CHAPTER 9
Cytochrome-lndependent Electron Transport Enzymes of Bacteria
Μ . I. D O L I N
I. Introduction 425 A. Electron Transport Not Coupled to Cytochrome 425
B. Development of Concepts regarding Flavoprotein Respiration 426
C. Nomenclature 430 II. Oxidases for Reduced Pyridine Nucleotides 430
A. Old Yellow Enzyme 430 B. D P N H Oxidase System of S. faecalis 431
C. Reduced Pyridine Nucleotide Oxidases of Clostridia 432
D . Ascorbic Acid-Dependent Reactions 436
III. Flavoprotein Peroxidases 436 A. Atypical Peroxidase Activity 436
B. D P N H Peroxidase 437 C. Distribution of Flavoprotein Peroxidases 441
IV. Diaphorases 441 A. Significance of Diaphorase Activity 441
B. Diaphorases of Microorganisms 442 C. Purified Diaphorase of S. faecalis 443 V. Direct Flavoprotein Oxidases 446
A. Pyruvate and Lactate Oxidases of L. delbrueckii 446
B. Lactate Oxidases of Mycobacteria 447
C. Other Direct Oxidases 448 VI. Dehydrogenase Activities 448
A. Coenzyme Level Dehydrogenases 449 B. Substrate Dehydrogenases 450 VII. Flavoproteins Concerned with Reduction of Nitrate, Nitrite, Hydroxyl-
amine, and Organo-Nitro Compounds 452 VIII. Phosphorylation Coupled to Anaerobic Electron Transport 453
IX. Significance of Flavoprotein Respiration in Anaerobes and Lactic Acid
Bacteria 454 A. Physiological 454 B. Evolutionary 456 References 457
I. Introduction*
A . ELECTRON TRANSPORT N O T COUPLED TO CYTOCHROME
T h e known mechanisms of electron transport used b y cytochrpme-con- taining bacteria have been discussed in Chapter 6. I n addition to the partic-
* Abbreviations used: P N , P N H , oxidized and reduced pyridine nucleotide; D P N , D P N H , oxidized and reduced diphosphopyridine nucleotide; T P N , T P N H , oxidized
425
426 Μ. I. DOLIN
ulate oxidases described there, various soluble systems for the reoxidation of reduced pyridine nucleotide are known. These systems, together with some indication of their distribution m a y be summarized briefly as follows:
(a) Flavoproteins (bacteria, yeast): These enzymes m a y catalyze either two- or four-electron transfer reactions [(equations (1) and (2)]. T h u s far,
P N H + H+ + 02 PN+ + H202 (1) 2PNH + 2H+ + 02 2PN<" + 2 H20 (2) four-electron transfer by flavoproteins has been demonstrated only in
bacteria.1 I n addition to the flavoproteins t h a t react directly with O2, bacteria, yeast, animals, and plants contain flavoproteins (diaphorases) which are reoxidized by artificial electron acceptors (Section IV). (b) Hydroperoxidases (plants): Under certain conditions, this enzyme m a y catalyze four-electron reduction of O2 to water with reduced diphospho- pyridine nucleotide ( D P N H ) or reduced triphosphopyridine nucleotide ( T P N H ) as. reductants.2 (c) Ascorbic acid-dependent enzymes (plants, animal systems, some bacteria): Certain oxidases for reduced pyridine nucleotide require the presence of ascorbic acid plus an ascorbic acid oxi
dase (see Section I I , D ) .
Among the enzymes which catalyze direct substrate oxidation, there are flavoproteins (and the Cu-enzyme, uricase) which catalyze two-elec
tron transfer reactions and copper enzymes such as tyrosinase (polyphenol oxidase) which catalyze four-electron transfer.3 Although there are early reports in the l i t e r a t u r e4 , 5 t h a t tyrosinase activity is found in certain bacteria, there is as yet no evidence t h a t tyrosinase or an equivalent enzyme is involved in any main-line respiratory process in bacteria. There is also no evidence t h a t the hydroperoxidase reaction occurs in bacteria.
Bacterial pigments such as pyocyanine or toxoflavin are redox systems,6 but they have not yet been implicated in any physiological electron trans
port mechanism. Available evidence indicates t h a t in cytochrome-free bacteria (e.g., lactic acid bacteria, Clostridia) flavoprotein systems are the major link between substrate and oxygen. Such systems m a y be phys
iologically useful for certain of the cytochrome-free facultative anaerobes (Section I X ) .
B . D E V E L O P M E N T S CONCEPTS REGARDING FLAVOPROTEIN RESPIRATION Wieland's dehydrogenatipn theory7 called for the activation of substrate by a specific catalyst followed by transfer of the "activated" hydrogen and reduced triphosphopyridine nucleotide; FP, F P H2 , oxidized and reduced flavo
protein; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; TPP, co- carboxylase; lip(SS), lip(SH)2 , oxidized and reduced lipoic acid; CoA, coenzyme A;
ADP, adenosine diphosphate; ATP, adenosine triphosphate; NMNH, reduced nico
tinamide mononucleotide.
