1.1. Folyadékkromatográfia 11
id˝oinek valószín˝uségi s˝ur˝uségfüggvénye:
P(n,τs,t)= r n
tτse−n−τst I1
Ãs4nt τs ,
!
(1.21)
aholI1(x) az ún. els˝ofajú, els˝orend˝u módosított Bessel-függvény.
Hanmegfelel˝oen nagy, az (1.21) egyenlet az alábbi Gauss görbével közelíthet˝o:
P(t)= 1 2τsp
πne−(τst −n)2
4n . (1.22)
A sztochasztikus elmélet szerintτma várható id˝otartam, amit egy részecske vándorlása során a mozgófázisban tölt egy deszorpció és az azt követ˝o adszorpció közt (repülési id˝o, fly-time), míg várhatóanτs adszorpciós id˝ot tölt az állófázis felületén egy-egy megköt˝odés alkalmával. A Frenkel-egyenletnek [11] megfelel˝oenτs közvetlenül kapcsolatba hozható az adszorpciós energiával (Ea) , mígnaz anyagátadási egységek számával analóg [6, 12]:
τs=τ0eRTEa, (1.23)
aholτ0konstans, értéke∼1,6×10−13s szobah˝omérsékleten.
A modell hátránya, hogy nem veszi figyelembe az állófázisban található olyan pórusokat, melyekben stagnál a mozgófázis, és ha egyszer egy részecske egy ilyen pórusban csapdába kerül, akkor nagy számú adszorpción és deszorpción megy át, mire kijut a pórusból. Mind- emellett az elmélet nem veszi figyelembe a mozgófázis hatásait sem a kialakuló kromato- gráfiás csúcsra.
A sztochasztikus modell alapján a kromatográfiás csúcs normalizált-centralizált mo- mentumai (µ) az alábbi általános kifejezés segítségével számíthatók [8]:
µk=k!nτks. (1.24)
Ennek megfelel˝oen a korrigált retenciós id˝o, ami a részecskék által az állófázison megköt˝od- ve eltöltött id˝o a
tR0 =µ1=nτs, (1.25)
a kromatográfiás csúcs varianciája a
σ2=µ2=2nτ2s, (1.26)
míg ferdesége (skew), ami a csúcs aszimmetriáját jellemzi az
S= µ3 µ3/22 = 3
p2n (1.27)
összefüggésekkel számítható.
1.2. Az ionkromatográfia
Ionkromatográfia alatt nem egy konkrét kromatográfiás technikát értünk, hanem több, egy- mástól teljesen eltér˝o retenciós mechanizmusú módszert. Az ionkromatográfia magába fog- lalja az összes, ionos vagy ionizálható komponens analízisére szolgáló folyadékkromatográ- fiás módszert. Tehát az ionkromatográfiát a többi kromatográfiás technikától nem a reten- ciós mechanizmus, hanem az elválasztott mintakomponensek min˝osége különbözteti meg.
Ionkromatográfiával az alábbi komponensek határozhatók meg:
1. szervetlen anionok: halogenidek (F–, Cl– stb.), oxoanionok (SO2 –4 , NO3–, ClO4–, BrO3–, PO3 –4 stb.)
2. szerves anionok: kis molekulatömeg˝u (vízoldható) mono-, di- és trikarbonsavak (formiát, acetát, oxalát, fumarát, citrát, EDTA stb.), szulfonsavak, beleértve a detergenseket is 3. szervetlen kationok: alkáli- és alkáliföldfémek (Li+, Na+, Mg2+stb.)
4. szerves kationok: kis molekulatömeg˝u (vízoldható) aminok (metil-, etil-, propilamin stb.) 5. ionos organo-metallo komplexek (fém-kelátok, tributil-ón stb.)
6. aminosavak (glicin, alanin stb.)
7. szénhidrátok (glükóz, fruktóz, szacharóz stb.)
1.2.1. Az ionkromatográfia típusai
Ioncsere-kromatográfia (HPIC: High Performance Ion-Exchange Chromatography) Az elválasztás mechanizmusát a mozgófázis és az állófázison kötött ioncserél˝o csoportok közti ioncsere folyamatok szabályozzák. Az ioncsere az egyik legrégebben bemutatott elvá- lasztási mechanizmus az irodalomban [13]. Polarizálható ionok esetén egyéb, nem ionos adszorpciós folyamatok is szerepet játszhatnak. Az állófázis ioncserél˝o csoportokkal mó- dosított sztirol-divinilbenzol kopolimer. Az ioncsere-kromatográfia egyaránt használható
1.2. Az ionkromatográfia 13
szerves és szervetlen anionok és kationok elválasztására is. Anionok elválasztását kvaterner ammónium csoportokat, kationok elválasztását pedig szulfoncsoportokat tartalmazó gyan- tán lehet kivitelezni.
