Contents lists available atScienceDirect
International Journal of Mass Spectrometry
j o u r n a l h o m e p a g e :w w w . e l s e v i e r . c o m / l o c a t e / i j m s
Dynamics of proton transfer from ArH + to CO
Björn Bastian
a, Eduardo Carrascosa
a,1, Alexander Kaiser
a, Jennifer Meyer
a, Tim Michaelsen
a, Gábor Czakó
b, William L. Hase
c, Roland Wester
a,∗aInstitutfürIonenphysikundAngewandtePhysik,UniversitätInnsbruck,Technikerstraße25/3,6020Innsbruck,Austria
bInterdisciplinaryExcellenceCentreandDepartmentofPhysicalChemistryandMaterialsScience,InstituteofChemistry,UniversityofSzeged,RerrichBéla tér1,SzegedH-6720,Hungary
cDepartmentofChemistryandBiochemistry,TexasTechUniversity,Lubbock,TX79409,UnitedStates
a r t i c l e i n f o
Articlehistory:
Received14August2018
Receivedinrevisedform3December2018 Accepted4December2018
Availableonline13December2018
Keywords:
Reactiondynamics Protontransfer Isomers Astrochemistry Crossedbeams Velocitymapimaging Directdynamicssimulations
a b s t r a c t
ThereactionofArH+withCOisafastprotontransferreactionthatcanformtwodifferentisomers,HCO+ andHOC+.Ithasbeeninvestigatedinacrossedbeamexperimentandwithdirectdynamicssimulations atcollisionenergiesrangingfrom0.4to2.4eV.Imagesofthedifferentialcrosssectionsrevealdominant forwardscattering,whichisevidencefordirectdynamics.Themeasuredproductinternalenergiesare primarilydeterminedbythereactionenthalpyandonlyatlargescatteringanglesdependnoticeably onthecollisionenergy.Thecomputationalresultsagreewellwiththemeasuredinternalenergyand scatteringangledistributionsandwiththepreviouslymeasuredtotalrateconstant.Thedirectreaction dynamicswithdominantforwardscatteringarewellreproducedbythealmoststeplikeopacityfunctions.
TheHCO+/HOC+branchingisfoundtobecloseto2:1inthesimulationsat0.83eVand2.37eVcollision energy.Amode-specificvibrationalanalysisprovidesfurtherinsightintotheisomerspecificdistribution oftheproductinternalexcitation.
©2018ElsevierB.V.Allrightsreserved.
1. Introduction
Ion-moleculereactions,oftennearthecollisionrate,compete withneutralreactionsatlowtemperatures[1].Theyaretherefore important to understand the composition and observed abun- dancesofmoleculesin coldplanetaryorinterstellargasclouds.
Protontransferisaprominentmechanismthatisknowntoproceed rapidlyinexothermicreactions[2].Arichnetworkofion-neutral reactionsintheinterstellarmediumisinitiatedbyprotontrans- ferreactionsofH+3 whichhasa lowerprotonaffinitythanmost interstellaratomsandmolecules[3,4].AmongthemisCO,thepre- cursorfortheformylcationHCO+thatinmanycasesisthemost abundantmolecularion[5].Alsothemetastableisoformylcation HOC+isformedinthisreactionandhasbeenobservedindense molecularclouds[6,7].TheHCO+/HOC+branchingratioindifferent environmentsandfuturedetectionsaresubjecttoongoinginterest [8–11]andcomparisontomodelssuggeststhatHOC+mustbecon- sideredinreliableabundancecalculations[12].AlsotherapidHOC+
∗Correspondingauthor.
E-mailaddress:roland.wester@uibk.ac.at(R.Wester).
1 Currentaddress:SchoolofChemistry,TheUniversityofMelbourne,Parkville, 3010VIC,Australia.
toHCO+conversionbyH2wassubjecttoacontroversialdiscussion [9,13].
Theproductbranchingofthemostimportantformationpath- wayH+3+COisnotyetfullyclarified[12,14].Investigationofthe simplerfouratomsystemArH++COcancontributetounderstand thegeneralcharacteristicsofprotontransferreactionsthatproduce differentisomers.ArH+hasbeenidentifiedintheCrabNebulain 2013asthefirstnoblegascompounddetectedinspace[15].Further detectionsindifferentgalacticandextragalacticsourcesfollowed in2015[16,17].Argoniumisa goodtracerofthealmostpurely atomic, diffuse interstellar medium witha low fractionalH2/H abundanceinthe10−4–10−3range[16,17].Isotopic36Ar/38Arratios aresignificantlylowerthanthesolarvalueandfutureobservations ofisotopicratioswithredshiftmayprovideusefulconstraintsfor nucleosynthesismodels[17].
InterstellarArH+ isformedviaionisationofatomicargonby cosmicraysfollowedbythereactionwithmolecularH2,andis mainlydestroyedbyphotodissociationorprotontransfertoneu- tralmolecularhydrogenoratomicoxygen[16].Chemicalmodels oftheMartianatmospherealsoincludefastprotontransferfrom argoniumtoseveralotherneutrals [18,19]. Theprotontransfer reaction
ArH++CO→Ar+HCO+ (rH=−2.33eV) (1) https://doi.org/10.1016/j.ijms.2018.12.004
1387-3806/©2018ElsevierB.V.Allrightsreserved.
ArH++CO→Ar+HOC+ (rH=−0.592eV) (2) isexothermicforboththeformylandisoformylcation.Thereac- tionenthalpiesrHarederivedfromtheprotonaffinitiesofthe neutral species [20–24]. The total ratecoefficientfor ArH++CO is1.25(35)×10−9cm3s−1.It hasbeenmeasuredin a drifttube from the thermal range to about 2eV and is nearly indepen- dentofthekineticcenter-of-massenergywhichistypicalalsofor otherexothermicprotontransferreactions[25].Thecapturerate kc=8×10−10cm3s−1issmallerthanthereportedvaluebutwithin twostandarddeviationsoftheexperimentalresult.Whilerecent models[18,19]assumeHCO+ astheonlyproduct,experimental ortheoreticalevidencefortheproductpartitioningintoHCO+and HOC+isstillmissing[26].