9. CYTOCHROME-INDEPENDENT ELECTRON TRANSPORT 427
to any of a number of unspecific, unactivated oxidants. With O2 as ac
ceptor, H2O2 was considered to be the obligatory reduced product [equation (3)]. Since the formation of stoichiometric amounts of peroxide was con-
H*
/
A + 02 - > A + H202 (3)
\
H*
H* = "active" hydrogen
sidered to be crucial for the validity of the theory, m a n y a t t e m p t s were made to establish the stoichiometry of reaction (3). Catalase-free bacteria were commonly used as experimental material. Early studies had shown t h a t pneumococci, streptococci,8 , 9 and various anaerobes1 0 could form some peroxide in the presence of O2. However, the first quantitative exper
iments were reported b y Bertho and Gluck,1 1 who showed t h a t during glucose oxidation b y cell suspensions of Lactobacillus delbrueckii or L.
acidophilus there was a 1:1 equivalence between 02 u p t a k e and peroxide formation as required b y equation (3). These d a t a were thought to con
firm t h e Wieland theory. Similar results were later reported for L. del
brueckii,12 L. bulgaricus,n and pneumococci.1 4 Simultaneous with this work, however, the first flavoprotein, the old yellow enzyme of yeast, was dis
covered.1 6 This enzyme catalyzes C N - and CO-insensitive T P N H oxidation as shown in equations (4a) and (4b). These reactions demonstrated t h a t
T P N H + H+ + FP -> ΤΡΝ+ + F P H2 (4a)
F P H2 4- 02 - * FP + H202 (4b)
hydrogen peroxide is formed via the two-electron reduction of 02 b y re
duced flavoprotein and not b y direct reaction between " a c t i v e " hydrogen and molecular O2. I t was also reported1 5 t h a t intact cells of L. delbrueckii contained a flavoprotein system t h a t could account for the entire respi
ration of the organism ( Q o2 = 12, with glucose-6-phosphate as substrate).
T h e system resembled the old yellow enzyme in turnover, insensitivity to C N and CO, and in formation of H202 as the reduced product. T h e turn
over of the old yellow enzyme (recent v a l u e1 6: 35 min.""1) is, however, m u c h too low to account for the respiratory activity of yeast. Further
more, t h e old yellow enzyme is unaffected b y C N and CO, b o t h of which inhibit yeast respiration. T h e concept arose, therefore, t h a t the only phys
iological p a t h w a y for flavoprotein reoxidation b y 02 was mediated b y C N - and CO-sensitive iron carriers.1 5 This belief was strengthened b y the finding t h a t in intact yeast cells tHe rate of cytochrome reduction is equal to the over-all respiratory r a t e ,1 7 and b y the subsequent isolation of flavoproteins
428 Μ. I. DOLIN
t h a t catalyzed rapid cytochrome c reduction b y reduced pyridine nucle
otides (Chapter 6).
Flavins are widely distributed among cytochrome and noncytochrome- containing o r g a n i s m s .1 5 , 1 8* Since the characteristics of m a n y isolated flavo
proteins resemble those of the old yellow e n z y m e1 9 it has been assumed t h a t the old yellow enzyme is a general model for all flavoprotein systems t h a t react directly with 02. T h e often reiterated properties of flavoprotein respiration are (a) high O2 requirement,2 0 (b) low turnover, (c) stoichio
metric peroxide formation. These characteristics are the basis for calling flavoprotein respiration "unphysiological." (In cytochrome- and catalase- free lactic acid bacteria, in which flavoprotein respiration does account for the observed respiratory rate, toxic concentrations of peroxide were considered to be t h e invariable reduced product.)
There have long been indications, however, t h a t this description of flavoprotein catalysis is not entirely valid. For instance, experiments with resting cells of Streptococcus faecalis21 indicated t h a t rapid oxidations could be achieved and t h a t excessive 02 concentrations were not required. Table I shows t h a t the respiratory activity of various cytochrome- and catalase- free bacteria is equal to or exceeds t h a t of baker's yeast, and t h a t peroxide need not be the reduced product. Subsequently, these results were con
firmed and extended when potent, soluble D P N H oxidases were isolated from S. faecalis,29'30 Clostridium kluyveri,*1 and C. perfringens1'32· 3 3 a«b I n crude extracts of C. perfringens, D P N H oxidase activity is as great, on a specific activity basis, as in crude extracts of the strict aerobe Azotobacter vinelandii*3* For the flavoprotein D P N H oxidase system of S. faecalis*0 for KB for 02 has been estimated3 4 as 2 Χ 10~6 Μ, a value which is of the same order of magnitude as t h a t found for various typical cytochrome- containing respiratory preparations, and some 30 times lower t h a n the KB for several classic flavoprotein oxidases.2 0
Although pyruvate can occur as an intermediate during the oxidation of glucose by lactic acid bacteria, the known spontaneous reaction between pyruvate and peroxide3 5 does not account for the fact t h a t some lactic acid bacteria, do not form peroxide as a product of glucose or alcohol oxidation (Table I ) . However, enzymatic catalysis of peroxide reduction in t h e presence of oxidizable substrate has been demonstrated with rest
ing cells of s t r e p t o c o c c i2 6·3 6 and lactobacilli.3 7-3 8 These peroxidase activi
ties were recognized as being atypical since they apparently did not in
volve heavy metal catalysts. T h e enzyme responsible for the reaction in
* The naturally occurring nucleotides are FAD and FMN. However, L. lactis cells, grown on lyxoflavin, synthesize lyxoflavin mono- and dinucleotides in place of ribo
flavin nucleotides. Lyxoflavin nucleotides are less active than FMN and FAD in the appropriate enzymatic test systems.1 8 e
9. CYTOCHROME-INDEPENDENT ELECTRON TRANSPORT 429
T A B L E I
OXIDATIONS CATALYZED BY CELL SUSPENSIONS OP CYTOCHROME AND CATALASE FREE BACTERIA (COMPARED WITH YEAST) Organism Substrate Reduced
product Qo," Ref
erence Baker's yeast Glucose H20 40-90 15, 22 Lactobacillus delbrueckii Glucose H202 10-20 12
Lactate H202 30-120
Lactobacillus casei Glucose — 3 23
Streptococcus faecalis (strain 24) Glycerol H202 70-110 21 Streptococcus faecalis, 10C1 Glucose H20 90 24 Pyruvate H20 80 25
Streptococcus faecalis (strain Β 33a) 26
Aerobically grown Glucose H20 100 Anaerobically grown Glucose H202 100 Streptococcus mastitidis Alcohol H20 60-170 27
Pneumococci Glucose H202 30-100 14
Clostridium saccharobutylicum Glucose H202 doubt
ful or nega
tive
20-40 28
β Qo* = μ1· 02 per hr. per mg. dry wt. of bacterial suspension (based on initial rates). A common feature of the reactions that result in H202 formation is that the respiratory rate decreases with time and may become negligible within 40 to 60 min.
