Á TMENETIFÉM ÉS RITKAFÖLDFÉM KOMPLEXEK REZGÉSI SPEKTROSZKÓPIAI SZERKEZETKUTATÁSA
Doktori (PhD) értekezés
Készítette:
Hajba László
okleveles vegyészmérnök
Témavezető:
Dr. Mink János
egyetemi tanár
Készült a Pannon Egyetem
“Környezettudományok” Doktori Iskolájához tartozóan
Pannon Egyetem
Mérnöki Kar Veszprém
2008
ÁTMENETIFÉM ÉS RITKAFÖLDFÉM KOMPLEXEK REZGÉSI SPEKTROSZKÓPIAI SZERKEZETKUTATÁSA
Értekezés doktori (PhD) fokozat elnyerése érdekében Írta:
Hajba László okleveles vegyészmérnök
Készült a Pannon Egyetem “Környezettudományok” Doktori Iskolájához tartozóan.
Témavezető: Dr. Mink János egyetemi tanár
Elfogadásra javaslom igen /nem
……….
(aláírás) A jelölt a doktori szigorlaton ... % -ot ért el,
Az értekezést bírálóként elfogadásra javaslom:
Bíráló neve: Dr. Kollár László igen /nem
……….
(aláírás) Bíráló neve: Dr. Stirling András igen /nem
……….
(aláírás)
A jelölt az értekezés nyilvános vitáján …...% - ot ért el
Veszprém, ……….
a Bíráló Bizottság elnöke A doktori (PhD) oklevél minősítése…...
………
az EDT elnöke
Kivonat
Több átmenetifém-komplex teljes színképi hozzárendelését és erőállandó számítását elvégeztem. Kutatásaim célja az általam vizsgált komplexek az irodalomban hiányos vagy igen régen publikált, illetve eddig még nem publikált IR és Raman-színképeinek felvétele, a meglévő rezgési adatok kiegészítése és a színképek teljes hozzárendelése. A szükséges kísérleti és elméleti módszerek – a vizes oldatokkal, illetve a sötét színű anyagokkal történő mérések nehézségeire hangsúlyt fektetve – szintén bemutatásra kerültek.
Első alkalommal végeztem el a M2(OCOCH3)4X2 típusú (M = Re(VII), Os(III); X = Cl, Br) komplexek teljes rezgési (infravörös, távoli-infravörös és Raman) spektroszkópiai vizsgálatát. Először regisztráltam a Re2(OCOCD3)4Cl2 komplex rezgési színképét. Az Os(OCOCD3)4Cl2 származékkal együtt az izotóp eltolódások és normálkoordináta számítások segítségével elvégeztem a színképek teljes hozzárendelését. A rendkívül intenzív Raman- sávok 282, 278 és 229 cm-1) alapján 3,228, 3,337 és 3,568 Ncm-1 fém-fém vegyérték erőállandókat kaptam sorrendben a Re(OCOCH3)4Cl, Re(OCOCH3)4Br és Os(OCOCH3)4Cl komplexekre. Igazoltam, hogy bármely egyszerűsített modell jelentősen felülértékeli a fém- fém kötési erőállandót (4,36 – 6,78 Ncm-1). A rénium-rénium kötések jellemző paraméterei (erőállandó, kötéshossz, vegyértékrezgési hullámszám) között empirikus összefüggéseket állapítottam meg, melyek segítségével analóg komplexek fontos szerkezeti paraméterei becsülhetők.
Az oktaéderes szerkezetű hexaakva komplexek ([Mg(OH2)6]2+, [Zn(OH2)6]2+, [Cd(OH2)6]2+, [Hg(OH2)6]2+, [Al(OH2)6]3+, [Ga(OH2)6]3+, [In(OH2)6]3+ és [Tl(OH2)6]3+) esetében a vizes oldatok távoli infravörös színképét először regisztráltam és a korábbi Raman- méréseket modern eszközökkel megismételtem. Erőállandó számítások eredményeként a fém- oxigén erőállandók és a Z1/2/R02
paraméter között (ahol Z – a komplex töltése, R0 – a fém- oxigén kötéstávolság Å-ben) jó közelítéssel lineáris összefüggést kaptam.
A sajátos szerkezetű nonaakva lantanoida komplexek többségénél a rezgési spektroszkópiai (infravörös, távoli-infravörös és Raman) méréseket és az erőállandó számításokat elsőként végeztem el. Az Ln(OH2)93+ (ahol Ln = La, Pr, Nd, Sn, Gd, Tb, Dy, Ho, Er, Tm, Yb és Lu) összetételű komplexeknél a lantanoida kontrakció következményeként, mind az ekvatoriális mind a prizmatikus Ln-O erőállandó növekszik a rendszám növekedésével, illetve az ion rádiusz, azaz a Ln-O kötéshossz csökkenésével.
Távoli infravörös és Raman-spektroszkópiás mérésekkel kiegészítettem a Co3(CO)9- µ3X típusú karbonil klaszterek korábbi adatait. Különös figyelmet szenteltem a Co3(CO)9- µ3CCl, Co3(CO)9-µ3S, Co2Fe(CO)9-µ3S és a Co3(µ3S)C6F5(CO)9 komplexek vázrezgéseire.
A normálkoordináta számítások eredményeként kapott fém–fém erőállandók és a kötéshossz között felállított empirikus összefüggés segítségével más hasonló szerkezetű karbonil klaszter
Abstract
Vibrational Spectroscopic Study of Transition and Rare Earth Metal Complexes
Complete assignments of the vibrational spectra and force constant calculations were performed for several transition metal-complexes. The main aim of the research was to complete the existing data and observe new spectroscopic data of the selected complexes. The applied experimental and theoretical methods are described, with special respect to measurements of aqueous solutions and dark coloured samples.
Full vibrational (infrared, far-infrared and Raman) spectroscopic studies of M2(O2CCH3)4X2 type complexes (where M=Re(VII), Os(III); X = Cl, Br) have been performed for the first time. The spectra of Re2(O2CCD3)4Cl2 complex were recorded at first.
Complete assignments of the vibrational spectra have been proposed on the basis of isotope shifts and results of normal coordinate calculations for Re and Os complexes. Very strong Raman features at 282, 278 and 229 cm-1 lead to 3.228, 3.337 and 3.568 Ncm-1 metal-metal stretching force constants for Re2(O2CCH3)4Cl2, Re2(O2CCH3)4Br2 and Os2(O2CCH3)4Cl2
complexes, respectively. It has been clearly demonstrated, that any simplified model of force constant calculations lead to considerable overestimation of metal-metal stretching force constants (4.36-6.78 Ncm-1). The characteristic parameters of Re-Re bonding (force constants, bond lengths and stretching wavenumbers) exhibit empirical correlations which can be used for prediction several structural parameters.
Far-infrared spectra in aqueous solutions of octahedral hexaaqua complexes namely for [Mg(OH2)6]2+, [Zn(OH2)6]2+, [Cd(OH2)6]2+, [Hg(OH2)6]2+, [Al(OH2)6]3+, [Ga(OH2)6]3+, [In(OH2)6]3+ and [Tl(OH2)6]3+, have been recorded first time, and their Raman spectra were updated with more advanced experimental facilities. The calculated metal-oxygen stretching force constants exhibited a satisfactory linear correlation with the parameter Z1/2/R02
(where Z is the charge of the metal atom and R0 is the metal-oxygen bond distance).
Vibrational (infrared, far-infrared and Raman) spectroscopic and force field studies of lanthanide nonaaqua complexes, of very special structure, have been performed for the first time. It was obtained that for Ln(OH2)93+ complexes (where Ln = La, Pr, Nd, Sn, Gd, Tb, Dy, Ho, Er, Tm, Yb and Lu) due to the so called lanthanide-contraction both the capping and prismatic LnO stretching wavenumbers and force constants are continuously increasing with increasing atomic number and decreasing ionic radii or LnO bond distance.