Herewepresentacombinedexperimentalandcomputational investigationofthetitlereactioninthe0.4–2.3eVenergyrange.
Thesestudiesextendexperimentalinsightintothetitlereaction toreactiondynamicsandproductbranching.Withitsfocusonthe HCO+/HOC+ratio,thisworkisinlinewithpreviouscrossedbeam experimentsontheprotontransferreactionoftheimportantinter- stellarmoleculesH+3 andHOCO+withCO[14,27].
2. Methods
2.1. Crossedbeamimaging
Angle-andenergydifferentialcrosssectionsofthetitlereac- tionhavebeenrecordedinacrossedbeamimagingsetupatfour differentcollisionenergiesfrom0.4to2.4eV.Werefertoprevi- ouspublicationsforadetaileddescriptionofthesetup[28,29].As aprecursor,argonwasionisedinaplasmadischargeandtrapped inaradiofrequencyoctupoleiontrapforthermalisationatroom temperature.Molecularhydrogenwasadmixedtotheargonbuffer gasin thetrap(20%H2 inAr)toreactexothermicallywithAr+ toformArH+ (H=−1.5eV[30]).In 10ms,anArH+/Ar+ ratioof about2:1wasachievedandafter40mstrappingtime,noresid- ualAr+couldbedetected.TheextractedArH+beamwascrossed withaneutralCO beamformedinasupersonicexpansion(20%
COin Ar).The chargedproductswerethen mappedona posi- tionandtime sensitivedetectorwiththevelocitymap imaging technique[31,32].Foreachevent,thethree-dimensionalproduct velocityisreconstructedinthecenter-of-massframeofthecolli- sion.Velocitydistributionsarebinnedwithrespecttothevelocity componentsparallel(vx)andperpendicular(vr)tothecollisionaxis andweightedbytheinverseperpendicularvelocity.Theresulting imagescorrespondtoslicedistributionsofreactivescatteringthat arecomparabletootherexperiments.Alternately,1000foreground and 100 background loopswere recorded in order to subtract eventsthat arenot related to reactivecollisions.For theback- groundmeasurements,theneutralbeamwaspulsedatalatertime toavoidcrossingwiththeionbeam,whilemaintainingthetotal pressureinthescatteringchamber.N2contaminationorCOthat diffusesfromthescatteringchambertothetrapmayformN2H+or HCO+/HOC+backgroundionswiththesamenominalmasses.They interferewiththelargeangleregionofthescatteringimagesatthe twolargestcollisionenergies.
Velocitymapimagingisalsousedtocharacterisethereactant ionandneutralbeamsforwhichthelatterisionisedbyelectron impact.Theexperimentswereperformedationenergiesfrom1to 6.4eVwithastandarddeviationrangingfrom70to110meV.The meankineticenergyoftheneutralbeamwas117meVwithafitted standarddeviationof20meVthatcorrespondstoatranslational temperatureof 13K. Convolutionof both velocity distributions determinesthemeancollisionenergiesandupperlimitsfortheir spread(Gaussianstandarddeviation),thatis0.38(2)eV,0.83(4)eV, 1.59(4)eVand2.36(5)eV.Thebeamswerecrossedatanangleof68◦
Fig.1. SchematicenergyprofilefortheprotontransferfromArH+toCO.ThePBE0 energiesusingthe6-31G(d,p)basissetaregivenineVrelativetothereactants.Val- uesinbracketsincludezero-pointenergies.Thezero-pointenergyforthereactants amountsto0.31eV.TS1andTS2arethetransitionstatesoftheargoncatalysedand freeHCO+/HOC+isomerisation.
withanglespreadsof2.4◦fortheneutralbeamandlessthan0.5◦ (1◦atthelowestenergy)fortheionbeam.Theresultingenergyand angularresolutioninthecenter-of-massframewasestimatedas describedpreviously[28].ThestandarddeviationsfromGaussian errorpropagationarenotlargerthan90meVforthecenter-of-mass kineticenergyand2.1◦(3.3◦atthelowestenergy)forthescattering angle.
2.2. Quasiclassicaltrajectorysimulations
Directdynamics simulationsof thetitle reactionswereper- formed with VENUS/NWChem [33–35] at 0.83eV and 2.37eV collisionenergy.ThetrajectorieswereintegratedbytheVelocity Verlet algorithm using a 0.2fs timestepwith on-the-fly calcu- lations of the forces. All electronic structure calculations used density functional theory (DFT) with the PBE0 hybrid density functional [36] and 6-31G(d,p) basis set[37]. Thisrather small basis setwaschosen forcomputationalefficiencyof thetrajec- tory simulations. Stationary point and zero-point energies for thereactant system,product channels,intermediatesand tran- sition states are given in Fig.1.Going tothe largertriple-zeta 6-311G(d,p)basissetchangesthestationaryenergiesbytypically +0.1eVand+0.2eVfortheAr+HOC+asymptote.Thesmallerbasis setreproducestheexperimentalexothermicitiesinEqs.(1) and (2),derivedfromtheprotonaffinitiesoftheneutralspecies,up todeviationsof0.07eVand0.3eVwiththePBE0functional.The computedCOprotonaffinityat thecarbon endof6.16eVcoin- cides with theliterature value [20]. The value obtainedat the oxygenendis 0.23eVlarger(4.56eV) thanthepublishedvalue of4.42eV[20].
Initialconditionsforrotationandvibrationwerechosenfrom therotatingMorseoscillatormodelandsampledfromatempera- tureof13KforCOand295KforArH+toresembletheexperimental conditions.FitstoDFTpointsforCOandArH+dissociationdeter- minedtheparametersoftheMorsepotentials(De=15.7/3.76eV, ˇ=2.12/1.99 ˚A−1 and r0=1.14/1.29 ˚A for CO/ArH+). The impact parameterbwassampledfromahomogeneousdistributionwithin a circular discwithradius bmax=6.5 ˚A. Thehomogeneous sam- plingwasinsufficienttocalculatereliablereactionprobabilitiesfor
b<0.9 ˚A. Onlyforthis purpose,216and 220additionaltrajecto- rieswerecomputedforthetwocollisionenergies.Thediatomic reactantswereinitiallyseparatedby10 ˚Aalongthecollisionaxis plustheperpendicularseparationb,thatis(102+b2)0.5 ˚A.Prod- ucts were generally identified at 11 ˚A separation by distance criteria.