Ionkizárásos-kromatográfia (HPICE: High Performance Ion Chromatography Exclusion) Ionkizárásos-kromatográfiában az elválasztás mechanizmusát az adszorpció, a Donnan- ill.
a sztérikus kizárás folyamatai szabályozzák. Állófázisként els˝osorban teljesen szulfonált szti- rol-divinilbenzol alapú, nagykapacitású kationcserél˝ok jöhetnek szóba. Az ionkizárásos- kromatográfia gyenge szervetlen és szerves savak elválasztására és meghatározására szol- gál. Az er˝os savak retenciót nem szenvedve haladnak át az oszlopon, a holtid˝ovel eluálódva.
Megfelel˝o detektálási rendszerrel kombinálva a technika aminosavak, aldehidek és alkoho- lok elválasztására is alkalmas.
Ionpár-kromatográfia (MPIC: Mobile Phase Ion Chromatography)
Az elválasztást leginkább meghatározó mechanizmus az ionpár-kromatográfiában az ad- szorpció. Az állófázis teljesen semleges, nagy fajlagos felület˝u, kis polaritású, porózus szti- rol-divinilbenzol vagy C8/18-szilikagél. Az elválasztó oszlop szelektivitását f˝oként a mozgó fázis összetétele határozza meg. A mozgófázis szerves módosítót, ún. ionpárképz˝o reagenst tartalmaz, mely az elválasztandó ionokkal ionpárt képez. A technika f˝oként felületaktív an- ionok és kationok ill. átmenetifém-komplexek elválasztására alkalmas.
Alternatív módszerek
A fentikekben felsorolt három módszeren túl a fordított fázisú folyadék-kromatográfiában (RP-HPLC) is egyre gyakrabban választanak el poláros, és ionos komponenseket. Az alter- natív módszerek közé szokás sorolni emellett a kationok és anionok elválasztására egyaránt használható kelát-ionkromatográfiát (ld. 1.4.1. alszakasz), valamint a micella-kizárásos-kro- matográfiát is.
1.2.2. Az ionkromatográfia eszközei
Az ionkromatográfiás rendszer felépítése lényegében megfelel a hagyományos folyadékkro- matográfiás rendszernek. Lényeges különbség f˝oként az elválasztás mechanizmusában, az- az az állófázis tulajdonságaiban, és a detektálás módjában található.
Ionkromatográfiában, a f˝oként szilikagél alapú állófázisokat használó klasszikus HPLC technikákkal szemben, els˝osorban a szerves polimer alapú állófázisok terjedtek el köszön- het˝oen sokkal nagyobb pH stabilitásuknak. Míg a szilikagél alapú állófázisok csak egy igen
sz˝uk, 2–8 közötti pH tartományban használhatók, addig a modern, szerves polimer alapú állófázisok pH stabilitása garantált a teljes pH (0–14) tartományra. Mindemellett a szilikag- él állófázist tartalmazó oszlopok hatékonysága lényegesen nagyobb a polimer alapúakénál.
Az ioncserél˝o oszlopok egyik leglényegesebb tulajdonsága az ioncserekapacitás, mely az ál- lófázison található ioncserél˝o funkciós csoportok számát jelenti mequiv/g vagy mequiv/ml egységben. Az anionkromatográfiában használatos állófázisokról b˝ovebben az 1.4.1. alsza- kaszban (22–25. o.) lesz szó.
Az ionkromatográfiában f˝oként szupresszált vezet˝oképességi detektálás használatos, ha- bár az UV/Vis spektrofotometriás, az amperometriás és fluoreszcenciás detektálás is egyre nagyobb szerepet kap egyes komponensek meghatározása esetén (ld. még 1.4. ábra, 18. o.).
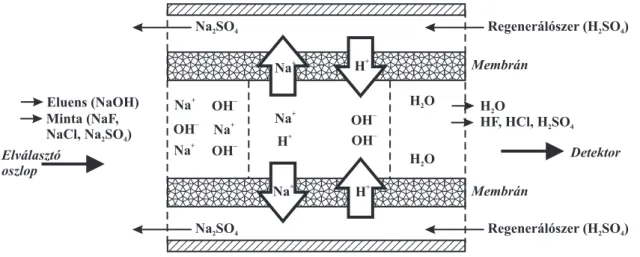
Szupresszált vezet˝oképességi detektálás esetén a detektor elé helyezett szupresszornak az a szerepe, hogy az ionok elúciójához használt elektrolit vezet˝oképességét kémiailag csök- kentse, és egyúttal a mintaionokét növelje, megnövelve ezáltal a hasznos analitikai jelet, és ezzel összefüggésben a detektor érzékenységét is. A szupresszorban egy ioncserél˝o memb- rán található. Kationkromatográfia esetén anion-, anionkromatográfia esetén kationcserél˝o membrán. El˝obbi esetben a mozgófázis anionjait hidroxidionokra, utóbbi esetben a mozgó- fázis kationjait protonokra cseréljük a szupresszorban, miáltal jelent˝osen csökken az eluens vezetése. Az ioncserél˝o membrán regenerálása folyamatos üzemmódban anionkromato- gráfiában kénsavval, kationkromatográfiában tetrametil-ammónium-hidroxiddal történik, ami biztosítja a reakcióhoz szükséges protonokat, ill. hidroxidionokat. A lejátszódó reakció nátrium-hidroxid eluens esetén az 1.3. ábrán látható.