In general, the reactions that do not yield peroxide follow zero-order kinetics.
S. faecalis (and presumably in t h e other lactic acid bacteria) was later identified as a flavoprotein D P N H p e r o x i d a s e .3 0 , 3 9 T h e combined action of two enzymes, a flavoprotein D P N H oxidase and t h e peroxidase, re
sulted in four-electron reduction of 02 t o water, as shown in equations (5a)-(5c). Another mechanism for catalyzing four-electron reduction of
D P N H + H+ + 02 -> DPN+ + H202 (5a) D P N H + H+ + H2Q2 -> DPN+ + 2H2Q (5b) Sum: 2 D P N H + 2H+ + 02 — 2DPN+ + 2 H20 (5c) 02 was discovered in C. perfringens.1' 32« 3 3 a«b I n this organism, reaction
(5c) is catalyzed b y a single flavoprotein; free peroxide is not a n inter
mediate.
Glycerol oxidation b y cells of S. faecalis,21 and p y r u v a t e oxidation b y cell-free extracts of S. faecalis*0 and L. delbrueckii*1 is coupled with t h e formation of "energy-rich'' phosphate bonds (at t h e substrate level). T h e utility of some of t h e oxidations is illustrated b y t h e fact t h a t some strains of S. faecalis (and various pediococci) will not utilize glycerol as a growth substrate except under aerobic conditions.4 2**1* Direct growth experiments
430 Μ. I. DOLIN
show t h a t the D P N H oxidation and peroxidation p a t h w a y in S. faecalis is physiologically u s e f u l2 6'3 0 (see Section I X , A). I t appears, therefore, t h a t whereas the classic characteristics of flavoprotein respiration m a y describe m a n y of the known enzymes, there are, among the cytochrome- free bacteria, flavoprotein systems which do not conform to the "classic"
unphysiological pattern. Detailed documentation for these statements is given in the succeeding sections.
C. NOMENCLATURE
A confusing array of terms is currently used to describe enzymes t h a t catalyze the oxidation of reduced pyridine nucleotides. I n this chapter, the following system is followed.
D P N H ( T P N H ) oxidase: These are enzymes t h a t catalyze t h e oxida
tion of reduced pyridine nucleotide according to equations (1) or (2). 02 is the electron acceptor.
Direct flavoprotein oxidases: These enzymes couple the oxidation of
"noncoenzyme" substrates to the reduction of 02. Free coenzymes do not mediate electron transport between the substrate and the oxidized enzyme.
(Direct oxidases of this sort are generally less useful t h a n the oxidases for reduced pyridine nucleotides. An organism whose aerobic respiration de
pends upon the function of one or more direct flavoprotein oxidases is obviously limited in its ability to use oxygen as a terminal electron ac
ceptor. On the other hand, an organism which can transfer electrons from D P N H and T P N H to oxygen can couple any of its pyridine nucleotide- linked dehydrogenases to molecular oxygen.)
Diaphorases: Diaphorase couples the oxidation of reduced pyridine nucleotide to the reduction of artificial electron acceptors. Enzymes t h a t use a physiological oxidant are named after the oxidant (i.e., nitrate re
ductase, cytochrome c reductase). Enzymes for which a physiological oxi
d a n t is known can also show "diaphorase activity" if artificial electron acceptors function as alternative oxidants.
Dehydrogenase: T h e term diaphorase is usually reserved for enzymes t h a t use reduced pyridine nucleotides as substrates. A dehydrogenase, in general, is an enzyme t h a t couples the oxidation of a substrate t o t h e re
duction of an acceptor other t h a n oxygen. Certain of the pyridine nucleo
tide-linked reactions involving thiol substrates are also best referred t o as dehydrogenase activities.
II. Oxidases for Reduced Pyridine Nucleotides
A. OLD YELLOW ENZYME
T h e old yellow enzyme of yeast,1 6 which catalyzes reaction (4a) and (4b), was the first flavoprotein isolated. T h e crystalline e n z y m e1 6 has a
9. CYTOCHROME-INDEPENDENT ELECTRON TRANSPORT 431 molecular weight of 100,000-105,000 and contains 2 moles of F M N per mole of enzyme. As mentioned previously, t h e turnover, 35 m i n .- 1, is too low t o account for a n y considerable proportion of yeast respiration. If there is an alternative physiological acceptor, it is as yet undiscovered.