Forthemode-specificvibrationalanalysisofthechargedprod- ucts,wecalculatedtheharmonicnormalmodesoftheequilibrium structuresofHCO+andHOC+withthesameleveloftheory.Each productmoleculeateachtimestepwasrotatedusingtheEckart transformation[38–40].Afterthisoperation,theproductmolecule hasa similarorientationcompared totheequilibrium structure anddisplacementsofatomsfromitsequilibriumpositionsbecome minimal. The Eckart transformed velocities and displacements were then transformed again using the transformation matrix betweenCartesianand normalmodecoordinatesfromthehar- monicfrequencyanalysis.Thesecoordinatesandvelocitiesyield mode-specificvibrationalenergiesintheharmonicapproximation asdescribed in more detail in [40].In geometries farfromthe equilibriumstructure,theharmonicpotentialenergymaylargely deviatefromthetruepotentialenergyoftheanalyzedproductrel- ativetoitsequilibriumpotentialenergy.Tosuppressunphysically largeharmonicactions,itwassuggestedtoextractthegeometry withtheminimumpotentialenergyfromthelastvibrationalperiod [41].Inasimilarway,weappliedtheanalysistothefinalprod- uctinthelastfiftytimestepsofeachtrajectoryandfilteredoutall timesteps,forwhichthetotalharmonicpotentialenergydeviates morethan0.2eVfromthecorrectcalculatedpotentialenergy.With thisapproach,accuratemodeenergieswereobtainedbyprimarily usingthekineticenergy.
3. Resultsanddiscussion 3.1. Experimentalresults
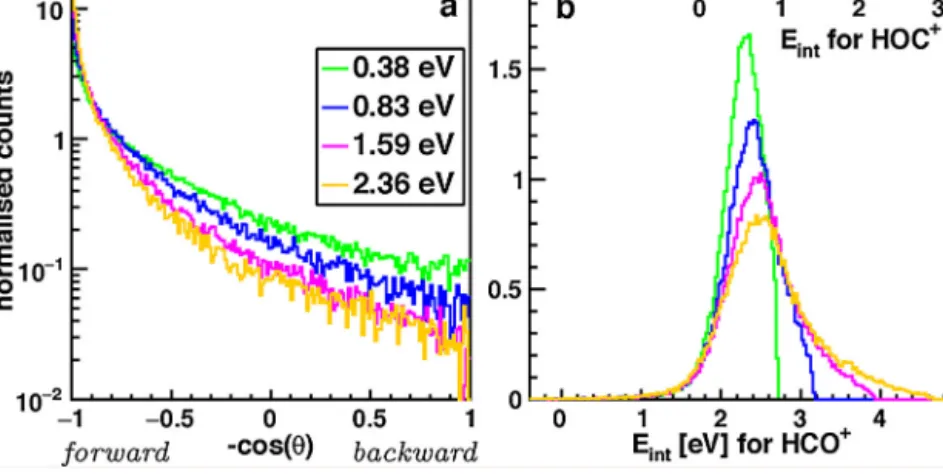
DifferentialcrosssectionsoftheprotontransferreactionofArH+ withCOhavebeenmeasuredatfourdifferentcollisionenergies 0.38(2) eV,0.83(4)eV,1.59(4)eVand2.36(5)eV.Theproduction velocitydistributionsarepresentedinFig.2togetherwiththedis- tributionsoftherelativekineticenergybeforethecollision(orange, denotedasthecollisionenergy)andaftercollision(blue)andthe derivedenergyofinternalexcitationoftheproducts(black).
Atallenergies,theprotontransferdominantlyproceedsinfor- warddirectionoftheproductionrelativetotheneutralreactant withsmallangulardeflection.Theforwarddirectionalityismore pronouncedathighercollisionenergies,seeFig.3a.Largenoise fromthebackground subtractionat about180◦ backward scat- teringatthetwohighestcollisionenergiesisduetobackground ions.
Theamountofenergy attributedtotherovibrational excita- tion(internalenergy)oftheproductscanbecalculatedfromthe productionvelocitiesbymeansofmomentumandenergyconser- vation.Thetotalavailablecenter-of-massenergyisthesumofthe collisionenergy andreactionexothermicity(disregardinginitial thermalexcitationofArH+)andreferredtoasthekinematiccutoff.
Itcorrespondstoproductsintheirgroundstateandismarkedby theoutermostblackandredcirclesfortheHCO+andHOC+products inFig.2(middlepanel).Theinteriorringsindicateenergydiffer- encestothecutoffinstepsof1eV.Becausetheneutralproduct isararegasatom,thedifferencescanbefullyattributedtointer- nalexcitationofthechargedproducts.Thekinematiccutoffand
Fig.2.ProductionvelocityandinternalenergydistributionsoftheprotontransferreactionArH++COatfourdifferentcollisionenergies.Upperpanel:Newtondiagram ofthecollisionincenter-of-massframeillustratingtheorientationandscatteringangle.Middlepanel:Experimentalproductionvelocitydistribution.TheNewtonrings correspondtothekinematiccutoffforHCO+(black)/HOC+(reddashed)and1eVspacings.Lowerpanel:Internalenergydistributions(black)withtheNewtonringenergies asinsetaxis.Largeanglescattering(>15◦)distributionsareshowningreenforthecutsdepictedbydashedgreenlinesinthevelocityimages.Additionally,therelative kineticenergiesbefore(orange)andafter(blue)thecollisionareshown.(Forinterpretationofthereferencestocolorinthisfigurelegend,thereaderisreferredtotheweb versionofthisarticle.)
Fig.3.Collisionenergydependenceofscatteringanglesandproductexcitation.(a)Distributionsofthecosineofthescatteringangle.Thenoiseatlargeanglesisdueto backgroundions.(b)InternalenergydistributionswithtwoaxesrelativetotheHCO+andHOC+groundstates.