aaaaaaaaaaaaaaaaaaaa aaaaaaaaaaaaaaaaaaaa aaaaaaaaaaaaaaaaaaaa aaaaaaaaaaaaaaaaaaaaNa+
Na+
H+
H+ Na+
H+
OH– OH–
H O2
H O2
Na+ Na+ Na+
OH– OH–
OH–
Regenerálószer (H SO )2 4
Regenerálószer (H SO )2 4
Membrán
Membrán Na SO2 4
Na SO2 4
Eluens (NaOH) Minta (NaF, NaCl, Na SO )2 4
H O2
HF, HCl, H SO2 4
Detektor Elválasztó
oszlop
1.3. ábra:A szupresszor m˝uködésének vázlata NaOH eluens használata esetén
1.2. Az ionkromatográfia 15
1.2.3. Az ionkromatográfia el˝onyei
Ionos komponensek meghatározása vizes oldatokban klasszikus analitikai probléma, mely- nek megoldására sokféle módszer és technika jöhet szóba. Miközben a kation analízis terü- letén egyaránt gyors és érzékeny módszerek állnak rendelkezésre (atomabszorpciós-, atom- emissziós spektrometria, indukált csatolású plazma, polarográfia stb.), anionok meghatáro- zására kevés megfelel˝o, nagy érzékenység˝u módszer létezik. A hagyományos nedves analiti- kai eljárások, a titrálás, fotometria, gravimetria, turbidimetria és kolorimetria, mind eszköz- és id˝oigényes módszerek. Ezzel szemben az ionkromatográfia az alábbi mérési jellemz˝ok- ben kínál jelent˝os el˝onyöket:
(1) gyors analízis, (2) nagy érzékenység, (3) kiváló szelektivitás, (4) szimultán detektálás,
(5) stabil kromatográfiás oszlop.
Gyors analízis
Az analízisid˝o csökkentése egyre inkább fokozódó követelmény, ugyanis a vizsgálandó min- ták száma jelent˝osen n˝ott az utóbbi id˝oben köszönhet˝oen egyrészt az ipari termékek min˝o- ségével kapcsolatos megnövekedett igényeknek, másrészt a környezeti és biológiai analitika fejl˝odésének.
Az elmúlt id˝oszakban megjelent nagyhatékonyságú elválasztó oszlopokkal ma már mindössze három perc alatt, tökéletes felbontás mellett, meghatározható a legfontosabb hét anion [14]. Azaz a hagyományos nedves analitikai eljárások id˝oszükségletének tört része alatt min˝oségi és mennyiségi információval szolgálhat az ionkromatográfia.
Nagy érzékenység
A mikrocsip technológia bevezetésének és az egyre hatékonyabb állófázisok kifejlesztésé- nek köszönhet˝oen a mai ionkromatográfiás gyakorlatban mintael˝okészítés nélkül is rutin feladatnak számít egyes anionok ppb tartományú detektálása [15]. 50µl-es huroktérfogat esetén a legtöbb egyszer˝u ion esetében a kimutatási határ∼10 ppb. A mintaionok teljes in- jektált mennyisége ezekben az esetekben ng tartományban van. Ultratisztaságú vizek ppt koncentrációtartományú ion-analízisére is az ionkromatográfia kínálja a legkedvez˝obb al- ternatívát, azonban ezekben az esetekben mintael˝okészítés (koncentrálás) is szükséges.
Aminosav analízis területéno-ftálaldehiddel (OPA) történ˝o, oszlop utáni származékkép- zést és fluoreszcenciás detektálást alkalmazva pmol nagyságrend˝u érzékenység érhet˝o el.
Kiváló szelektivitás
Az ionkromatográfia ill. általánosságban véve a kromatográfia egyik legnagyobb el˝onye a hagyományos és modern analitikai technikákkal szemben, hogy a különböz˝o típusú minta- komponensekre nagyfokú szelektivitást mutat. A klasszikus analitikai gyakorlat egyik nagy problémája, hogy az adott módszer szempontjából hasonló kémiai viselkedés˝u mintakom- ponensek egymás jelenlétében történ˝o meghatározása, pusztán az adott technikát alkal- mazva, gyakran nem kivitelezhet˝o. Klorid-szelektív elektród például jóval nagyobb szelekti- vitást mutat az oldatban jelenlev˝o bromidionra, mint a kloridra, emiatt kis mennyiség˝u bro- mid is jelent˝osen zavarhatja a kloridionok ionszelektív elektróddal történ˝o meghatározását.