Far-infrared and Raman spectroscopic measurements have been completed for Co3(CO)9-µ3X type carbonyl cluster complexes. Special attention has been paid for skeletal vibrations of Co3(CO)9-µ3CCl, Co3(CO)9-µ3S, Co2Fe(CO)9-µ3S and Co3(µ3S)C6F5(CO)9 complexes. The calculated metal-metal force constants showed an empirical correlation with the bond lengths, which can be useful to predict bond distances from force constants for other
РЕЗЮМЕ
Для ряда комплексных соединений переходных металлов выполнено полное отнесение колебательных спектров и проведен расчет силовых постоянных. Описаны используемые экспериментальные и расчетные методы, особое внимание уделено измерениям в водных растворах и работе с темными образцами.
Впервые выполнено полное спектроскопическое исследование (КР, ИК, дальний ИК) комплексов типа M2(O2CCH3)4X2, (где M = Re(VII), Os(III); X = Cl, Br). Спектры комплекса Re2(O2CСD3)4Cl2 измерены впервые. На основании анализа значений изотопных сдвигов и результатов нормально-координатного анализа комплексов Re и Os предложено полное отнесение полос в колебательных спектрах. Полосы с высокой интенсивностью в спектре КР при 282, 278 и 229 см-1 соответствуют колебаниям связи металл-металл с силовыми постоянными 3.228, 3.337 и 3.568 Н см-1 для Re2(O2CСН3)4Cl2, Re2(O2CСН3)4Br2 и Os2(O2CСН3)4Cl2, соответственно. Показано, что использование упрощенных моделей для расчета силовых постоянных приводит к существенному завышению соответствующих силовых постоянных (4.36 – 6.78 Н см-1).
Эмпирически установлена корреляция между характеристическими параметрами связи Re-Re (силовые постоянные, длины связей и частоты валентных колебаний), которая может быть использована для предсказания ряда структурных параметров.
Впервые получены спектры в дальней ИК-области для октаэдрических аквакомплексов в водных растворах ([Mg(OH2)6]2+, [Zn(OH2)6]2+, [Cd(OH2)6]2+, [Hg(OH2)6]2+, [Al(OH2)6]3+, [Ga(OH2)6]3+, [In(OH2)6]3+, [Tl(OH2)6]3+), с использованием современного оборудования уточнены спектры КР этих частиц. Рассчитанные силовые постоянные связей металл-кислород проявляют почти линейную корреляцию с параметром Z1/2/R02 (где Z – заряд иона металла, R0 – длина связи металл-кислород).
Методами колебательной спектроскопии (КР. ИК, дальний ИК) впервые исследованы нонааквакомплексы лантаноидов, выполнен расчет силовых постоянных. Показано, что для комплексов Ln(OH2)93+
(Ln = La, Pr, Nd, Sm, Gd, Tb, Dy, Ho, Er, Tm, Yb и Lu) вследствие так называемого лантаноидного сжатия наблюдается монотонное увеличение как частот валентных колебаний, так и силовых постоянных LnO для обоих типов связей с увеличением атомного номера и уменьшением ионного радиуса или длины связи LnO.
Для кластерных карбонильных комплексов типа Co3(CO)9-µ3X выполнены дополнительные измерения Раман и дальних ИК спектров. Особое внимание было уделено скелетным колебаниям комплексов Co3(CO)9-µ3CCl, Co3(CO)9-µ3S, Co2Fe(CO)9- µ3S и Co3(µ3S)C6F5(CO)9. Установлена корреляция между рассчитанными силовыми постоянными связей металл-металл и длинами связей, что может оказаться полезным для предсказания длин связи на основании величин силовых постоянных для других карбонильных кластеров с аналогичной структурой.
Tartalomjegyzék
BEVEZETÉS...8
1. SZAKIRODALMI ÖSSZEFOGLALÓ...9
1.1.FÉM-FÉM KÖTÉSEKET TARTALMAZÓ ÁTMENETIFÉM KOMPLEXEK...9
1.1.1. Fémorganikus klasztervegyületek ...11
1.2.KOORDINÁCIÓ TÍPUSAI A FÉM-AKVA KOMPLEXEKBEN...11
1.2.1. Hatos koordináció ...11
1.2.2. Kilences koordináció ...12
1.3.FÉM-FÉM VEGYÉRTÉKREZGÉSEK SAJÁTOSSÁGAI...13
1.4.REZGÉSELMÉLET...14
1.4.1. Kétatomos molekulák rezgése...14
1.4.2. Többatomos molekulák rezgése ...17
1.4.2.1. Kinetikus energia... 17
1.4.2.2. Belső koordináták és a G-mátrix... 18
1.4.2.3. Potenciális energia ... 20
1.4.2.4. Normálkoordináták ... 20
1.4.2.5. Szimmetria koordináták ... 22
1.4.3. Izotópeffektus...23
2. KÍSÉRLETI RÉSZ...24
2.1.AZ ALKALMAZOTT MŰSZEREK, MÓDSZEREK...24
2.1.1. Távoli infravörös spektroszkópia...24
2.1.2. Raman-mikroszkópia ...25
2.1.3. Depolarizációs arány...26
2.2.MINTAELŐKÉSZÍTÉS...27
2.3.VIZES OLDATOK SPEKTROSZKÓPIÁJA...29
2.4.NORMÁLKOORDINÁTA ANALÍZIS...30
3. EREDMÉNYEK...31
3.1.KÉTMAGVÚ ÁTMENETIFÉM-ACETÁTÓ KOMPLEXEK REZGÉSI SPEKTROSZKÓPIAI VIZSGÁLATA [S1,S4]...31
3.1.1. Pontcsoport analízis ...31
3.1.2. Rezgések hozzárendelése...32
3.1.2.1. A karboxilátion-csoport (COO-) rezgései ... 38
3.1.2.2. CC vegyértékrezgések... 38
3.1.2.3. Fém-oxigén vegyértékrezgések... 39
3.1.2.4. Fém-fém vegyértékrezgés ... 39
3.1.2.5. Fém-halogén vegyértékrezgések... 40
3.1.3. Erőállandó számítások...40
3.2.FÉM-AKVA KOMPLEXEK REZGÉSI SPEKTROSZKÓPIÁJA [S2,S3] ...53
3.2.1.1. Vázrezgések hozzárendelése ... 54
3.2.1.2. Erőállandó számítások ... 58
3.2.2. Nonaakva komplexek ...60
3.2.2.1. Normálkoordináta analízis... 61
3.2.2.2. Rezgések hozzárendelése ... 61
3.3.TÖBBMAGVÚ ÁTMENETIFÉM-KARBONIL KLASZTEREK REZGÉSI SPEKTROSZKÓPIÁJA [S5]...66
3.3.1. A klaszterváz rezgéseinek hozzárendelése...67
3.3.2. Erőállandó számítások...70
4. ÖSSZEFOGLALÁS ...74
FELHASZNÁLT IRODALOM ...76
AZ ÉRTEKEZÉS TÉMÁJÁHOZ TARTOZÓ TUDOMÁNYOS KÖZLEMÉNYEK JEGYZÉKE ...82
EGYÉB TUDOMÁNYOS KÖZLEMÉNYEK...82
TUDOMÁNYOS ELŐADÁSOK ...84
A DOKTORI (PHD) ÉRTEKEZÉS TÉZISEI...86
THESES ...88
KÖSZÖNETNYILVÁNÍTÁS ...90
Bevezetés
Az átmenetifém-komplexek egyre nagyobb jelentőséggel bírnak mind az alapkutatásban, mind a gyakorlatban, például a katalitikus kémia területén. Kémiai viselkedésük megértéséhez nagyon fontos a molekulaszerkezetük ismerete. A röntgendiffrakciós szerkezet meghatározási módszerek mellett egyre nagyobb szerepe van a rezgési, azaz az infravörös- és Raman-spektroszkópiai szerkezetvizsgálatnak. A kísérleti adatok alapján elvégzett erőállandó számítások további hasznos információval szolgálnak a kémiai kötések természetéről, a kötéserősségről.