1eVspacingsarealsoshownbytheblackandredinsetaxesinthe internalenergydistributionsinthelowerpanelofFig.2.Atallcol- lisionenergies,thedistributionsrisenearthekinematiccutoffof theHOC+productandextendasfarastheavailableenergyallows.
Unexpectedly,thedifferenceinexothermicitydoesnotletusdis- criminatethereactionchannels(1)and(2):thereactivecollisions mayeitherbeattributedtoHCO+productswithinternalexcitation above2eVortoHOC+productswithsmallerinternalexcitation.In otherwords,itisnotobservedthattheadditionalexothermicity thatisavailableforHCO+comparedtoHOC+isconvertedintorel- ativekineticenergy.Instead,thisadditionalexothermicityleadsto higherinternalexcitation.
Overall,thecollisionenergy essentiallydeterminestheaver- agerelativekineticenergyoftheproducts(seeFig.2,lowerpanel), whilethemaximaoftheinternalexcitationareclosetothereaction exothermicityat2.33eVforHCO+(0.59eVforHOC+)asisseenbest inFig.3b.Energytransferbetweencollisionandinternalenergy andbetweenexothermicityandrelativekineticenergyoftheprod- uctsseemstobesmall.Incontrasttothesmallshiftofthemean internalexcitationwiththecollisionenergy,thefulldistributionis relativelynarrowandsymmetricat0.38eVbutbroadensstrongly towardshigherexcitationwhenthecollisionenergyisincreased.
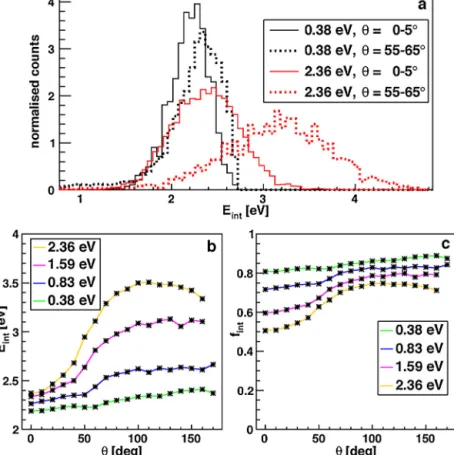
Todifferentiatethesetwofeatures,itisinstructivetoconsiderthe distributionsrestricted toangular deflectionslargerthan 15◦ in thelowerpanelofFig.2(greenlines),whereitisseenthatthe largeanglescatteringaccountsforthemajorpartofthebroaden- ingathighercollisionenergies.Thesamecanbeseendirectlywith smalland largeanglecutsappliedtotheinternal energydistri- butionsinFig.4a.The0–5◦sliceddistributionshiftsonlyslightly withincreasingcollisionenergywhilethe55–65◦distributionlies atsignificantlyhigherenergiesasfarasthetotalavailableenergy allows.
Toquantifythisbehaviour,themeaninternalenergywascal- culatedforthe±5◦ slicedistributionsfrom0to110◦ instepsof 10◦andispresentedforallcollisionenergiesinFig.4b.Smallangle scatteringischaracterisedbyameanproductinternalenergyclose tothereactionexothermicityalmostindependentlyofthecollision energy.Thisisequivalenttosimilarinitialandfinalkineticenergies.
Thetransferofcollisionenergyintointernalexcitationbecomes importantonlyforlargerscatteringangles.Atanglesabove50◦, a substantialamount of kineticenergy is converted intointer- nalenergy.Inordertoquantifythelatterbyarelativemeasure, weconsiderthefractionfintoftheinternalenergyrelativetothe totalavailableenergyinFig.4c.Whiletheinternalenergyatsmall scatteringanglesisalwaysclosetotheexothermicity,theinternal energyfractionfordifferentcollisionenergiesapproachessimilar valuesinthe74to87%rangeatlargescatteringangles.Whilethe
energyscalesinFig.4assumetheexothermicityforformationof HCO+,thesamegraphsfortheHOC+channellookverysimilar;i.e.
thedifferenceoftheexothermicitiesleadstoashiftoftheEintaxis.
Thefint axisisshiftedandrescaledasafunctionofthecollision energy.
Thefindingsdescribedaboveareinstrongcontrasttotheproton transferfromHOCO++COthatwereinvestigatedatenergiesrang- ingfrom0.3to2.3eVinapreviouspublication[27].Inthiscase, theformationofHOC+isendothermicby1.18eVandtheinternal energydistributionsriseatthekinematiccutofffortheexother- micformationofHCO+(rH=−0.55eV).Uptothehighestcollision energy,themajorpartoftheinternalenergydistributionislocated belowtheHOC+ formationthreshold.TheHOC+ productisonly accessibleinthehighenergyrangethatis,similarlytotheresults presentedhereforArH+,dominatedbylargeanglescattering.The ArH++COsystemwithlittleexchangebetweenkineticandinternal energyfeaturessimilarinternalenergiesforbothisomericproducts withrespecttotheHCO+groundstate.Ontheotherhand,thefor- mationofHOC+fromHOCO+requiresthetransferofasubstantial amountofthecollisionenergyintopotentialenergy.Assuming,for bothisomers,identicalfractionsoftheinternalenergyrelativeto thetotalavailableenergyfortherespectiveisomer—areasonable approachforthereactionwithHOCO+butnotforthereactionwith ArH+—anupperlimitof<2%fortheHOC+fractionwasobtained fromatwo-isomerfitin[27].
Another reaction for which the formation of both product isomers is exothermic, is the proton transfer from H+3 to CO (rH=−1.76/−0.13eV).Previouscrossedbeamexperiments[14]
in the 0.2 to 4.3eV collision energy range reveal monomodal internaldistributionsasinthereactionswithHOCO+ andArH+. Substantialpartsofthedistributionresidebelowthekinematic thresholdof HOC+ formation butat 1.8eV collisionenergy and above,thedominantpartallowsforbothisomersandrangesinto theautoisomerisationdomainathigherenergies.Inthisrespect, wemayregardtheH+3+COreactionasanintermediatecasewhich isalsojustifiedbythereactionenthalpybetweenthemoreandless exothermicreactionswithArH+andHOCO+.