I t h a s been reported4 3 t h a t a t atmospheric O2 tension and in t h e presence of cytochrome c, about 5 0 % of t h e reducing equivalents from reduced old yellow enzyme are transferred t o cytochrome c and 50 % t o 02. W i t h the discovery of T P N H - c y t o c h r o m e c reductase (Chapter 6) (turnover, 650 min."1 for a two-electron reaction)4 4 it was suggested4 5 t h a t t h e cyto- chrome c reaction of t h e old yellow enzyme m a y have been caused b y slight contamination with cytochrome c reductase. T h e acceptor speci- ficity of crystalline old yellow enzyme h a s n o t yet been reported.
When old yellow enzyme is partially reduced b y hydrosulfite in t h e presence of excess T P N ,4 6 a a red color is formed. T h e color was originally attributed t o t h e formation of a n enzyme-pyridine nucleotide complex, with t h e flavin stabilized as t h e red semiquinone. Recent experiments4 6 b have shown t h a t a radical is, in fact, detectable (by paramagnetic reso- nance methods) on reduction of old yellow enzyme b y T P N H ; however, it appears t h a t t h e radical is n o t identical with t h e red compound. T h e latter m a y be a flavoprotein-TPN complex. These spectrophotometric experiments are, rather puzzling a t present, since t h e red compound is not always detected.4 7
B . D P N H OXIDASE SYSTEM OP S. faecalis
S. faecalis contains a t least five separate flavoproteins t h a t catalyze D P N H oxidation2 9' 3° ·3 9·4 8 ( T P N H is n o t a substrate for a n y of these reactions). These enzymes are (a) a D P N H oxidase [equation (5a)], (6) a D P N H peroxidase [equation (5b); Section I I I , B], (c) a cytochrome c reductase (Chapter 6), (d) a diaphorase t h a t uses 2,6-dichlorophenol- indophenol, ferricyanide, and a series of quinones as oxidants (Section I V ) , and (e) a menadione reductase t h a t is n o t identical with a n y of t h e first four enzymes.
These enzymes m a y be separated from each other b y classic fraction- ation procedures followed b y zone electrophoresis. T h e enzymes also show different sensitivities t o ultraviolet irradiation.3 0 These results indicate t h a t t h e practice, which is sometimes followed, of attributing all D P N H oxidase activities t h a t are observed in crude extracts t o t h e function of a single " D P N H oxidase" is not warranted.
As mentioned in Section I, B , S. faecalis catalyzes t h e four-electron reduction of 02 t o water via reactions (5a) a n d (5b), which are catalyzed by D P N H oxidase and peroxidase, respectively. I n initial extracts, t h e activity of t h e oxidase a n d peroxidase system ( ~ 0 . 1 5 Mmoles D P N H ox-
4 3 2 Μ. I. DOLIN
idized per min. per mg. protein) can account for all t h e respiration of intact cells (Table I ) . T h e cell-free oxidase, unlike the peroxidase, is u n stable on storage and has not been purified. However, D P N H oxidase apoenzyme can be prepared either from flavin-deficient cells3 0 or b y t h e classic acid resolution procedure.4 9 Activity is restored specifically by flavin adenine dinucleotide ( F A D ) . A requirement for Mn++ can also be demonstrated. Under conditions in which t h e peroxidase activity is made limiting, peroxide can be demonstrated as t h e reduced product.2 9
C . REDUCED PYRIDINE NUCLEOTIDE OXIDASES OF CLOSTRIDIA
1. OXYGEN UPTAKE BY CLOSTRIDIA
Although Clostridia cannot grow aerobically, cell suspensions of m a n y clostridial species are able t o catalyze rapid oxygen u p t a k e .2 8 , 5 0· 5 1 · 5 2 Per
oxide m a y or m a y n o t be formed in these oxidations, depending upon the nature of t h e enzymes i n v o l v e d .1 , 3 2 , 3 3 a T h e literature dealing with peroxide formation b y anaerobes has been reviewed.5 3 Since growth studies5 3 suggest t h a t peroxide formed through respiratory reactions m a y account in large measure for the inability of anaerobes t o use 02 as a physiological oxidant, the nature of t h e oxidative catalysts present in Clostridia becomes an important physiological consideration.
2 . D P N H OXIDASE OF C. perfringens1'32,33a,b
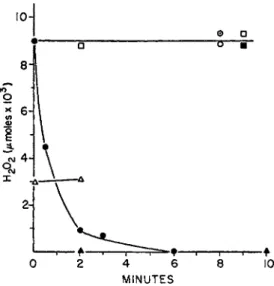
a. Isolation, Prosthetic Group. C. perfringens extracts catalyze the four- electron reduction of 0 2 t o water, with D P N H as electron donor [equation (5c)]. Free peroxide is not an intermediate in t h e reaction, as shown in Fig. 1. T h e mechanism of t h e oxidation is thus different b o t h from t h a t of the old yellow enzyme and the oxidase and peroxidase systems of S.
faecalis. I n crude extracts, t h e specific activity for D P N H oxidation (Table I I ) equals t h a t shown b y extracts of t h e strict aerobe, Azotobacter vinelandii.