Munkám során három átmenetifém-komplex vegyületcsoport infravörös- és Raman- spektroszkópiai mérését végeztem el. Az első csoportba tartoznak a fém-fém kötéseket tartalmazó tetraacetátó komplexek. A második csoportot a különböző hatos és kilences koordinációjú fém-akva komplexek alkotják. Az utolsó csoport pedig a fém karbonil klaszterek. A dolgozat kísérleti részében a mérések során felmerülő nehézségekre, illetve az esetleges problémákra és azok korszerű megoldásaira térek ki, különös tekintettel a távoli infravörös és a Raman-spektroszkópia technikai nehézségeire. A Raman-spektroszkópiai technikák közül nagy jelentőséggel bír a Raman-mikroszkópia, mert a minta mérete minimálisra redukálható, és különösen a hő, illetve fotonérzékeny, vagy fluoreszkáló minták vizsgálatánál előnyös.
Kutatásaim célja a vizsgált komplexek a szakirodalomban hiányos vagy igen régen publikált, illetve eddig még nem publikált IR és Raman-színképeinek felvétele korszerű mérési berendezésekkel (FT-IR, FT-FIR, FT-Raman, Raman-mikroszkóp), a meglévő rezgési adatok kiegészítése és a színképek teljes hozzárendelése az elvégzett normálkoordináta számítások segítségével. Továbbá hasznos összefüggéseket állapítottam meg a kötések különböző paraméterei (kötéshossz, vegyérték erőállandók, vegyértékrezgési hullámszámok) között.
1. Szakirodalmi összefoglaló
1.1. Fém-fém kötéseket tartalmazó átmenetifém komplexek
A periódusos rendszer alacsony oxidációfokú fém elemeinek általános sajátsága, hogy fém-fém kötéseket tartalmazó komplexeket képeznek. Ezek a komplexek π-donor ligandumokkal, mint például halogénekkel és alkoxidokkal stabilizálhatók. A fém-fém kötés jelenléte akkor teljesen egyértelmű, ha hídkötésű ligandumok nincsenek jelen a komplexben, mint például a [Re2Cl8]2- esetében [1]. A d-csoport bizonyos elemei (mint pl. Fe, Co, Ir, Rh) az ilyen típusú komplexek π-akceptor ligandumokkal, főképpen CO ligandumokkal stabilizálhatók.
Az átmeneti fémeknél a fém-fém kötéseket tartalmazó vegyületek stabilitása, illetve a fém-fém kötés erőállandója arra enged következtetni, hogy a periódusos rendszerben egy adott oszlopon belül a rendszámmal nő a fém-fém kötés erőssége. Ennek valószínű oka a d pályák nagyobb térbeli kiterjedtsége.
A tetragonális szerkezetű fém-fém kötéseket tartalmazó komplexek esetén, mint pl.
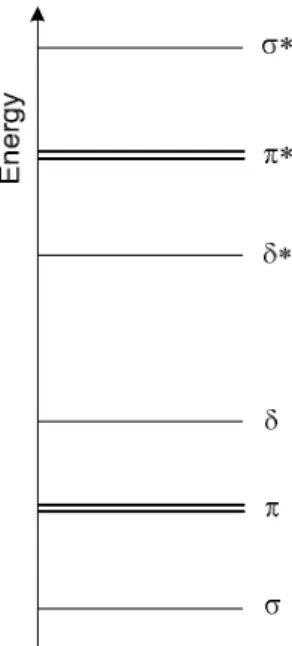
[Re2Cl8]2- és Mo2(OCCH3)4, a kötés rendje 1 és 4 közötti. Ha az összes kötő orbitál betöltött, a „konfiguráció” σ2π4δ2 (1.1. ábra) és négyes fém-fém kötés jön létre. Az egyik legismertebb négyes kötést tartalmazó komplex a dimolibdén-tetraacetátó komplex [2].
1.1. ábra: A fém-fém kölcsönhatás egyszerűsített molekulapálya-energia diagramja
Sokkal gyakoribb azonban a háromszoros fém-fém kötés. Ez abban az esetben jön létre, ha mind a δ-kötő, mind a δ*-lazító orbitál teljesen betöltött. A magasabb energiaszinten lévő π*-orbitál betöltöttsége a kötésrendben további csökkenéshez vezet. A kötésrend természetesen függ a ligandum típusától is. Tekintsük a Re2L10 típusú komplexek példáját, ahol két oktaéderes szerkezetű ReL5 fragmentum orbitáljai kölcsönhatnak egymással, hogy fém-fém kötést hozzanak létre. Abban az esetben, ha a Re alacsony oxidációfokú (elektronban gazdag) és az L ligandum π-akceptor típusú, akkor a π és δ pályák energiaszintje jóval a σ orbitálok alatt található és a komplexet a fémtől a ligandum felé történő viszont-koordináció stabilizálja. Ha a ligandum π-donor, akkor σ orbitál energiaszintje nem sokkal van a π és δ orbitálok felett. A Re2(CO)10 és a [Re2Cl10]2- komplexek esetén a fém-fém kötések molekulapálya diagramja a 1.2. ábrán látható. A karbonil komplexnél, ahol a Re 0 oxidációfokú, a fém-fém kötés egyszeres σ kötés. Ezzel ellentétben a hasonló szerkezetű kloro-komplexnél, ahol a Re +4-es oxidációfokú, négyes fém-fém kötés jön létre (1σ kötés, 2π kötés, 1δ kötés).
1.2 ábra: Az Re2L10 és két ReL5 fragmentum molekula orbitáljai közötti korreláció
1.1.1. Fémorganikus klasztervegyületek
A fémorganikus vegyületek definíciójához szükséges feltétel, hogy legalább 1 darab fém- szén kötést kell tartalmazniuk. Így tehát ebbe a vegyületcsoportba soroljuk például a Ni(CO4)4, [Fe4(CO)13]2-, illetve a Fe(C5H5)2 komplexeket. A hexacianoferrát-ion tartalmaz ugyan fém-szén kötéseket, mégsem soroljuk a fémorganikus vegyületek közé. Ezzel ellentétben a karbonil ligandumot tartalmazó komplexek a fémorganikus vegyületek csoportjába tartoznak.
Fém klaszterekről akkor beszélünk, ha a fém-fém kötések háromszöget vagy nagyobb zárt struktúrát alkotnak. A híd ligandum jelenléte a klaszterben növeli a lehetőségét annak, hogy a két fématomot a ligandum tartja össze, nem a fém-fém kötés. A fém-fém kötéshossz ismerete segít ennek a szerkezeti problémának a tisztázásában, mert ha a fém-fém távolság sokkal nagyobb, mint a fém atomrádiusz kétszerese, akkor okunk van feltételezni, hogy a fém-fém kötés vagy nagyon gyenge, vagy nincs jelen. Például az irodalomban [3-5] mai napig vita van a Fe-Fe kötés kialakulásáról a Fe2(CO)9 klaszter esetében.
1.2. Koordináció típusai a fém-akva komplexekben
A komplexek primer koordinációs szférájába tartoznak a központi fémionhoz közvetlenül rögzített ligandumok. A koordinációs térben a központi atomhoz közvetlenül kapcsolódó atomok számát nevezzük koordinációs számnak. Abban az esetben, ha egy komplex kationhoz, mint például [Mn(OH2)6]2+, egy anionos ligandum (pl. SO42-
) kapcsolódik elektrosztatikusan anélkül, hogy lecserélődtek volna a meglévő ligandumok, külsőszféra- komplexről beszélünk.