Közel azonos protonálódási tulajdonságokkal rendelkez˝o gyenge savak sem határozhatók meg egymás jelenlétében klasszikus, sav-bázis titrálás segítségével. Az atomspektroszkópia, annak ellenére, hogy igen érzékeny, önmagában nem használható módosulat analitikára, a módszer nem képes különbséget tenni az adott elem kémiai módosulatai (oxidációfok, komplex forma stb.) között. Helyesen megválasztott ionkromatográfiás rendszerrel (moz- gófázis-összetétel, elválasztóoszlop, detektálási mód) azonban a fent említett szelektivitás- beli problémákkal nem kell számolni. Ma már rutin feladatnak számít a halogenid ionok, különböz˝o oxidációfokú és komplexformájú nehézfémek, ill. szerves savak egymástól való elválasztása ionkromatográfiás módszerrel.
Szimultán detektálás
Az ionkromatográfia egy másik nagy el˝onye a hagyományos, nedves analitikai technikákkal szemben a különböz˝o komponensek együttes meghatározásának lehet˝osége, azokat az ese- teket kivéve, mikor a különböz˝o minta komponensek koncentrációja extrém módon tér el egymástól adott mintán belül. Ilyen esettel lehet találkozni a legtöbb szennyvíz ill. csurga- lékvíz analízisekor [16, 17]. Ezekben az esetekben két külön analízisre van szükség a nagy és a kiskoncentrációjú komponensek kimutatására. Azonban még ekkor is néhány perc alatt megkaphatjuk a minta teljes anion- és kationösszetételét.
Kromatográfiás oszlop stabilitása
Az ionkromatográfiás oszlopokban alkalmazott töltetek nagy pH stabilitása lehet˝ové teszi er˝os savak és lúgok eluensként való felhasználását. Mindemellett a szerves polimer alapú oszlopok nem érzékenyek komplex mátrixú mintákra, szennyvizekre, ételmintákra, testfo- lyadékokra sem. Ilyen esetekben a mintael˝okészítés gyakran csak egyszer˝u hígítás. A szer- ves polimerek hátránya, hogy gyakran korlátozott a stabilitásuk szerves oldószerekkel szem- ben, így nem használhatók szerves szennyez˝ok eltávolítására. Köszönhet˝oen a polimerké- mia utóbbi években tapasztalható nagymérték˝u fejl˝odésének, ma már egyre több gyártó lép
1.3. Az ionkromatográfia alkalmazása a környezeti analízisben 17
ún. oldószer kompatibilis tölteteket tartalmazó oszlopokkal a piacra.
Kis mintaszükséglet
A klasszikus analitikai eljárások során gyakran több 10 ml mintamennyiség szükséges az ol- dat összetételének meghatározásához. A nagy precizitású mintabeviteli rendszerek és az érzékeny detektálás következtében az ionkromatográfiában az egy elemzés során vizsgált mintamennyiség mindössze néhány 10−100µl, azaz három nagyságrenddel kisebb, mint a klasszikus módszerek esetén.
1.2.4. Elválasztási és detektálási rendszer választása
Ahogy azt az 1.2.1. alszakaszban (12 .o.) láthattuk, ionkromatográfia alatt több, mecha- nizmusában egymástól teljesen eltér˝o módszert értünk. Ennélfogva megfelel˝o elválasztási rendszer összeállításánál az állófázis és a megfelel˝o detektálási módszer megválasztása na- gyon lényeges az analízis szempontjából.