T h e clostridial enzyme can be purified b y isoelectric precipitation, followed by zone electrophoresis a t or near the isoelectric point. With purified frac
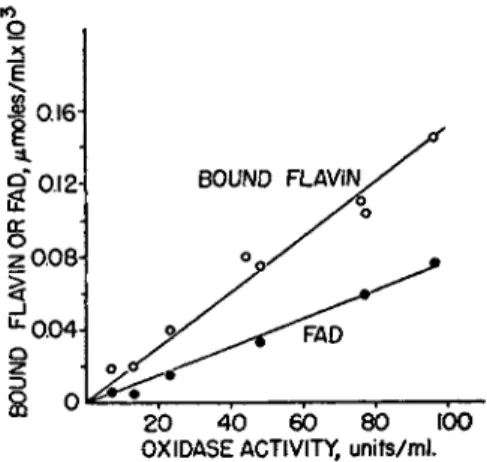
tions, t h e bound flavin or F A D content is directly proportional t o oxidase activity (Fig. 2 ) . On a molar basis, only one-half of t h e bound flavin is F A D , which suggests t h a t t h e enzyme contains two flavin prosthetic groups.
T h e turnover of t h e purified enzyme, unlike t h a t of t h e old yellow enzyme, compares very favorably with t h e flavoprotein turnovers of some typical aerobic respiratory preparations (Table I I ) .
b. Inhibitors. T h e oxidase is not inhibited b y 0.01 Μ C N , b u t is sen
sitive to peroxide and p-chloromercuribenzoate and is inhibited 5 0 % b y 0.01 Μ azide. Although stoichiometric peroxide concentrations are n o t formed during D P N H oxidation, traces ( ~ 1 Χ 10~β M) m a y be detected.
9. CYTOCHROME-INDEPENDENT ELECTRON TRANSPORT 433
•OH
•
©
•σ •
ΙΟ
g
X
f 1 Τ — 1
2 4 6
MINUTES
6 10
0 8
FIG. 1. Difference in role of H202 in D P N H oxidase systems of Streptococcus fae
calis (10C1) and Clostridium perfringens. Anaerobic experiment (details in reference 33a). Complete system: 0.066 Μ potassium phosphate, pH 7.0; D P N H , 0.3 Mmoles;
H202 , 0.009 Mmoles. Final volume 3 ml.; Temp. 23°C. S. faecalis: crude extract con
taining D P N H oxidase and peroxidase, 4 units of D P N H oxidase activity: # , com
plete system; O, without D P N H . C. perfringens: crude extract, 11 units of D P N H oxidase activity: • , complete; • > without D P N H ; Δ , complete, except used 0.003
Mmoles H202 ; A, without H202 . Θ, H202, 0.009 Mmoles in complete system without enzymes. The experiment demonstrates that the D P N H oxidase and peroxidase sys
tem of S. faecalis utilizes coenzyme levels of H202 in an enzyme and D P N H dependent reaction, whereas the C. perfringens enzyme does not. The free H202 (approx. 1 X 10~β M) formed during D P N H oxidation by C. perfringens extracts is, therefore, not an intermediate in the over-all reaction [equation (5c)]. Reprinted with permission from the Journal of Bacteriology.
These small concentrations cause, in the presence of D P N H , a first-order decay of enzyme activity (k = 0.13-0.15 min."1)) which m a y be reduced 3-fold in the presence of catalase (fc = 0.049 m i n r1) .3 3* Concentrations of D P N H in excess of 3 Χ 10~4 Μ cause immediate inhibitions which are a direct linear function of the D P N H concentration. This type of inhibi
tion, which m a y reflect the binding of an additional molecule of D P N H to the enzyme, is not prevented b y catalase.
c. Specificity. T P N H is not a substrate for the enzyme. Crude extracts contain enzymes which catalyze D P N H - d e p e n d e n t reduction of menadione and free flavins. These reactions are not catalyzed b y t h e four-electron oxi
dase. Aerobically, the menadione- and FAD-dependent reactions result in peroxide formation (Chapter 6, Section IV, C ) .
d. Relation to Cytochrome c Reductase. Evidence has been p r e s e n t e d1-3 3 b
TABLE II OXIDASES FOR REDUCED PYRIDINE NUCLEOTIDE: COMPARISON OF PARTICULATE CYTOCHROME CONTAINING ENZYMES AND SOLUBLE FLAVOPROTEIN OXIDASES TON' Preparation Source Composition /uMoles PNH oxidized per min. per mg. protein MMoles PNH oxidized per min. per μπιοΐβ bound flavin
Reference "Closed" DPNH oxidase Beef heart Flavin and cytochrome- containing particu late oxidase
1.0 (23°C.) 2600 54 Electron transport particle (DPNH oxidase) Azotobacter vinelandii Flavin and cytochrome- containing particu late oxidase (1) crude enzyme (2) purified particles 0.8 (35°C.) 6.8 (35°C.) 6800 55 Old yellow enzyme (TPNH oxi dase) Bottom yeast Crystalline flavoprotein (FMN) 0.35 (38°C.) 18 16 DPNH oxidase Clostridium per- fringens Flavoprotein (FAD + unidentified flavin) (1) crude enzyme (2) purified enzyme 0.7-0.9 14 4500 1, 32, 33a,b α The oxidant is air for all preparations except the old yellow enzyme, in which case it is pure 02 .
434 Μ. i. DOLIN
9. C Y T O C H R O M E - I N D E P E N D E N T E L E C T R O N T R A N S P O R T 435
Ο 25 Ε
OXIDASE ACTIVITY, units/ml .