1.2.1. Hatos koordináció
A periódusos rendszer d-csoport fémjei leggyakrabban hatos koordinációjú komplexeket alkotnak. Az akva komplexeknél is a hatos koordináció érvényesül. 1H-es és 17O-NMR mérések alapján a hat vízmolekula oktaéderesen helyezkedik el a fém kation körül a primer koordinációs szférában, melynek oxidációfoka +2 vagy +3 [6].
A poláris vízmolekulák főként elektrosztatikus kölcsönhatás révén kapcsolódnak a központi Mn+-ionhoz. A Oδ-–Hδ+ kötések polaritása miatt, a vízmolekulák hidrogén kötések révén kapcsolódnak az elsődleges koordinációs szférában lévő vízmolekulákhoz, ezzel kialakítva egy másodlagos koordinációs szférát.
Mn+
O O
O O
O O
O
O H
-
- -
+
H
+
H
+
H
+
Elsődleges koordinációs
szféra
Másodlagos koordinációs
szféra
H+ H+
A d-csoport fémjei esetén a vízmolekulák az elsődleges koordinációs szférában olyan erős kölcsönhatásban vannak a központi fématom d orbitáljaival, hogy a d orbitálok kristálytér-felhasadást szenvednek, emiatt a hidratált fémionok oldatban általában színesek, pl. a [Ti(H2O)6]3+ lila, a [Fe(H2O)6]2+ zöld.
1.2.2. Kilences koordináció
A lantanoidák és aktinoidák csoportjába tartoznak azok az elemek, amelyek a kívülről számított harmadik (n-2) elektronhéjban f-elektronokat is tartalmaznak. Ezért is nevezik más néven az f-mező elemeinek őket. A 14 lantanoida elem fizikai és kémiai sajátságai közelítőleg megegyeznek. Ez abban nyilvánul meg, hogy oxidációfokuk +3. Csak néhány elemnél fordul
Komplexképzés során az f-pályák nem vesznek részt a kötések kialakításában. Itt is a d orbitáloknak van elsődleges szerepe a kötések kialakításában, csakúgy, mint az átmenetifémek esetében. A lantanoidáknál leggyakrabban a 9-es koordináció fordul elő. Az összes lantanoida (Ln) 9-es koordinációjú akva komplexeket képez [Ln(H2O)9]3+. Ezt röntgendiffrakciós vizsgálatokkal is igazolták, hogy a bromát és etil-szulfát sók hidratációjával 9-es koordinációjú akva komplexek képződnek [8-10].
1.3. Fém-fém vegyértékrezgések sajátosságai
A fém-fém vegyértékrezgés sávjai általában 300-100 cm-1 közötti tartományban jelennek meg, mivel a fématomok tömege viszonylag nagy. Ismeretes, hogy a fém-fém rezgések intenzív sávokat adnak a Raman-spektrumban, ami elsősorban a rezgés során a fém-fém kötés polarizálhatóságában történő jelentős változásnak köszönhető [11].
Ezzel ellentétben a távoli infravörös spektroszkópiával nagyon nehéz detektálni ezeket a rezgéseket, mert ha az azonos fémekből álló fém-fém kötésű rendszer középpontosan szimmetrikus, akkor a fém-fém vegyértékrezgés az infravörös spektrumban inaktív. Ha a szimmetriacentrum megszűnik, pl. ha két különböző fém kapcsolódik össze, akkor is csak nagyon gyenge infravörös sávot eredményez [12].
1.4. Rezgéselmélet
1.4.1. Kétatomos molekulák rezgése
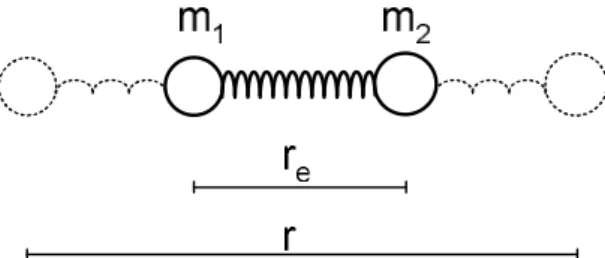
A rezgő mozgás természetének megértéséhez először két tömegpontból álló rendszert vizsgálunk. A modell nagyon egyszerű, 2 atomból (tömegük m1 és m2), valamint egy súlytalan, merev rugóból áll. A rugó egyensúlyi hossza re, megfeszített állapotban pedig r.
1.3. ábra: Kétatomos molekula modellje
Az egyensúlyi pozícióból történt elmozdulást a rezgés belső koordinátájának (q) segítségével írhatjuk le, amely
q = r – re (1.1)
A molekula potenciális energiája (V) a következő Taylor-sor segítségével írható le [13]:
2 3
2 3
2 3
0 0
( ) 1 ( ) 1 ( )
( ) (0) ...
2 6
dV q d V q d V q
V q V q q q
d q d q d q
= + + + +
(1.2)
ahol a 0 index az egyensúlyi állapotban (re) vett deriváltakat jelenti.
V(0) értéke zérusnak választható ugyanúgy, ahogy az egyensúlyi állapotban vett első derivált értéke is. A köbös, illetve az annál nagyobb hatványú tagok értéke pedig elhanyagolható. Az előző egyenlet tehát a következőképpen egyszerűsödik le:
2
2 2
2 0
1 ( ) 1
( ) 2 2
d V q
V q q k q
d q
= =
(1.3)
ahol k a kötés erőállandója.
A fenti összefüggés a harmonikus potenciál függvény. A harmonikus közelítésben az atomokra ható erő a Hooke-törvény segítségével írható le:
F = -k q (1.4)
Mivel a belső koordináta a molekula tömegközéppontjára vonatkozik, leegyszerűsíthetjük a problémát arra, hogy egy részecske oszcillál a tömegközéppont körül. Így a részecske tömege az ún. redukált tömeg lesz, amit a következőképpen definiálhatunk:
1 2
1 2
m m
m m
µ=
+ (1.5)
A Newton-féle mozgásegyenletet alkalmazva kapjuk:
2
0, azaz d q2
q k q k q
µ&&+ = µ⋅d t = − (1.6)
A fenti egyenlet írja le a harmonikus oszcillátort, megoldása:
q = q0 sin (2 π ν t + φ) (1.7)
ahol q0 – az amplitúdó φ – a fáziseltolódás
ν – a rezgési frekvencia (s-1).
A rezgési frekvenciát a következőképpen számolhatjuk:
1 2 ν k
= π µ (1.8)
Továbbá a rezgési energianívók (Ev) v rezgési kvantumszámtól való függését a következő összefüggés adja meg:
v v 1 E = hν
2
+
(1.9)
ahol v – 0, 1, 2, … a rezgési kvantumszám h – Planck-állandó
ν – a rezgési frekvencia (s-1).
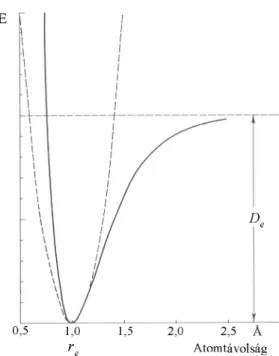
A valódi potenciál függvény – különösen nagy amplitúdójú rezgéseknél – eltér a harmonikustól (anharmonikus). Erre a potenciál függvényre jó becslést ad a teljesen empirikus Morse-függvény [14] (1.4. ábra):
( ) 2
. 1 a re r
anh e
V =D −e − (1.10)
ahol De – a disszociációs energia
a – egy adott molekulára jellemző állandó.