Giventhemeasuredinternalenergydistributions,wegainno directinsightintotheproductbranchingoftheArH++COreaction fromtheexperimentaldataalone.Tocorrectlydisentanglethetwo reactionchannels,wethereforeusedirectdynamicssimulations.
3.2. Simulationresults
Direct dynamics simulations have been performed for the ArH++COsystematcollisionenergiesof0.83eVand2.37eV.For eachenergy,morethan5500trajectorieswerestartedwithimpact
Fig.4. Angle-differentialmeaninternalenergies.EnergiesareshownrelativetotheHCO+groundstate.(a)Scatteringangleslices=0–5◦and55–65◦fortwocollision energies.(b)Meaninternalenergyand(c)fractionofthemeaninternalenergyrelativetothetotalavailableenergyasafunctionofthescatteringangleforfourcollision energies.Thelargestanglesatthetwohighestcollisionenergiesweredominatedbybackgroundionsandomitted.
Table1
Totalnumberofsimulations,totalandproductspecificnumberofreactivetrajecto- riesandthemaximumimpactparameterforsimulationsattwocollisionenergies.
Energy bmax Total Reactive HCO+ HOC+
0.83eV 6.5 ˚A 5607 1740 1159 581
2.37eV 6.5 ˚A 5982 1097 724 373
Table2
Simulatedreactivecrosssectionsandrateconstants.Theinitialcollisionenergies andrelativevelocitiesaregiveninthefirsttwocolumns.Fromthereactionproba- bilityprandmaximuminitialimpactparameterbmax,thereactivecrosssectionr
andrateconstantkwerecalculatedasr=prb2maxandk=vrelr.
Energy vrel[m/s] pr r[ ˚A2] k[cm3s−1]
0.83eV 3133 31.0% 41.2 1.29×10−9
2.37eV 5244 18.3% 24.3 1.28×10−9
parametersbwithinacirculardiscwithb<bmax=6.5 ˚A.Thehomo- geneoussamplingwithinthediscisconfirmedbythehistogramof thesampledimpactparametersbinFig.6(graypointswithalin- earfit).Table1summarisesthenumberofsimulatedandreactive trajectories.Withtheratioprofthereactivetototalnumberoftra- jectoriesandtherelativevelocityofthereactantsvrelweobtainthe reactivecrosssectionrandreactionratek=1.29×10−9cm3s−1 (1.28×10−9cm3s−1at2.37eVcollisionenergy)inTable2.
The reaction rate only negligibly depends on the colli- sion energy and compares well with the experimental value 1.25(35)×10−9cm3s−1byVillingeretal.[25]Botharelargerthan thecapturetheoryratekc=8.0×10−10cm3s−1calculatedfromthe meanpolarizability˛¯=13.08a30ofCOreportedbyDiercksenetal.
[42] Thisisreflected by thereactivecrosssectionsof 41.2and
24.8 ˚A2thatarelargerthanthecapturecrosssectionsof25.8and 15.3 ˚A2,respectivelyat0.83and2.37eVcollisionenergy.
We obtaintheHCO+/HOC+ productbranching ratioof about 2:1fromTable1(1.99:1and1.94:1at0.83and2.37eV,respec- tively).Theanalysis ofthereactionprobability asa functionof theinitialorientationofthebimolecularreactantswithrespectto theirrelativevelocityrevealsthattheArH+orientationonlyslightly influencesthetotalreactivity.TheCOorientationhasastrongeffect ontheoutcomeofasinglereaction(seeFig.5).At0.83eVcollision energy,onlyHOC+isformediftheoxygenatomisorientedtowards ArH+uptoanangleof45◦ withrespecttotherelativevelocity.
TheprobabilityofHCO+thenriseslinearlywithlargeranglesand theoppositeistrueifthecarbonatomisorientedtowardsArH+. Thelineardependenceisalsoseenat2.37eVcollisionenergybut withoutthepronouncedthresholdbehaviour.
Wenowgiveacomparisontotheexperimentaldataandthengo moreintodetailonthesimulationresults.Thestatisticsofthedirect dynamicssimulationsareinadequateforadirect comparisonto themeasuredtwo-dimensionaldifferentialcrosssections.Instead, wecomparethewellsampledone-dimensionalangleandinternal energydistributions.
Themeasuredandsimulatedscatteringangledistributionsin Fig.7comparewellonalogarithmicscaleuptolargescattering angles.Themeasureddistributiondropsfasteratanglesnearand above90◦ especiallyatthehighercollision energy.Thesimula- tionsdonotrevealstrikingdifferencesbetweenthetwoproduct channelsgiventhesmallnumberofreactivetrajectoriesforlarge scatteringangles.
The calculation of the internal energy using the reaction exothermicitydependsonthefinalproducts.Becausetheisomers cannot beseparated inthe crossedbeamexperiment, we refer totheinternalenergy Eint relative totheHCO+ groundstate.It
Fig.5.ReactionprobabilityasafunctionoftheinitialCOorientationanglewithrespecttotherelativevelocityvrelofthereactants.Anangleof0◦correspondstotheoxygen atombeingalignedtowardstheArH+reactantion.
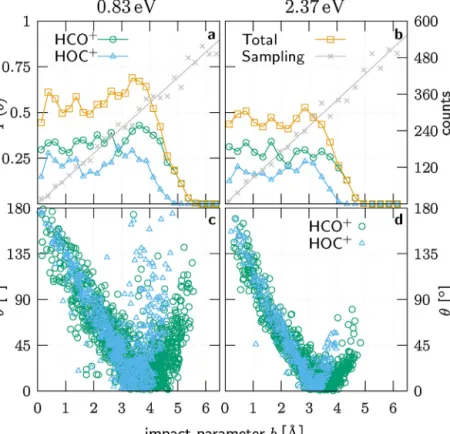
Fig.6. SimulatedopacityfunctionP(b)andscatteringanglesasafunctionoftheimpactparameterb.ThetotalreactionprobabilityandtheseparateHCO+andHOC+ contributionsareshownfor(a)0.83eVand(b)2.37eVcollisionenergy.Thenumberofsimulatedtrajectoriesineachintervalisgivenbythealternatey-axis.Scattering anglesat(c)0.83eVand(d)2.37eVcollisionenergyaredistinguishedforthetworeactionchannelsbydifferentsymbols.