FIG. 2. D P N H oxidase, Clostridium perfringens. Relation between D P N H oxidase activity and flavin content of the purified enzyme (text). Bound flavin is the total flavin content, corrected for the amount of flavin that is fluorescent before hydrolysis of the enzyme with trichloracetic acid. F A D was determined enzymically. See refer
ence 33b. Reprinted with permission from the Journal of Bacteriology.
which strongly suggests t h a t the native D P N H oxidase is also a cyto
chrome c reductase. Spontaneous or ultraviolet-induced loss of the four- electron transfer reaction leads to conversion of the oxidase to a cytochrome c reductase devoid of oxidase activity. T h e altered enzyme has different physical properties from the native oxidase.
e. Michaelis Constants for the Native Oxidase (pH 7.0, 23° C). W i t h 02 as acceptor, KB ( D P N H ) = 2.5 Χ 1 0 "5 M; 7m ax . = 4500 moles [2H]
transferred per mole bound flavin per minute. With cytochrome c as ac
ceptor, KB (cyt. c) = 4.2 Χ 10~5 M\ Vm&%. = 3700 moles [2H] transferred per mole bound flavin per minute. T h e Ks for 02 was not determined;
however, oxygen is an efficient electron acceptor since reaction (5c) pro
ceeds at nearly the same rate at atmospheric 02 tension or in solutions t h a t have been gassed either with N2 or 02 for several minutes.
/. Summary. Despite the fact t h a t C. perfringens contains a very active four-electron D P N H oxidase, 02 is not a physiological electron acceptor since (a) traces of peroxide formed during D P N H oxidation contribute to the decay of oxidase activity, (b) other DPNH-oxidizing enzymes of the organism, which use autoxidizable acceptors such as F A D or mena
dione, cause t h e rapid formation of growth-inhibitory concentrations of peroxide.
3. D P N H O X I D A S E O F C. kluyveri
Extracts of C. kluyveri catalyze D P N H3 1 and T P N H5 6 oxidation with 02 as electron acceptor. W i t h D P N H as reductant, t h e extracts also
436 Μ . I . D O L I N
catalyze reduction of cytochrome c3 1 and free flavins.57 T h e relationship between these various reactions is unknown. B y analogy to known systems, the bound prosthetic group of the D P N H oxidase is thought to be a flavin.
With extracts from which the free flavins have been removed by dialysis, D P N H oxidation proceeds according to equation (5a) (two-electron reduc
tion of 02) .3 3 a
D . A S C O R B I C A C I D - D E P E N D E N T R E A C T I O N S
Certain D P N H oxidases of peas,5 8 p o t a t o e s ,5 9 suprarenal microsomes,6 0 and microorganisms6 1 require the presence of ascorbic acid plus some type of ascorbic acid-oxidizing system. T h e mechanism of these reactions is not completely understood; however, it is thought t h a t ascorbic acid is oxidized to the one-electron intermediate, monodehydroascorbic acid, and t h a t the latter serves as an electron acceptor for D P N H oxidation. Among the microorganisms, ascorbic acid-ascorbic acid oxidase-dependent D P N H oxidase systems have been found in yeast, E. coli, and A. aerogenes*1 A D P N H oxidase of M. denitrificans*2 is markedly stimulated b y ascorbic acid in t h e absence of an added ascorbic acid oxidase system. I n yeast,6 1 the D P N H oxidase portion of the over-all system has been purified and shown to be a flavoprotein, which, after acid resolution, can be reactivated either b y F A D or F M N . T h e results with Μ. denitrificans suggest t h a t ascorbic acid-dependent D P N H oxidases m a y occur normally in bacteria, even though bacteria have not been shown to possess ascorbic acid oxidase.
I t is possible t h a t other reactions m a y be able to replace the typical Cu- containing ascorbic acid oxidase. As shown for pea extracts,5 8 CU++ or F e+ + + or, as suggested for the suprarenal microsome system,6 0 cytochrome b6, m a y be able to function as the ascorbic acid oxidase portion of the over-all D P N H oxidase system.
T h e stoichiometry for ascorbic acid-dependent D P N H oxidation has not been reported.
III. Flavoprotein Peroxidase
A. A T Y P I C A L P E R O X I D A S E A C T I V I T Y
Some catalase- and cytochrome-free lactic acid bacteria can catalyze the reduction of peroxide to water with oxidizable substrates as electron donors; therefore 02 uptake in these organisms does not result in peroxide accumulation (Table I ) . Such peroxidase reactions have been demonstrated with cell suspensions of the following organisms (substrates indicated in parenthesis): S. mastitidis (alcohol);3 6 S. faecalis, strain B33a (glucose, glycerol, l a c t a t e ) ;2 6 S. mitis, sulfathiazole-resistant strain (glucose);6 3 Lactobacillus brevis (lactate);3 7 Leuconostoc mesenteroides, strain 548 (fruc-
9. C Y T O C H R O M E - I N D E P E N D E N T E L E C T R O N T R A N S P O R T 437 tose, glucose, gluconate).3 8 T h e reaction is adaptive in S. faecalis, strain B33a; peroxidase activity is demonstrable only in cells t h a t have been grown aerobically2 6 (cf. Table I ) . T h e nature of the organisms which catalyze the peroxidations listed above and the fact t h a t at least two of the reactions are C N - i n s e n s i t i v e3 6 , 3 7 indicated t h a t the catalysts were not typical iron-protoporphyrin enzymes. Growth studies6 4 suggested the func- tion of a flavoprotein in the S. faecalis system.