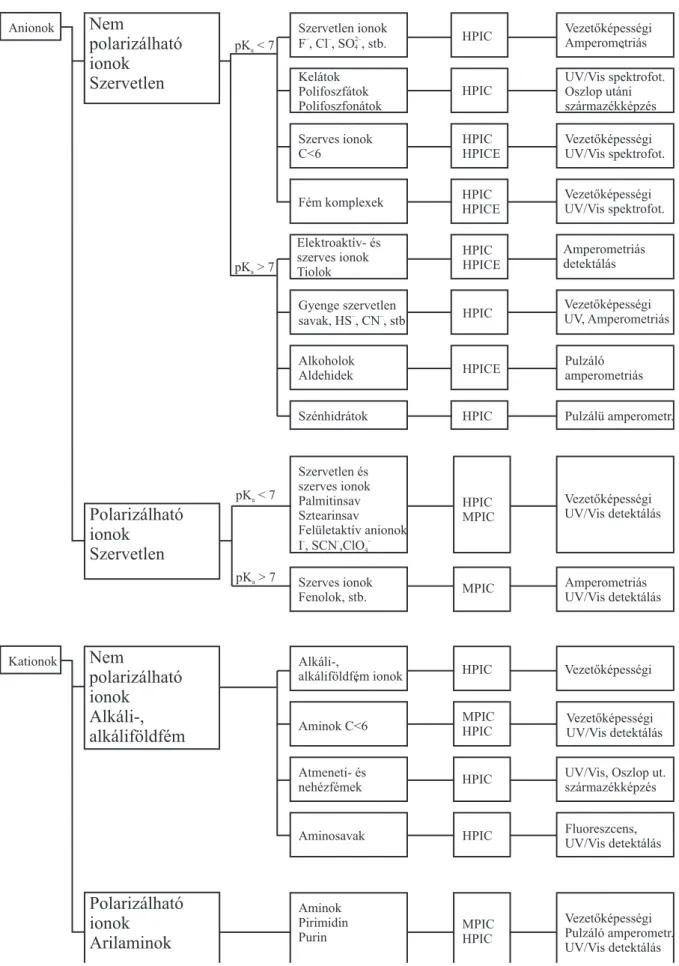
Az analitikus általában rendelkezik el˝ozetes információkkal a vizsgálandó minta összeté- telér˝ol (szerves, vagy szervetlen ionokat tartalmaz-e), felületi aktivitásáról, töltésér˝ol, savas ill. bázikus tulajdonságairól. Ezen információk alapján az 1.4. ábra segítségével könnyen kiválasztható az analízis szempontjából legel˝onyösebb állófázis és detektálási rendszer. Sok esetben több kombináció is szóba jöhet az adott minta elemzésére. Ilyen esetekben a dön- tés során figyelembe kell venni a minta mátrixot, a módszer bonyolultságát és az analízis költségeit.
1.3. Az ionkromatográfia alkalmazása a környezeti analízis- ben
A kromatográfiás elválasztási módszereket széles körben alkalmazzák különböz˝o környezeti minták elemzése során. Az ionkromatográfia egyaránt alkalmazható ivóvizek és szennyvi- zek összetételének meghatározására, talajok, iszapok, csurgalékok, szilárd hulladékok anali- tikai vizsgálatára is [18]. A technikának els˝osorban anionok meghatározásában van kiemelt jelent˝osége.
Az ivóvízmin˝osítés területén az ionkromatográfia igen elterjedt, ugyanis lehet˝oséget nyújt az ivóvízben található szervetlen ionok többségének nagy hatékonyságú elválasztá- sára. Ennek köszönhet˝oen több szabványos mérési módszer is rendelkezésre áll a vízmin˝o- sítés területén. Ilyenek például hazánkban az MSZ EN ISO 10304-1:1998 [19], az MSZ EN ISO 10304-2:1999 [20] és az MSZ EN ISO 10304-3:1999 [21] szabványok. A sorozat negyedik
Alkáli-,
alkáliföldfém ionok4
Nem
polarizálható ionok
Szervetlen
Polarizálható ionok
Szervetlen
Kelátok Polifoszfátok Polifoszfonátok Szerves ionok C<6
Fém komplexek Elektroaktív- és szerves ionok Tiolok
Gyenge szervetlen savak, HS , CN , stb– – Alkoholok Aldehidek
Szénhidrátok pK > 7a
pK < 7a
pK > 7a
Szervetlen és szerves ionok Palmitinsav Sztearinsav Felületaktív anionok I , SCN ,ClO- - 4-
Szerves ionok Fenolok, stb.
Kationok Nem
polarizálható ionok
Alkáli-,
alkáliföldfém Aminok C<6 Atmeneti- és nehézfémek
Aminosavak
Polarizálható ionok
Arilaminok
Aminok Pirimidin Purin
HPIC HPICE
HPIC
HPICE
HPIC HPIC HPICE
HPIC HPICE
HPIC MPIC
MPIC
MPIC HPIC
HPIC
HPIC
MPIC HPIC
HPIC Vezetőképességi
UV/Vis spektrofot.
Oszlop utáni származékképzés Vezetőképességi UV/Vis spektrofot.
Amperometriás detektálás
Pulzáló amperometriás
Pulzálü amperometr.
Vezetőképességi UV/Vis detektálás
Amperometriás UV/Vis detektálás
Vezetőképességi UV/Vis detektálás UV/Vis, Oszlop ut.
származékképzés
Fluoreszcens, UV/Vis detektálás
Vezetőképességi Pulzáló amperometr.
UV/Vis detektálás Vezetőképességi UV/Vis spektrofot.