1.4. ábra: A Morse-függvény (folyamatos vonal) és a harmonikus potenciál függvény (szaggatott vonal) összehasonlítása
Az anharmonikus potenciált a Schrödinger-egyenletbe helyettesítve a rezgési energianívókra a
2 v
1 1
(v ) (v ) ...
2 2
e e e
E =hω + −hω x + + (1.11)
ahol ωe – a harmonikus frekvencia (s-1) xe – anharmonikus konstans.
Összességében elmondhatjuk, hogy harmonikus közelítés esetén a rezgési energianívók egymástól egyenlő távolságra helyezkednek el és a molekula képtelen disszociálni.
Anharmonikus közelítés esetén pedig a rezgési energianívók sűrűsödnek a v növekedésével, illetve magas vibrációs gerjesztés esetén a molekula disszociálhat.
1.4.2. Többatomos molekulák rezgése
Egy izolált molekula rezgési energiája a kinetikus (T) és potenciális (V) energiák összegeként írható fel [16]:
Evib= +T V (1.12)
1.4.2.1. Kinetikus energia
N pont helyzetét 3N koordinátával tudjuk leírni. Jelöljük xi-vel a Descartes-koordinátákat, mi-vel a molekulán belüli atomok tömegét, ahol i az atomok indexe. A kinetikus energia a következőképpen írható fel [17]:
2 1
1 x , x
2
N
i i i
i
T m ahol dx
= dt
=
∑
& & = (1.13)Mátrix formában felírva az egyenletet:
1 1 1
x x
2 2
T T
T = r& M⋅ =r& p Mr ⋅ − pr (1.14)
ahol xr& a Descartes-koordináták idő szerinti deriváltjainak vektora, a T felsőindexszel jelölt pedig a transzponáltja
M – az atomtömegek diagonális mátrixa
M-1 – az inverz atomtömegek diagonális mátrixa
pr – az impulzus-momentum vektor, a T felsőindexszel jelölt pedig a transzponáltja.
1.4.2.2. Belső koordináták és a G-mátrix
A rezgések leírására célszerű olyan koordinátarendszert választani, mely az atomok relatív elmozdulásait írja le. Ezek az ún. belső koordináták, amelyek kémiai jelentőséggel bírnak.
A molekulák rezgéseinek jellemzésére mindössze az alábbi négy belső koordinátatípus definiálása szükséges:
1. Vegyértékkoordináta (kötéstávolság változás)
r
2. Szögdeformációs koordináta α
3. Síkra merőleges deformáció (negyedik atom kimozdulása a három másik atom által alkotott síkból)
ρ
Sík szerkezetű molekuláknál, pl. etilén, benzol fordul elő.
4. Torziós koordináta (két sík elfordulása a metszésvonalak körül) 4
2 3 1
χ
Az 1,2,3 és a 2,3,4 atomokból álló síkok 2- 3 tengely körüli elfordulása, azaz a köztük lévő térszög változása.
A Descartes- és belső koordináták között az alábbi lineáris összefüggést adhatjuk meg mátrixos formában:
x
Rr=Br (1.15)
ahol Rr
– a belső koordináták vektora B – a transzformációs mátrix.
Az impulzus momentumot is átírjuk belső koordinátákra a következő lineáris transzformációval (T-index transzponáltat jelöl), ahol Pur
jelöli a belső koordinátákban megadott impulzus momentum vektort:
pr=B PT r
(1.16) A kinetikus energiára vonatkozó (1.14) egyenletbe behelyettesítve a következőt kapjuk:
1 1 1
2 2
T T T
T= P B Mr − B Pr= P G Pr r
(1.17) ahol a G-mátrixot a következőképpen definiálhatjuk: G = B M-1 BT.
A G-mátrix tehát az atomtömegek és a molekula geometria (kötéshosszak, kötésszögek, stb.) ismeretében meghatározható. A kinetikus energia természetesen kifejezhető a belső koordináta vektor idő szerinti deriváltjával is, úgy hogy Rr& =G Prismeretében (az összefüggés az impulzus momentum definíciójából és a korábbi egyenletekből levezethető), mivel G négyzetes és nem szinguláris mátrix, P G Rr= −1r& és a transzponáltja pedig PrT=R Gr&T −1, ezeket behelyettesítve az (1.17) egyenletbe a következőt kapjuk:
1 1
2
T= R G Rr&T − r& (1.18)
1.4.2.3. Potenciális energia
Egy többatomos molekula potenciális energiáját – figyelembe véve az egyensúlyi állapotot, illetve a köbös és a magasabb rendű tagok elhanyagolását – a belső koordináták segítségével a következőképpen írhatjuk fel:
3 2 3
,
, ,
0
1 1
( ) 2 2
N N
i j i j i j
i j i j i j
V R V R R f R R
R R
∂
=
Σ
∂ ∂ =Σ
(1.19)ahol fi, j– az erőállandó.
Mátrix formában felírva:
1 2
V = R F RrT r
(1.20) ahol F – a szimmetrikus erőállandó mátrix.
Habár az erőállandók kiszámíthatók a molekuláris hullámfüggvényekből, nagyon fontos ezen adatok számítása a kísérleti spektroszkópiás adatokból is. Egyrészt azért is, hogy teszteljük az ab initio és egyéb elméleti számítások helyességét, másrészt az egyszerűsített formájában hasonló típusú modellek esetén kémiailag jól összehasonlítható erőállandókat kapunk, viszonylag egyszerű számításokkal.
1.4.2.4. Normálkoordináták
Definiáljuk a normálkoordinátákat, amit a következő lineáris transzformáció kapcsol össze a belső koordinátákkal:
Rr = LQr
(1.21) A kinetikus és a potenciális energia a következőképpen fejezhető ki a normálkoordináták segítségével:
1 1
1 1 1
2 2 2
T T T T
T = R G Rr& − r& = Q L G LQr& − r& = Q E Qr& r& (1.22)
1 1 1
2 2 2
T T T T
V = R F Rr r = Q L F LQr r = Qr ΛQr
(1.23) ahol E – egységmátrix
Λ – a sajátérték diagonális mátrixa.
Az átalakítás eredményeként, az „off-diagonális” elemeket is eltávolítottuk az egyenletekből.
A rezgési frekvencia számításának utolsó lépéseként számoljuk ki egy többatomos molekula rezgési energiáját az (1.22) és (1.23) számú egyenletek segítségével:
1 1
2 2
T T
Evib = T+V = Q E Qr& r& + Qr ΛQr (1.24)
Mivel LT G-1 L = E és LT F L = Λ, illetve az LT = L-1 G, felírható a következő mátrix egyenlet:
L-1 G F L = Λ (1.25)
G F L = Λ L (1.26)
A fenti egyenlet tulajdonképpen nem más, mint a G F mátrix sajátérték egyenlete, ahol L a sajátvektor mátrix, Λ pedig a diagonális sajátérték mátrix.
A sajátérték probléma megoldható, ha a következő szekuláris egyenletnek léteznek gyökei:
│G F – E λi│ = 0 (1.27)
ahol λi-k – a szekuláris egyenlet megoldásai.
Az egyenletnek 3N-6 nem zérus gyöke van, azaz annyi, amennyi alaprezgése van egy többatomos molekulának.
A sajátértékek és az alaprezgések között a következő összefüggést írhatjuk fel:
λi= 4 π2 νi2
(1.28)
Áttérve hullámszámra (cm-1) és ha a tömegeket atomtömeg egységben (amu) az erőállandót Ncm-1 egységben vesszük figyelembe, a következő összefüggést kapjuk [11]:
2 i i
λ = 0,589148 ν 1000
% (1.29)
Összefoglalva, a rezgési problémát leegyszerűsítettük sajátértékproblémára a normál- koordináták segítségével. A sajátrezgések számítása pedig leegyszerűsödött G és F mátrixok meghatározására. A módszer alkalmazásával több számítógépes program is készült [18-22].
