III./3.2.3.4 Diagnosis of dystonia
Diagnosis is based primarily on a thorough history and physical
examination. Information about family history, mode of inheritance, and the onset and progression of the disease is crucial. It is also important to rule out other neurological symptoms and to determine which muscles are affected, in order to be able to classify the disease as focal,
segmental or generalized dystonia.
Wilson’s disease should be ruled out by specific laboratory tests (CBC, liver enzymes, ceruloplasmin and copper in 24-hour urine sample).
Furthermore, neuroacanthocytosis should be excluded by differential blood test.
Certain rare metabolic diseases may cause generalized dystonia, such as methylmalonic-acidemia, gangliosidosis GM1, homocysteinemia type 1 or glutaracid acidemia type 1.
To determine whether the dystonia is primary or secondary, magnetic resonance imaging (MRI) has to be performed to detect or rule out structural brain damage.
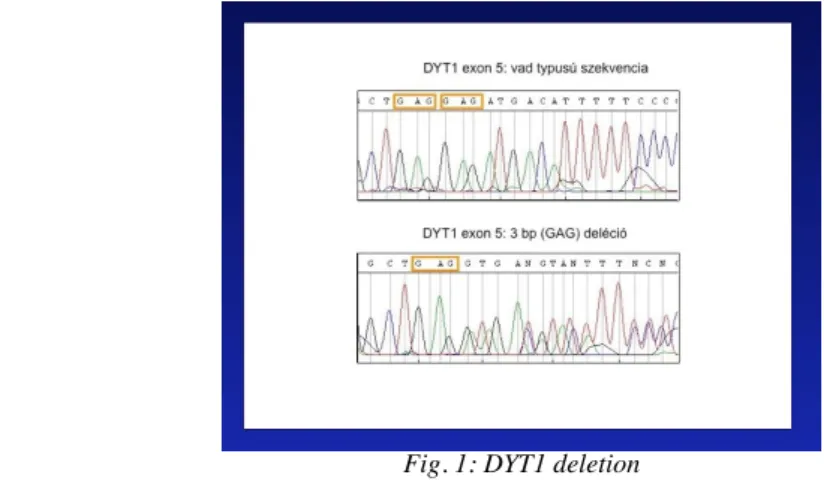
Genetic testing for DYT1 is positive in about 50% of early onset progressive generalized dystonia. It is an inexpensive, reliable and fast method, therefore it should be always asked for. In focal or segmental dystonia, DYT1 genetic testing is recommended in early onset cases (younger than 26 years). In elderly patients, genetic testing is only recommended if family history is positive.
Fig. 1: DYT1 deletion
Further genetic testing is available for rare diseases, such as DYT5 (DRD) and DYT11 dystonia. The sequencing of the large DYT5 gene is time-consuming and expensive, thus the clinical levodopa test is the first step in the diagnostic process. Generalized dystonia, myoclonus and positive response to alcohol are the typical features of DYT11 alcohol responsive myoclonic dystonia. Genetic testing of the ε-sarcoglycan gene confirms the diagnosis.