Vezetőképességi UV, Amperometriás HPIC
Anionok Szervetlen ionok
F , Cl , SO , stb.– – 2–4
pK < 7a HPIC Vezetőképességi
Amperometriás4
1.4. ábra:Elválasztási és detektálási rendszer választásának sémája az ionkromatográfiás analízisben
1.3. Az ionkromatográfia alkalmazása a környezeti analízisben 19
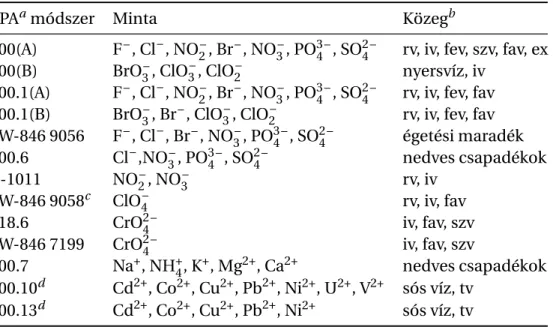
tagját, az ISO 10304-4:1997 jel˝ut [22] hazánk még nem vette át. Az Egyesült Államok Környe- zetvédelmi Hivatala - EPA - által alkalmazott US-EPA Method 300 jelzés˝u szabványsorozat is meglehet˝osen sok ionkromatográfiás módszert ajánl anionok meghatározására (1.1. táblá- zat).
1.1. táblázat:Természetes vizek és szennyvizek meghatározására kidolgozott ionkromato- gráfiás szabványok az Amerikai Egyesül Államokban
EPAamódszer Minta Közegb
300(A) F–, Cl–, NO2–, Br–, NO3–, PO3 –4 , SO2 –4 rv, iv, fev, szv, fav, ex 300(B) BrO3–, ClO3–, ClO2– nyersvíz, iv
300.1(A) F–, Cl–, NO2–, Br–, NO3–, PO3 –4 , SO2 –4 rv, iv, fev, fav 300.1(B) BrO3–, Br–, ClO3–, ClO2– rv, iv, fev, fav SW-846 9056 F–, Cl–, Br–, NO3–, PO3 –4 , SO2 –4 égetési maradék 300.6 Cl–,NO3–, PO3 –4 , SO2 –4 nedves csapadékok
B-1011 NO2–, NO3– rv, iv
SW-846 9058c ClO4– rv, iv, fav
218.6 CrO2 –4 iv, fav, szv
SW-846 7199 CrO2 –4 iv, fav, szv
300.7 Na+, NH+4, K+, Mg2+, Ca2+ nedves csapadékok 200.10d Cd2+, Co2+, Cu2+, Pb2+, Ni2+, U2+, V2+ sós víz, tv
200.13d Cd2+, Co2+, Cu2+, Pb2+, Ni2+ sós víz, tv
aEnvironmental Protection Agency – Egyesült Államok Környezetvédelmi Hivatala
brv – reagens víz, iv – ivóvíz, fev – felszíni víz, fav – felszín alatti víz, szv – szennyvíz, ex – extraktum, tv – tengervíz
ctervezet
daz ionkromatográfia a spektrometriai vizsgálat el˝otti mintael˝okészítést szolgálja
A víztisztítás során alkalmazott fert˝otlenítés anionos melléktermékeinek kimutatására is el˝onyösen alkalmazható az ionkromatográfia. Ilyen vegyületek például a bromát, klorit és a klorát. A bromát az ózonos fert˝otlenítés során képz˝odik a nyersvíz természetes bromidtar- talmából, míg a klorit és klorát a klór-dioxidos fert˝otlenítés mellékterméke. Ezek az ionok igen súlyos egészségügyi kockázatot hordoznak már néhányµg/dm3-s koncentrációnál is.
Például a bromát er˝osen karcinogén hatású, az EPA becslése szerint a potenciális rák kocká- zata 10−4már 5µg/dm3koncentráció esetén is, hosszú távú fogyasztás esetén [23]. Bromát ion meghatározását az EPA Method 300.1(B) szabvány szabályozza. Az elválasztást Dion- ex IonPac AS9-HC típusú oszlopon, 9 mmol/dm3-s nátrium-karbonát eluens és szupresszált vezet˝oképességi detektálás alkalmazásával hajtják végre. A módszer során használt nagy ka- pacitású oszlop növeli a bromátion kloridiontól való elválasztásának hatékonyságát [15].
A klórral való fert˝otlenítés során a vízben lév˝o szerves anyagokból halogénezett szerves szennyez˝ok keletkezhetnek, melyek közül legnagyobb mennyiségben a halogénezett ecet- savak találhatók meg az ivóvizekben. Attól függ˝oen, hogy a nyersvíz milyen mennyiségben
tartalmaz bromidot, klórozott- ill. brómozott-klórozott ecetsavak különböz˝o variációi kelet- kezhetnek [24]. Ezek a vegyületek er˝osen toxikus hatásúak, feltételezhet˝oen rákkelt˝ok is, bár ez utóbbi hatás egyel˝ore még nem bizonyított [25, 26]. Ivóvízben, köszönhet˝oen meglehet˝o- sen alacsony pKa értéküknek, disszociált formában, azaz anionokként vannak jelen. Ionkro- matográfiás meghatározásuk nehézségét els˝osorban az okozza, hogy a különböz˝o mérték- ben halogénezett ecetsavak nagyon eltér˝o módon köt˝odnek az állófázishoz, így analízisük f˝oként gradiens technika és induktív csatolású plazma tömegspektometria (ICP-MS) detek- tálás alkalmazásával oldható meg [24, 27].