1.4.2.5. Szimmetria koordináták
Ha a molekula szimmetriával bír, a szekuláris egyenlet megoldása leegyszerűsíthető azáltal, hogy a belső koordinátákat egy transzformációs mátrix (U) segítségével szimmetria koordinátákká alakítjuk [23]:
S = U Rr r
(1.30) A szimmetria koordináták tulajdonképpen a megfelelő belső koordináták lineáris kombinációi. Normalizált szimmetria koordináták (Sr
) esetén, az U transzformációs mátrix ortogonális, tehát UT = U-1.
Hasonló transzformáció elvégezhető a G és F mátrixokra:
FS = U F UT (1.31)
GS = U G UT (1.32)
A szimmetria transzformáció az F és G mátrixokat blokkokra bontja, megkönnyítve ezzel a diagonalizációt, illetve a szekuláris egyenlet megoldását. Ugyanis a blokkok így külön-külön is diagonizálhatók. Minden egyes blokk általában egy-egy szimmetria típusnak felel meg.
1.4.3. Izotópeffektus
Abban az esetben, ha egy molekulában az egyik atomot egy másik izotópjára cseréljük, az elektronszerkezet és az erőállandó is – igen jó közelítésben – változatlan marad, de a redukált tömegek különbözősége miatt a rezgési frekvencia megváltozik. Az izotóp eltolódásra a következő összefüggést írhatjuk fel kétatomos molekula esetében:
ν = µ
ν µ
∗
∗
%% (1.33)
ahol a *-al jelölt adatok az izotópcsere utáni redukált tömegre vonatkoznak.
Több atomos molekulákra a Teller-Redlich szorzat szabály vonatkozik:
1 2 1 2
...
...
n n
G
= G
ν ν ν ν ν ν
∗ ∗⋅ ∗ ∗
⋅
% % %
% % % (1.34)
ahol ν%n∗ – az izotóp-helyettesített molekula rezgési hullámszámai cm-1-ben.
A fenti (1.34) egyenlet minden szimmetria blokkra érvényes. Az izotóp-helyettesítéses technika nagyban elősegíti a sávok hozzárendelését, ugyanis csak azokban a rezgésekben történik eltolódás, amiben az izotóp-helyettesített atom részt vesz. Emiatt a módszert előszeretettel alkalmazzák a rezgési spektroszkópiában [24-29].
Ezen kívül az izotóp-helyettesítés segíti az ún. „kísérleti” erőállandók meghatározásának pontosságát és növeli a meghatározható (kiszámítható) erőállandók számát.
2. Kísérleti rész
2.1. Az alkalmazott műszerek, módszerek
A kísérleti munkák laboratóriumi részét a Pannon (Veszprémi) Egyetem Analitikai Kémia Tanszékén, az MTA Kémiai Kutatóközpont, illetve a Svéd Királyi Technológiai Főiskola (Stockholm) laboratóriumaiban végeztük.
A közép infravörös színképek felvételéhez Digilab (Bio-Rad) FTS-60A típusú spektrométert használtunk, amely KBr hordozós Ge-fényosztóval és Peltier-hűtésű DTGS detektorral van felszerelve. A távoli infravörös spektrumok felvételéhez Digilab (Bio-Rad) FTS-40 spektrométert használtunk, ami nagynyomású higanygőz lámpával, fémhálós (Wire mesh) fényosztóval és polietilén ablakos DTGS detektorral van ellátva. A Raman-méréseket Digilab FT-Raman spektrométerrel végeztük. Gerjesztő lézerként egy 6 W maximális teljesítményű, a közeli infravörös tartományban működő, Nd/YAG lézert használtunk. A sötétebb színű, termikusan instabil minták esetén a Raman-színképek felvételéhez Renishaw- 1090 típusú Raman-mikroszkópot alkalmaztunk, 633 nm (vörös színű) He-Ne lézergerjesztéssel.
2.1.1. Távoli infravörös spektroszkópia
A távoli infravörös spektroszkópia fontos eszköz a szerkezetkutatásban, főként a fém- komplexek és egyéb, nehéz vagy gyengén kötött atomokat, illetve ligandumokat tartalmazó molekulák esetén. Számos fémorganikus és szervetlen molekula alaprezgése jelentkezik ebben a tartományban. A távoli infravörös spektroszkópia művelésekor sok gyakorlati problémával kell szembenéznünk. Nagyon kevés fényforrás használható ebben a tartományban, és általában nagyon gyenge az intenzitásuk. A diszperzív spektrométerek alacsony optikai áteresztőképessége szinte lehetetlenné tette a mérések kivitelezését. A gyakorlatban használt izzó feketetestek emissziója a távoli infravörös tartományban aszimptotikusan a nullához közeledik. Nem véletlen tehát, hogy a Fourier-transzformációs technikát az elsők között alkalmazták ebben a méréstartományban. Az interferométerek nagy optikai áteresztőképessége párosult azzal a sajátossággal, hogy viszonylag kis mechanikai pontosság szükséges a hosszú hullámok tartományában, és ez a fizikai tény egyszerűbbé teszi a mozgótükör szabályozását. Tehát a távoli infravörös tartomány tanulmányozása ily módon
az FT-FIR technika segítségével már a Fourier-spektroszkópia kezdeti szakaszában is sikeresen kivitelezhetővé vált.
2.1.2. Raman-mikroszkópia
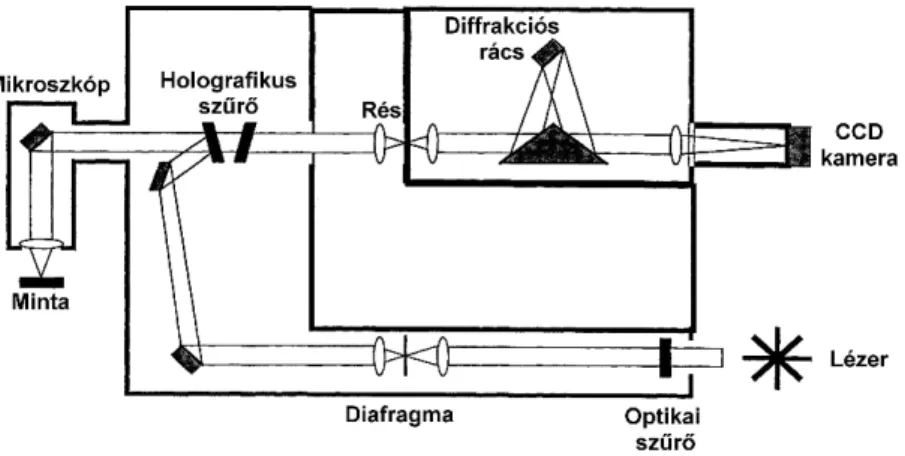
A Raman-spektroszkópia jelentős elterjedését nagymértékben elősegítette az FT-Raman technika alkalmazása mellett a Raman-mikroszkóp megjelenése. A CCD detektorok gyors technikai fejlődése tette lehetővé a Raman-mikroszkópia korszerű megvalósítását.