Fig.7. Scatteringangledistributionsfromexperimentsandsimulationsat(a)0.83eVand(b)2.37eVcollisionenergy.Separatedistributionsofthesimulatedproductsare shownaswell.
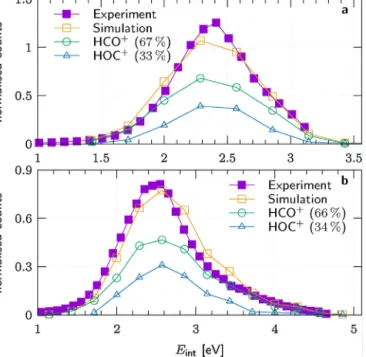
Fig.8.Distributionoftheproductinternalenergyat(a)0.83eVand(b)2.37eVcol- lisionenergy.Thetotaldistributionfromthedirectdynamicssimulations(orange squares)isthesumofthedistributionsoftheproducts(greencirclesandbluetrian- gles)andisnormalisedtoatotalareaofone.TheinternalenergyforbothHOC+and HCO+isgivenrelativetotheHCO+groundstate.(Forinterpretationofthereferences tocolorinthisfigurelegend,thereaderisreferredtothewebversionofthisarticle.)
includesboth rovibrational excitation andisomerisation energy whenrelatedtotheHOC+products,whichareclearlydistinguished inthesimulations.Asopposedtotheapproximateinternalenergy derivedfromexperimentaldata,thesimulationresultsaddition- allyincludethesmallandalmostnegligibleinitialexcitationofthe reactants.ThesimulatedtotalEint distributionsmatchcloselyto themeasureddistributionsatbothsimulatedcollisionenergiesin Fig.8.At0.38eVcollisionenergy,thetotalEintdistributionsepa- ratesinto67%oftheHCO+and33%oftheHOC+productwithvery similarmeanandspread,2.4±0.4and2.4±0.3eV.Thisexplains theabsenceofaclearindicatorforasecondproductchannelinthe experimentaldata.Similarly,HCO+andHOC+contribute66%and 34%tothereactivityat2.37eVwithmeanandspread2.7±0.6and 2.7±0.5eV.
Twoidealisedmodelcasesofdistributingtheavailableenergy for thetwo isomersare (a)similarkinetic energy distributions and(b)similarfractionsfintoftheinternalenergyrelativetothe availableenergy(relativeenergylosses).Forcomparisonwithour
experimentaldata, we calculate fint=Eint/Emax.Neglecting ini- tialexcitationofthereactants,Emax=Erel+EexoandEint=Emax− Erel ,withthecollisionenergyErel,zero-pointcorrectedexother- micities Eexo fromFig. 1and the relative energy aftercollision Erel.Inthepresent work,wefounda situationthatcorresponds tocase(a)withsimilarkineticenergy distributionsforthetwo isomers.Theobtainedrelative energylossesforHCO+/HOC+are 0.72±0.11/0.49±0.17and0.56±0.13/0.35±0.15at0.83eVand 2.37eVcollisionenergy.
Incontrasttothepresentreactionsystem,thepreviouslystud- iedreactionofHOCO++COisincompatiblewithcase(a),because almostallproductshavekineticenergiesabovethekinematiccutoff fortheformationofHOC+[27].Instead,case(b)hasbeenassumed toestimateanupperboundoftheHOC+fraction.Thesamemodel wasusedfortheH+3+COreactiontoderiveupperboundsof24%at 1.8eVand10%atlowercollisionenergies[14].Inlightofthepresent resultsforArH+,itbecomesapparentthatenergydistributionscor- respondingmoretothecase(a)canactuallynotbeexcludedfor thereactionofH+3.ThiswouldyieldlargerHOC+fractionsforthe reactionofH+3+COthanpreviouslyestimated.SuchlargerHOC+ fractionsaresupportedbyrecentchemicaldynamicssimulations [43],eventhoughthesimulatedinternalenergydistributionsdo notquantitativelyagreewiththeexperiments.
Angledependentproductinternalenergieswiththeirstandard deviationsin10◦slicesarepresentedinFig.9.Thereisgoodagree- mentwiththeexperimentalresultsupto60–90◦.Despitelarge statistical fluctuations at thelarger collision energy, there is a cleartrendforlargerinternalexcitationatlargeranglesbutthe meaninternalenergyisslightlyunderestimatedbythesimulations mainlyduetotheHOC+products.Thisseemssurprisinggiventhe theoreticalzero-pointcorrected exothermicityof 0.89eVthatis largerthantheexperimentalvalueof0.6eV.Moresignificantisthe separationofthemeaninternalenergies(alwaysrelativetothe HCO+groundstate)ofthedifferentproductsatlargeanglesespe- ciallyat2.37eVcollisionenergy.Theinefficienttransferofkinetic intointernalenergy,measuredforlowscatteringanglesandsmall energies,suggestsaninfluencefromthedifferentexothermicities predominantlyatlargerscatteringangles.
Moreinsightintothedynamicsofthetitlereactionisgained from theopacity functions, which are presented in Fig.6.The reaction probabilitiesare coarsely constant between0.6 (lower energy)and0.5(higherenergy)onarangeofimpactparameters wellbeyondthecapturelimitsof2.86and2.20 ˚A.Thethermody- namicallyfavouredHCO+ isthemorelikelyandatlargeimpact parameters(>4 ˚Aat2.37eV)onlyproduct.Thisisunderstoodbythe dipolemomentofC–O+intheorderof0.12debyes[42]thatfavours theorientationof theCside towardsArH+.Since largerimpact
Fig.9. Experimentalandsimulatedangle-differentialmeaninternalenergiesat(a)0.83eVand(b)2.37eVcollisionenergy.SimulatedresultsforHCO+andHOC+products arealsoshownseparately.