A króm különböz˝o oxidációfokú formákban fordulhat el˝o a természetben. Míg a Cr(III) a cukor metabolizmus szempontjából alapvet˝o fontosságú nyomelem, addig a Cr(VI) vegyü- letei igen toxikusak, valószín˝uleg karcinogén hatással is rendelkeznek. A kromátion Dion- ex IonPac AS7 típusú anioncserél˝on és 1,5-difenil-karbonohidraziddal történ˝o származék- képzés utáni UV/VIS detektálással történ˝o meghatározásának kimutatási határa mindössze 0,018µg/l [28]. A módszer a hat vegyérték˝u króm ivóvízb˝ol, felszínalatti, vagy ipari szenny- vizekb˝ol történ˝o kimutatására egyaránt alkalmazható.
Hatékony ionkromatográfiás módszerek állnak rendelkezésre az er˝osen toxikus cianid kimutatására is, amelynek az ipar számos területe, a bányászat, a kohászat, és m˝uanyagipar is potenciális forrása. Az ionkromatográfiás elválasztás elektrokémiai detektálással akkor ajánlott, ha a minta tiocianátot [15], vagy egyéb kéntartalmú vegyületet tartalmaz, egyéb- ként mind a szupresszált vezet˝oképességi [29], mind az UV/Vis detektálás alkalmazható ki- mutatásukra [30].
Az ivóvíz-szennyez˝o fémek többsége az átmeneti fémek csoportjába tartozik, amelyek kimutatására spektroszkópiás módszereket alkalmaznak elterjedten. Léteznek azonban szabványosított ionkromatográfiás módszerek kationok, pl. ammónium-, nátrium-, kálium- , kalcium- és magnézumion szimultán elválasztására csapadék és es˝ovizekb˝ol, felszíni és felszín alatti vizekb˝ol, ivóvízb˝ol, vagy kevert kommunális és ipari szennyvizekb˝ol [31]. Külö- nösen el˝onyös a módszer ammónium- és egyéb kationok kimutatására aminok jelenlétében, mivel ezek a vegyületek zavarják a hagyományos kolorimetriás ammónia meghatározást.
Gyakran használják az ionkromatográfiát szennyvizek analitikai vizsgálatai során is, bár az ilyen minták esetében a mintael˝okészítésnek jelent˝os szerepe van. Az el˝okészítés álta- lában hígítást jelent, annak érdekében hogy a mér˝om˝uszer mérési tartományán tudjunk dolgozni. Az oszlop élettartamának megnövelése érdekében gyakran indokolt a hidrofób szerves anyagok sz˝uréssel történ˝o eltávolítása [32].
Az ionkromatográfiás analitikai módszereket az ivóvizek és különböz˝o szennyvizek vizs- gálata mellett széles körben alkalmazzák természetes eredet˝u vízminták, úgymint ásványvi- zek, mélységi talajvizek, termálvizek, fúrólyukak vízmintái, a talaj pórusainak vize, a felszíni vizek (álló- és folyóvizek), az es˝ovíz, savas es˝o, hó, vagy jéges˝o és jégtakarók elemzésére is.
1.3. Az ionkromatográfia alkalmazása a környezeti analízisben 21
Az es˝ovizek, és els˝osorban a savas es˝ok ionos összetev˝oinek meghatározása kiemelt fontos- ságú a savasodás a természetes és m˝uvi környezetre való hatásainak el˝orejelzésében . Lé- teznek eljárások a csapadékvízb˝ol, illetve atmoszferikus aeroszolokból kationok és anionok egyidej˝u elválasztására is gyengén savas kationcserél˝on [33].
Az ionkromatográfia területén napjainkban került el˝otérbe az igen toxikus perklorát fel- színi és felszín alatti vizekb˝ol történ˝o meghatározása [34, 35, 36]. A perklorát ion a szilárd rakéta hajtóanyagok kulcsösszetev˝oje, ezért els˝osorban olyan területeken szennyezheti a fel- színi és talajvizeket, ahol ilyen anyagokat, l˝oszereket, t˝uzijátékokat gyártanak, tesztelnek.