A módszer lényege, hogy egy holografikus szűrőkkel ellátott diszperzív Raman- spektrométerhez kapcsolunk egy fénymikroszkópot (2.1. ábra). A megfelelő optikával ellátott fénymikroszkóp segítségével a gerjesztőlézer jól fókuszálható, a visszaszórt fényt a nagy nagyítású és nagy numerikus apertúrával rendelkező objektívek hatékonyan gyűjtik össze. A kis hullámhosszú gerjesztőlézerek alkalmazásakor érhető el a legnagyobb térbeli felbontás, a diffrakciós limit Raman-mikroszkópnál kevesebb, mint 1 µm lehet. A konfokális optikai elrendezés is fontos sajátossága a berendezéseknek, azaz annak a képessége, hogy mennyire hatékonyan tudjuk kiszűrni, illetve eltávolítani a számunkra nem fontos mintaterületről szóródott fényt. Az itt használható 785 nm-es közeli infravörös lézerek alkalmazásával a Raman-hatáskeresztmetszet is 3,5-szerese egy FT-Raman rendszerhez képest (1064 nm). A Raman-mikroszkóp nagyérzékenységű többcsatornás CCD detektorral van ellátva, ami az FT- Raman spektrométerekhez képest még kisebb lézerintenzitások (1-2 mW) esetén is képes színképet regisztrálni, ezért számos fény- és hőérzékeny anyag is mérhető. A Raman- mikroszkóp nagyon jól alkalmazható sötét színű és termikusan instabil (pl. biológiai) minták esetén is. A korszerű multidetektoros változatok képalkotásra is alkalmasak.
2.1. ábra: CCD detektorral működő Raman-mikroszkóp
Mind az FT-Raman, mind a Raman-mikroszkóp együttes hiányossága, hogy a Rayleigh- szórás sávszélessége a jelenlegi szűrőkkel nem csökkenthető, így kis energiájú rezgések (pl.
rácsrezgések) nem mérhetők. A rendelkezésünkre álló szűrőkkel 50-100 cm-1 tartomány alatt már nem tudunk mérni. Meg kell említeni azt is, hogy a Raman-mikroszkópok leginkább szilárd minták vizsgálatára alkalmasak. Folyadékok, oldatok a módszerrel nehezen mérhetők, így pl. a sávok depolarizációs arányának meghatározása gyakorlatilag nem lehetséges.
2.1.3. Depolarizációs arány
A Raman-spektroszkópia egy további értékes információval is szolgálhat a szerkezet- meghatározásokhoz, illetve a rezgési sávok hozzárendeléséhez, ez pedig a depolarizációs arány. A ma használatos folyamatos üzemű (CW) lézer fényforrások síkban polarizáltak. A szórt Raman-sugárzás – az aktív rezgés természetétől függően – különböző szögben polarizálható. A depolarizációs arány ρ a következőképpen definiálható [30]:
III
I⊥
=
ρ (2.1)
ahol I⊥ a gerjesztőlézer polarizációjára merőlegesen polarizált sávok Raman-intenzitása, III pedig a gerjesztőlézer polarizációjával párhuzamosan polarizált Raman-sávok intenzitása.
A depolarizációs arány a rezgés szimmetriájáról szolgáltat számunkra információt, ezzel megkönnyítve a Raman-sávok hozzárendelését. Például, ha egy molekula gömbszimmetrikus és a rezgés teljesen szimmetrikus, akkor a gerjesztőlézer polarizációja megmarad, így a depolarizációs arány nagyon kicsi, 0 közeli értéket vehet fel, mivel a merőleges polarizált sáv intenzitása közel 0 lesz (polarizált sávok). Ellenkező esetben, amikor a rezgés során csökken a szimmetria vagy a molekula eleve nem szimmetrikus, jelentős depolarizáció lép fel. A nem totálszimmetrikus rezgéseknél a ρ ≈ 0,75. A totálszimmetrikus rezgések esetén 0 ≤ ρ < 0,75.
A depolarizációs arány például szerves komplex molekuláknál is szerkezeti információval szolgál, miközben a szubsztituenseket szisztematikusan változtatják [31]. A kisebb szimmetriájú molekuláknál is segítséget nyújthat a ρ ismerete, mivel az egyes csoportok (molekulafragmensek) lokális szimmetriája felismerhető, továbbá segíthet annak eldöntésében például, hogy a molekula rendelkezik-e szimmetriasíkkal vagy sem [32].
2.2. Mintaelőkészítés
A dolgozatban tárgyalt átmenetifém-komplexek többsége szilárd halmazállapotú. Az infravörös méréseknél az ismert, mátrixba való ágyazást, pasztillázást alkalmaztunk. A közép infravörös tartományban KBr-t, a távoli infravörös tartományban pedig polietilén mátrixot használtunk. Gyakran előfordulhat, hogy a KBr pasztilla préseléskor reakcióba lép a fémkomplexszel (kation/anion csere), ezért gyakran használtunk még a mintából készített paraffinolajos emulziót két KBr ablak között elhelyezve. Távoli infravörös színképek is készültek emulzió formájában (akár paraffinolajban, akár vazelinban), de ekkor a mintát polietilén ablakok közé tettük. Az emulziós módszer hátránya, hogy a pasztillába való ágyazáshoz képest a nagyobb mintaigénye.
Ha a szilárd mintánk kristályos anyag, akkor a kristályrácsban a molekulák közötti kölcsönhatások miatt a sávok pozíciója eltolódik – plusz sávok jelenhetnek meg a rezgési spektrumban – ami megnehezítheti az hozzárendelést. A probléma megoldásaként a mintánkat oldatba visszük, így a kristályrácsban lévő kölcsönhatások megszüntethetők (2.2. ábra).
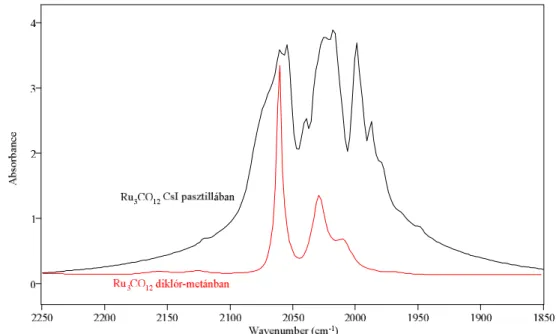
2.2. ábra: Ru3CO12 infravörös színképe a karbonil vegyértékrezgések tartományában Szerves oldószerekben történő méréseknél KBr optikai ablakkal ellátott, az oldószertől függően 0,006mm–1mm-es rétegvastagságú folyadékcellákat használunk. A távoli infravörös tartományban polietilén vagy szilícium optikai ablakkal ellátott folyadékcellákat alkalmazunk (2.3. ábra).
2.3. ábra: Si-ablakos folyadékcella (balra). A jobb oldalon lévő Si-küvetta ablak a Si reflexiós tulajdonságát illusztrálja.
A Si alapú küvettáknál a szilícium nagy törésmutatója miatt számolnunk kell jelentős reflexiós veszteséggel. Ez a küvettára eső fény 50 %-át is elérheti.
2.3. Vizes oldatok spektroszkópiája
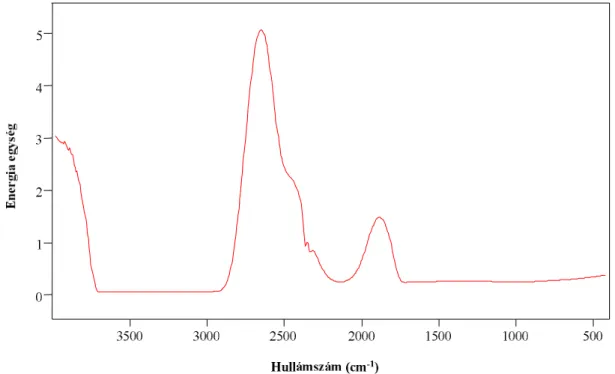
A vizes oldatok infravörös spektroszkópiája nem tartozik a rutinszerűen kivitelezhető mérések közé. A vizes oldatok mérése során több probléma merül fel [33]. Egyrészt, a víznek az infravörös tartományban még igen kis rétegvastagság esetén is széles és intenzív elnyelési sávjai vannak. A 2.4. ábrán látható, hogy ~3700-2800 cm-1 tartományban és 1750 cm-1 alatt a 0,127 mm rétegvastagságú vízminta nem ereszt át IR fényt.