There are indications t h a t nonhematin peroxidases m a y exist even in organisms containing the usual iron catalysts. I n E. coli, for instance, there appears to be a C N - and azide-resistant peroxidase activity which can be demonstrated in cell suspensions with H2, succinate, or glucose as the electron d o n o r s .6 5
B . D P N H P E R O X I D A S E 1. I S O L A T I O N A N D P R O P E R T I E S
A flavoprotein D P N H peroxidase, catalyzing reaction (5b), has been isolated from S. faecalis, strain 1 0 C 1 .2 9-3 0 ·3 9 ·6 6 This enzyme accounts for t h e peroxidase activity exhibited by cell suspensions of S. faecalis and m a y be a model for the other peroxidase activities described in Section I I I , A.
T h e enzyme in crude sonic extracts of S. faecalis has been purified 2000- fold b y conventional fractionation procedures followed by zone electro- phoresis on starch. Properties of the purified e n z y m e3 9 are described in the following sections.
a. Prosthetic Group. T h e enzyme contains 0.66% F A D , which is equiva- lent to a minimum molecular weight of 120,000. There are no hematin components present and no metals have been found in amounts equivalent to the bound flavin.
b. Substrate Specificity. D P N H is the only physiological reductant known for the peroxidase activity of the enzyme. Molecular oxygen can serve as a weak electron acceptor for the peroxidase. I n oxygen saturated solution, D P N H and T P N H are oxidized b y molecular oxygen at 3^oo of the rate at which D P N H is oxidized enzymically b y peroxide. A wide variety of compounds including cytochrome c, free flavins, and dyes have been tested as electron acceptors, but the only alternative oxidants found are ferricyan- ide, menadione, and 1,4-naphthoquinone. T h e rates of oxidation with the latter three compounds are, respectively, one-half, one-third, and one-fifth of those obtained when H2O2 serves as oxidant.
c. pH Optimum. W i t h H202 as acceptor, the optimum p H is 5.4 in acetate buffer. T h e rate is still % maximal at p H 7, if a mixture of phosphate and acetate is used at the latter p H .
d. Activators and Inhibitors. E n z y m e activity is stimulated b y anions
438 Μ . I . D O L I N
of the lower fatty acids (C1-C4) and b y high concentrations of phosphate.
T h e enzyme is not inhibited b y 0.01 Μ C N , or 0.05 Μ azide, is not apprecia
bly sensitive to — S H reagents, such as p-chloromercuribenzoate, selenite, and arsenite, and shows striking stability towards peroxide. Ag+, Pb++, and C u+ + are effective inhibitors. Incubation of the enzyme with D P N H causes first-order decay of enzyme activity (fc = 0.066 m i n . "1) . T h e inhibition is almost completely prevented b y the cosubstrate, H2O2. Kinetic d a t a sug
gest t h a t H202 exerts its protective effect b y maintaining t h e enzyme in t h e form of a catalytically active ternary complex (composed of flavoprotein, D P N H , and H202) which does not undergo inactivation.
e. Michaelis Constants and Turnover (pH 54, 26° C.): W i t h H202 as oxi
dant, Ks ( D P N H ) = 1.4 X 10^5 M\ #S( H202) = 2 Χ ΙΟ"4 M; 7m a x = 6 Χ 103 moles D P N H oxidized per mole bound F A D per minute. W i t h ferricyanide as oxidant, KB ( D P N H ) = 6 X 10~6 Μ; KB (ferricyanide) = 1.1 X 10~4 M) Fm ax = 2.96 Χ 103 moles D P N H oxidized per mole bound F A D per minute.
2. S P E C T R U M O F E N Z Y M E A N D E V I D E N C E F O R E N Z Y M E - S U B S T R A T E C O M P L E X F O R M A T I O N3 9 , 6 6· 6 7 A«B
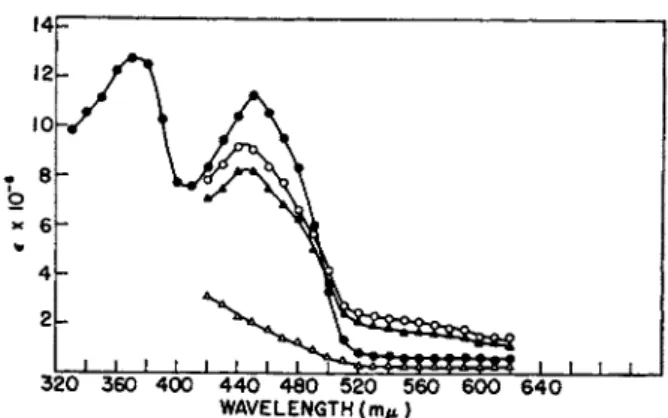
T h e spectrum of the enzyme is shown in Fig. 3. T h e oxidized enzyme has a typical flavin spectrum with no anomalies visible. On chemical reduction b y hydrosulfite the flavoprotein undergoes typical bleaching; however, reduction of the enzyme b y D P N H causes only partial decrease in height of the 450-ιημ band and results in the formation of a new band in the long wavelength region (520-600 ιημ). Addition of hydrosulfite to D P N H - reduced enzyme causes little further change in absorption. These results indicate t h a t D P N H combines with the flavoprotein to form a complex t h a t cannot be dissociated b y reducing equivalents from hydrosulfite. T h e long wavelength absorption change shown in Fig. 3 is somewhat similar to t h a t seen on substrate reduction of D P N H6 8* or T P N H4 4 cytochrome c reductase; substrate reduction of acyl-CoA dehydrogenase4 7 causes spectral changes t h a t are strikingly similar to those observed with D P N H peroxi
dase. I n the acyl-CoA dehydrogenase system, the long wavelength absorp
tion band has been attributed to a substrate-stabilized flavin radical.4 7 W i t h D P N H peroxidase, it is difficult to reconcile t h e absorption changes with radical formation, since the addition of fully reduced substrate
( D P N H ) to chemically reduced enzyme results in immediate reformation of the 450-mu band and the reappearance of t h e long wavelength b a n d . (The same spectrum is produced whether hydrosulfite is added before D P N H or D P N or after D P N H . ) Present evidence, therefore, suggests a fully reduced structure for the visible complex. T h e broadness of t h e 540- m/i band, as well as the lack of detail in the band, suggests t h a t t h e spec-
9. CYTOCHROME-INDEPENDENT ELECTRON TRANSPORT 439
320 360 400 440 480 520 560 600 640 WAVELENGTH (m/t)
FIG. 3. Spectrum of D P N H peroxidase. Enzyme, specific activity 9000, 3.3 mg.