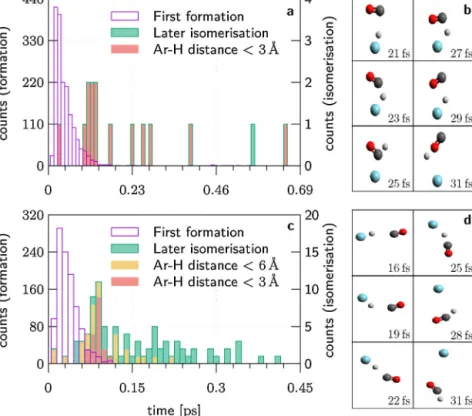
Fig.10.ProductformationtimesrelativetothemomentofnearestapproachofthereactantsArH+andCOat(a)0.83eVand(c)2.37eVcollisionenergy.Differentscalesare usedforthecountsoffirstproductformation(leftaxis)andsubsequentisomerisationevents(rightaxis).Differentcolorsshowiftheargonatomisneartothehydrogen atomduringisomerisation.Examplecartoonsareshownfor(b)anargoncatalysedisomerisationwithbackwardscatteringat0.83eVand(d)adirectreactionwithforward scatteringat2.37eV.
parametersaremorefrequent,thelargeimpactparameterrangeis animportantcontributiontothetotalHCO+/HOC+branchingclose to2:1(seeTable1).
Theregionofsmall impactparametersis directlyconnected toproductsscatteredbackwardsintolargeanglesasdepictedin Fig.6candd.Thescatteringanglesdecreasewithincreasingimpact parametertoitsminimumatthegloryimpactparameterandthen riseduetothelong-rangeattractivepart[44]ofthepotentialuntil theopacityfunctionbecomeszero.Incasehydrogenapproaches theCsideofCOinareactiontowardsHCO+,thecarbonmonoxide isexposedtothedeeperpotentialwellsuchthatthekineticenergy willbelargerintheproximitytoargon.Thisisrelatedtoaglory impactparameterthatisonaverageslightlylargerforHCO+than forHOC+,asseeninFig.6candd.
Relatedtothescatteringangle,wenoteonepeculiarityinthe opacityfunctionatthelowercollisionenergyinFig.6a.Withvan- ishingangledeflectionneartherespectivegloryimpactparameter, thereactionprobabilityforbothHCO+andHOC+isenhancedatthe lowercollisionenergy.Thisleadstoamaximumopacityofabout 70%nearb=3.4 ˚A.Thisisnotthecaseatthehighercollisionenergy withlesstimeforreorientationoftheprotontowardsCO.Duetothe higherenergy,thetransferredprotonalsobouncesbacktoargon in3ofatotalof11nonreactivetrajectoriesnearb=3.3 ˚A.
InordertoidentifyifisomerisationfromHOC+ toHCO+con- tributes to the computed preference for HCO+ formation, we countedthenumberofinitiallyformedandfinalproductsindiffer- entimpactparameterranges.HCO+moleculesaredetectedinthe trajectorieswhen(i)theH–Odistanceisatleast1.2timeslarger thantheH–Cdistance,(ii)thesumofbothdistancesissmallerthan 5 ˚Aand(iii)thebendinganglerelativetothelineargeometryofthe moleculeissmallerthan70◦.Condition(i)isinvertedfordetection ofHOC+.Onlyabout0.5%ofthereactivetrajectoriesundergoiso- merisationaftertheinitialproductformationatimpactparameters above2 ˚Aandwerestrictthefollowingdiscussiontob<2 ˚A.
At0.83eVcollisionenergy,isomerisationoccursin3%ofthe reactionswithb<2 ˚A, butequally inboth directions.The inter- nalenergyofthefinalproductsrangesfrom2.4to3.2eV(mean 2.8±0.3eV)andallisomerisationeventsbelowtheautoisomerisa- tionthresholdof3.1eV(seeFig.1)arecatalysedbytheargonatom.
Incontrast,isomerisationoccursin13%ofthereactionsat2.37eV withb<2 ˚AsuchthattheinitialHCO+branchingof61%isincreased to65%.Thisisduetoisomerisationeventsinthepresenceofargon thatoccuratallinternalenergiesandleadtoHCO+formationin 14of16cases.Wenote,however,thatalmostallisomerisingtra- jectoriescomealongwithfinalinternalenergiesstartingnearthe autoisomerisationthreshold(meanandspread3.9±0.5eV),and thetwoisomerscannolongerbedistinguished.
Usingthecriteriaforproductformationthatweredescribedfor theisomerisationstatisticsabove,wecanalsodeducethetimes ofinitialproductformationandlaterisomerisationinalltrajec- tories.Theformationtimesrelativetothemomentofthenearest approachofthereactantsarepresentedinFig.10.Theprotontrans- fertypicallyoccurslessthan0.1psafterthenearestapproachand isfasteratthehighercollisionenergyinFig.10c.Acartoonofa fastdirectreactionwithlargeimpactparameterispresentedin Fig.10d. On average,at 0.83 and2.37eV,HCO+ isformedafter 4.2±4.0 and3.3±1.9psafterthenearestapproachwhile HOC+ asinitiallyformedproductrequires5.6±4.4and3.9±1.8ps.The reactiontimesincreaseonlyslightlywiththeimpactparameterin theorderof0.3to0.6psexceptat0.83eVwherethemeantimeof HOC+formationincreasesto7.7±3.7psforb>4 ˚A.
Thefewargoncatalysedisomerisationeventsatthelowcol- lisionenergy,Fig.10a,happentypicallyjustafter0.1psbutalso atearlier timesand up tomorethan 6ps later.An exampleat early times is given by the cartoon in Fig. 10b. At 2.37eV in Fig. 10c, no argon catalysed isomerisation was observed after 0.12psbutlaterautoisomerisationmayoccuratalltimesasenergy allows.