2.4. ábra: Tiszta víz egysugaras színképe CaF2 cellában (0.127 mm film)
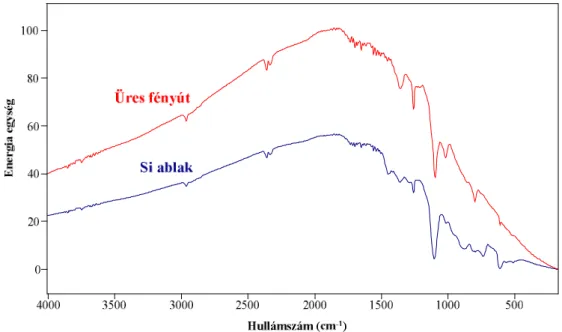
Másrészt, az infravörös spektroszkópiában általánosan alkalmazott optikai ablakok többsége vízoldható (KBr, NaCl, stb.). Ezért a vizes oldatok mérésénél Si-ablakos (teljes infravörös és távoli infravörös tartomány), illetve CaF2-ablakkal ellátott (közép infravörös tartomány) cellákat használunk. A CaF2 folyadékcellák általában 900-1000 cm-1 alatti tartományban átlátszatlanok. A víz erős elnyelése miatt a folyadékcella rétegvastagságát 6-12 µm-nek választottuk. Problémát okozhat még a Si-ablakok korábban már említett reflexiós vesztesége is (2.5. ábra).
2.5. ábra: A Si-ablak reflexiós vesztesége
A rendkívül kis rétegvastagságok miatt a vízben oldott minta nagy koncentrációja (2-10 mol/dm3) szükséges a mérések kivitelezéséhez [34]. Célszerű az oldat színképből az oldószer, azaz a tiszta víz színképének kivonása, hogy a számunkra megfelelő spektrális információk láthatóvá váljanak [35, 36].
Az infravörös spektroszkópiával ellentétben a Raman-spektroszkópiában a víz oldószerként való alkalmazása nem okoz gondot. Ugyanis a víz nagyon rossz Raman-szóró, ezért a Raman-spektroszkópiát előszeretettel használják vizes oldatok tanulmányozására.
Mintatartónak NMR csöveket vagy tetszőleges üveg edényeket, illetve üveg kapillárisokat használtunk.
2.4. Normálkoordináta analízis
Az erőállandó és normálkoordináta számításokat Wilson-féle GF-mátrix módszeren alapuló számítógépes program [18] segítségével végeztük el. A nagyméretű mátrixokkal elvégzendő algebrai műveletekhez az Octave és a Matlab programcsomagokat használtuk.
3. Eredmények
3.1. Kétmagvú átmenetifém-acetátó komplexek rezgési spektroszkópiai vizsgálata [S1, S4]
A többszörös fém-fém kötéseket tartalmazó komplexek nagyon jól alkalmazhatók heterogén katalitikus polimerizációs és hidrogénezési reakcióknál, mint iniciátorok [37]. Több közlemény tanúskodik arról, hogy például dimolibdén-acetátó komplexek jó hatásfokkal iniciálják a ciklopentadién kationos polimerizációját [38-42].
Az M2(O2CCH3)4X2 (ahol M=Re, Os; X=Cl, Br) típusú komplexek, illetve a fém-fém többszörös kötéseket tartalmazó molekulák jellemzésére már az irodalomban F.A. Cotton és munkatársai [2] több mint 50 dirénium-komplex röntgenszerkezeti vizsgálatát foglalták össze.
A dirénium-komplexek rezgési spektroszkópiai tanulmányozásával kapcsolatban ellenben kevés irodalmi hivatkozást találunk.
A Re2(O2CCH3)4X2 komplexekről már közöltek a szakirodalomban Raman- [43-45], illetve infravörös színképeket [45], ahol csak a Re-Re, Re-O és Re-Cl vegyértékrezgések hozzárendelését végezték el. A fém-fém vegyérték erőállandót többnyire kétatomos közelítés alapján számolták ki W.K. Bratton és munkatársai. Egy másik közelítés, amikor egy M2X8 egyszerűsített vázat alkalmaztak az erőállandó számításhoz [45]. Az Os-komplex esetén is történt már részleges asszignáció [46], de erőállandó számításokat még nem végeztek, ezért tűztük ki célul az M2(O2CCH3)4X2 típusú komplexek rezgési spektrumainak teljes hozzárendelését, illetve a vegyületek teljesebb geometriájának alkalmazásával az erőállandók meghatározását. Továbbá első ízben vizsgáltuk a komplexek deuterált analógjait, melyeknél az acetát metil-csoportjának összes hidrogénjét kicseréltük deutériumra. A komplexek előállítását német együttműködő partnerünknél, a Müncheni Műszaki Egyetem Szervetlen Kémiai Intézetének laboratóriumában végezték a szakirodalomban leírtak alapján [47, 48].
3.1.1. Pontcsoport analízis
Az M2(O2CCH3)4X2 típusú molekuláknak 90 alaprezgése van. Figyelembe véve, hogy a metil-csoport rezgéseit könnyű értelmezni és nagy valószínűséggel ezek a rezgések nem befolyásolják, illetve nem csatolódnak a fém-ligandum és fém-fém rezgésekkel, ezért olyan egyszerűsítést alkalmaztunk, amelyeknél a metil-csoportokat pontszerű tömegnek vettük.
Elvégezve ezt az egyszerűsítést, az alaprezgések száma 54-re csökken. A D4h pontcsoportba tartozó molekulának az 54 alaprezgése a következőképpen oszlik meg a szimmetriablokkok között:
6 A1g + 2 A2g + 4 B1g + 3 B2g + 6 Eg + A1u + 5 A2u + 3 B1u + 2 B2u + 8 Eu
ahol az E szimmetriatípusok kétszeresen degeneráltak.
A rezgések közül 19 Raman-aktív (6 A1g, 4 B1g, 3 B2g, 6 Eg), 13 infravörös-aktív (5 A2u, 8 Eu) és 8 inaktív (2 A2g, A1u, 3 B1u, 2 B2u). Figyelembe véve a degenerált rezgéseket, a rezgési spektrumban 32 alaprezgési sávot kell kapnunk.
Mivel ezeknek a molekuláknak szimmetriacentruma van, érvényes rájuk az ún. alternatív tilalmi elv. Ez azt jelenti, hogy az infravörös és a Raman-sávok nem esnek egybe.
3.1.2. Rezgések hozzárendelése
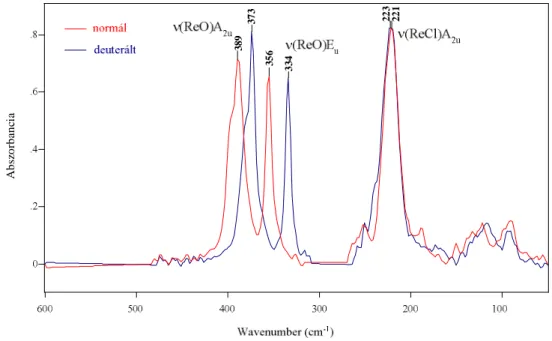
A rezgési színképek értelmezését nagyban segítették a korábbi részleges színképi hozzárendelések [45, 46], illetve az acetát-ion asszignációja [49]. A színképi hozzárendelést a deuterált változatok esetében tapasztalt izotópeltolódás tanulmányozása is elősegíti (3.1.
ábra).
Abszorbancia 389 373 356 334 223 221
3.1. ábra: A Re2(O2CCH3)4Cl2 molekula és deuterált változatának FIR színképe
A színképi hozzárendeléseket a 3.1. (Re-komplexek) és 3.2. (Os-komplexek) táblázatok tartalmazzák. A hozzárendelés részletezését főként a Re2(O2CCH3)4Cl2 komplexre, illetve