A felszíni vizek fémvegyületeinek meghatározásában is egyre nagyobb teret hódít az ion- kromatográfia. Nagy el˝onye, hogy mérhet˝ové teszi a különböz˝o oxidációs állapotok - pl.
a Fe(II) és Fe(III) - mennyiségét, a stabil fémkomplexeket – fém-cianidok –, vagy mind a szerves, mind a szervetlen arzén vegyületeket. Így lehet˝ové válik a fémvegyületek sorsának, transzportjának, toxicitásának el˝orejelzése [37].
A talajminták vizsgálata volt az ionkromatográfia egyik els˝o környezetanalitikai alkal- mazási területe. Hasonlóan a természetes vizekhez, a talajminták esetén is az a nagy el˝o- nye érvényesül, hogy a talaj extraktumokból a fémek különböz˝o oxidációfokú megjelené- si formái és a stabil fémkomplexek meghatározhatók ugyanúgy, ahogy a nitrogén, foszfor, kén mennyisége, valamint a hozzájuk tartozó oxoanionok, mint a nitrit-, nitrát-, foszfát- , vagy szulfátionok, amely komponensek igen fontosak a talaj min˝osége, term˝oképessége szempontjából [31]. Optimális esetben a mérés során injektált mintának alacsony a szer- ves anyag, er˝os sav és sótartalma, bár ez a talajminták extrakciója során hagyományosan alkalmazott módszerekkel nem igazán valósítható meg. Napjainkra már számos ionkroma- tográfiában jól alkalmazható extrakciós módszert dolgoztak ki, ami alapján a vizsgálandó vegyületekhez a megfelel˝o extraháló szer és eljárás kiválasztható [38].
Az iszapok, csurgalékok és szilárd hulladékok szervetlen iontartalmának meghatározása, alapjában véve nem tér el a talajmintáknál alkalmazott módszerekt˝ol [16]. Bár az ionkroma- tográfiát általánosan az anionok kimutatására használják ezeknél a mintáknál is, lehet˝oség van a csurgalékvizek illékony zsírsav tartalmának meghatározására, amit fontos információ a hulladéklerakó állapotára nézve [17].
Mivel környezeti minták analízisekor sok különböz˝o minta fordulhat el˝o, számtalan el- választási módszer jöhet szóba attól függ˝oen, hogy milyen összetétel˝u a minta, illetve me- lyek a meghatározni kívánt komponensek. Ezért az elválasztási módszert az adott problé- mához kell optimalizálni.
1.4. Anioncsere-kromatográfia
1.4.1. Állófázisok
Ahogy az 1.2.2. alszakaszban már említettem, az ionkromatográfiában els˝osorban szerves polimer alapú állófázisok terjedtek el. Kisebb mértékben azonban, de szervetlen alapú töl- teteket tartalmazó oszlopok is használatosak a gyakorlatban.
Szerves polimer alapú anioncserél˝ok
Sztirol-divinil-benzol, polimetakrilát és polivinil alapú ioncserél˝ok a legfontosabb szerves polimer alapú állófázisok az anionkromatográfiában. Ezek közül is a sztirol-divinil-benzol alapúak a legelterjedtebbek, köszönhet˝oen 0–14-ig terjed˝o pH stabilitási tartományuknak.
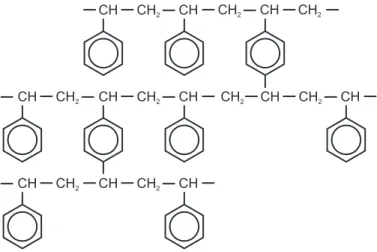
A sztirol kopolimerizációja divinil-benzollal szükségszer˝u a gyanta megfelel˝o mechanikai stabilitásának érdekében. A divinil-benzol, két funkciós csoportjának köszönhet˝oen, össze- köt két polisztirén láncot, így az állófázist térhálóssá teszi (1.5. ábra). Az ioncserél˝ok térhá- lósságát az el˝oállításukhoz felhasznált divinil-benzol százalékos mennyisége határozza meg.
A térhálósság befolyással van többek között a gyanta porozitására, szelektivitására, mecha- nikai stabilitására, ioncsere-kapacitására is [39]. Ez utóbbi, mely az egységnyi tömeg˝u vagy térfogatú gyantán található aktív csoportok számát jelzi az anioncserél˝o állófázisok talán legfontosabb tulajdonsága .
CH CH2 CH CH2 CH CH2
CH CH2 CH CH CH2 CH CH2
CH CH2
CH CH2 CH CH CH2
1.5. ábra:Sztirol-divinilbenzol szerves polimer alapú állófázis sematikus ábrázolása
A szerves polimer alapú állófázisokra az ioncserél˝o funkciós csoportokat két lépcs˝oben kötik. Az els˝o lépésben a gyanta aromás csoportjait klórmetilezik, a második lépcs˝oben pedig tercier-amin segítségével a klórmetil csoportot aminálják, így kvaterner-ammónium funkciós csoportokkal rendelkez˝o anioncserél˝ohöz juthatunk [39].