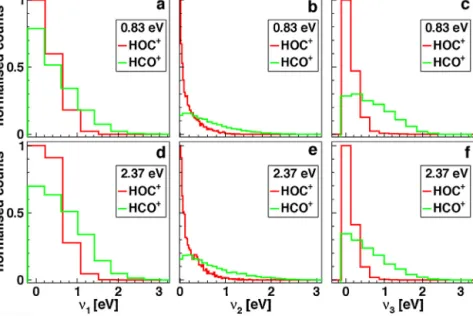
Fig.11.VibrationalenergydistributionsfromthenormalmodeanalysisfortheHOorHCstretching(v1),bending(v2)andCOstretching(v3)at(a)–(c)0.83eVand(d)–(f) 2.37eVcollisionenergy.ThedistributionsfortheHOC+andHCO+productsarenormalisedtoidenticalareasineachsubplot.Thebinwidthsaregivenbythevibrational energyquantainharmonicapproximation.
Table3
MeanvibrationalenergiesineVfromthenormalmodeanalysisfortheHCorHO stretching(v1),bending(v2)andCOstretching(v3).Inbrackets,thestandarddevi- ationsofthedistributionsaregiveninunitsofthelastdigit.Thetotalenergyis comparedtotheexperimentalmeaninternalenergyrelativetotheHCO+andHOC+ groundstateenergy,respectively.Finally,theratiosbetweenthehigherandlower collisionenergyvaluesaregiven.
Energy Product v1 v2 v3 Erot Total Exp.
0.83eV HCO+ 0.4(5) 0.6(5) 0.6(5) 0.7(5) 2.3(4) 2.4(4)
19% 24% 28% 29% 100%
HOC+ 0.2(3) 0.2(2) 0.1(2) 0.3(2) 0.8(3) 0.7(4)
29% 23% 14% 35% 100%
2.37eV HCO+ 0.6(6) 0.6(6) 0.6(5) 0.8(6) 2.6(6) 2.6(6)
23% 25% 22% 30% 100%
HOC+ 0.3(3) 0.2(3) 0.1(2) 0.5(4) 1.1(5) 1.0(6)
28% 20% 9% 41% 100%
Ratio HCO+ 1.35 1.14 0.88 1.17 1.12 1.10
Ratio HOC+ 1.32 1.15 0.88 1.63 1.32 1.31
Tolocatetheinternal energyoftheproducts, weperformed amode-specificvibrational analysisasdescribedin Section2.2.
Bothisomersfeaturetwo stretching(v1,v3)anda bending(v2) mode.Thevibrational frequenciesω1,ω2,ω3,atthePBE0level withthe6-31G(d,p) basis setare3261,869and 2294cm−1 for HCO+and3489,201and1998cm−1forHOC+.Thenon-integerhar- monicactionsarebinnedtointegervaluesandpresentedwiththeir correspondingenergyscalesinFig.11.Themeanrotationaland mode-specificvibrationalenergieswithpercentagesaretabulated inTable3alongsidetheexperimentalmeaninternalenergy.
Themoststrikingdifferencebetweentheisomersisthestrongly increasedexcitationoftheCOstretch(v3)intheHCO+ products inFig.11candf.Itsmeanfractionofthetotalinternalexcitation energyisabouttwiceaslargeasfortheHOC+products.Further- more,itstandsoutbecauseitspopulationisnotenhancedbythe largercollisionenergy.WeinferthattheCOstretchexcitation,the onlymodethatisonlyindirectlycoupledtothereactioncoordinate, isgovernedbythereactionenthalpy.Itisassumedtobedetermined bythegainofpotentialenergyduetothechangedC–Oequilibrium distance.
ThevibrationalenergiesintheHCO+productsaccountfor70%
ofthetotalinternalexcitationandarealmostequallydistributedto allthreemodesat2.37eVcollisionenergy.Mostoftheadditional 0.3eVinternalenergyatthelargercollisionenergyischannelled
intotheHCstretchmode(v1).Thisisalsotrueinrelativeterms givenbytheenhancementratiosinTable3.Theyareverysimilar forbothisomers(comparingtheHCtotheHOstretch)exceptfor theprominentincreasebyafactor1.63oftherotationalexcitation ofHOC+,thatisgenerallymoreimportantforthisisomerwitha percentageof35–41%asopposedto30%forHCO+.
Thetotalmeaninternalenergyagreeswiththeexperimental resultsupto0.1eV.Despitethesimulatedexothermicityof0.89eV asopposedto0.59eVascalculatedfromliteraturevalues,itisonly slightlyoverestimatedforHOC+.
4. Conclusion
The protontransfer reactionfrom ArH+ toCO with theiso- mericproductsHCO+andHOC+atcollisionenergiesrangingfrom 0.4to2.4eVhasbeeninvestigatedexperimentallyusingcrossed beamimagingandtheoreticallywithdirectdynamicssimulations.
Thereactionischaracterisedbyafastdirectmechanismwithlit- tletransferbetweenkineticenergyandinternalexcitation.Upon increasingthecollisionenergyfrom0.4to2.4eV,themeaninter- nalenergyrelativetotheHCO+groundstateincreasesfrom2.2to only2.6eVstayingclosetotheexothermicityof2.33eV.Theangle- differentialanalysisoftheexperimentaldatarevealsthatenergy transferfromthecollisionenergytointernalexcitationonaverage onlytakesplaceatlargescatteringangles.
ThesmallinfluenceofthedifferentexothermicitiesoftheHCO+ isomersontheproductinternalenergy,orequivalentlyontherel- ativeenergy afterthecollisionthat is directlyaccessiblein the experiments, makes it impossible tofind anindicator tosepa- ratetheisomericproductsfromthemeasuredcrosssectionsalone.
ThisisincontrasttotheHOCO++COreactionforwhichtheHOC+ productchannelisendothermic,andtheassumptionofidentical internalenergyfractionsofthetotalavailableenergyforbothiso- mersallowedtheestimationofanupperlimitof<2%fortheHOC+ fraction[27].Thepresentstudysuggests,thatsimilarestimatesfor theH+3+COreaction[14]mayhaveresultedintoosmalllimitsfor theHOC+fractionwhichissupportedbyrecentdirectdynamics simulations[43].
The computed rate constant close to 1.3×10−9cm3s−1 at both collisionenergies agrees wellwiththemeasuredvalueof 1.25(35)×10−9cm3s−1 from drift tube experiments [25]. The