of protein per ml., 0.2 ml. in 0.02 Μ potassium phosphate buffer, pH 6.5. DPNH, where used, 0.005 ml. of a 30-/xmole per ml. solution. Hydrosulfite, where used, 0.01 ml. of a 10% solution in 5% N a H C 03. Readings are corrected for dilution. See reference 39.
KEY: # , peroxidase; O, peroxidase plus D P N H ; Δ , peroxidase plus hydrosulfite;
A, peroxidase plus D P N H plus hydrosulfite. Reprinted with permission from the Journal of Biological Chemistry.
t r u m of D P N H - r e d u c e d enzyme m a y be t h a t of a charge-transfer com- p l e x .6 8 b
T h e spectral and kinetic properties of the complexes formed between D P N H peroxidase and a series of D P N H analogs (modified in the car- boxamide group and in other positions) have been i n v e s t i g a t e d .6 7 a b I t has been found t h a t those pyridine nucleotide analogs t h a t are peroxidase sub
strates are also able to cause the typical absorption changes; those nucleo
tides t h a t do not cause the typical absorption changes are not substrates.
T h e results m a y be summarized as follows: (a) Pyridine nucleotides t h a t cause t h e formation of both the 450- and 540-ηΐμ bands are peroxidase sub
strates. T h e nucleotides in this group contain an adenylic acid moiety, (b) Those nucleotides ( T P N H and N M N H ) t h a t cause the formation of only t h e 450-ηΐμ band are very poor peroxidase substrates, but in common with active nucleotides, they serve as hydrogen donors for the weak oxidase ac
tivity of D P N H peroxidase. Apparently the ability to generate the 540-ηΐμ band is characteristic of pyridine nucleotides t h a t contain an adenylic acid moiety t h a t can be bound to the enzyme. Correspondence between the kinetic and spectral properties of the complexes formed between D P N H peroxidase and either T P N H or N M N H suggests t h a t the 2'-phosphate of T P N H prevents the binding of the adenylic acid moiety, thus making T P N H and N M N H identical as substrates, (c) T h e simplest compound found t h a t can cause t h e formation of t h e 450-ηΐμ band is N M N H . On re
moval of the 5'-phosphate residue, the nucleotide loses most of its ability t o complex t h e enzyme.
14 12 10 Τ β
Ο
κ 6
4 2
440
Μ . I . D O L I NComplex formation can be followed by observation of the increase in absorbance at either 450 or 540 ιημ on titration of hydrosulfite reduced enzyme with graded levels of pyridine nucleotide analogs.
6 7 a'
bThe dissocia
tion constants calculated from these data have been correlated with the Michaelis constants derived from kinetic studies. In all cases there is good correspondence between the two constants, the average ratio of the dis
sociation constant (K) to the Michaelis constant (K
e) being 4. The physio
logical substrate of the enzyme, DPNH, shows the lowest dissociation from the enzyme (K = 1.6 Χ ΙΟ"
5M, pH 6.5), whereas TPNH and NMNH show the highest dissociation (K = 0.97 X 10~
3M, pH 6.5). Thus, all the criteria that have been employed lend strong support to the hypothesis that the visible intermediate formed in the DPNH peroxidase system is a kinetically active enzyme-substrate complex.
3. M E C H A N I S M A N D K I N E T I C C O N S T A N T S
Present kinetic experiments (unpublished experiments of the author) suggest the formation of a ternary complex between enzyme, DPNH and peroxide. One possible reaction sequence is shown in equations (6a), (6b),
FP + D P N H
kt F P D P N H (6a)
F P D P N H + H202
ki F P D P N H H202 (6b)
F P H202 + D P N H -* + F P D P N H H202 (6c) F P D P N H H202 + H+ FP + DPN+ + 2 H20 <6dJ
and (6d). A random binding sequence would involve, in addition, the forma
tion of an FPH
20
2complex (not shown) and the combination of DPNH with this complex [equation (6c)]. If a fully reduced enzyme-pyridine nucleotide complex is the intermediate, FP may be replaced by FPH
2in equations (6a) to (6d). The assumption of a random binding sequence for the mechanism shown above leads to Michaelis constants analogous in form to the Briggs-Haldane relation.
680The rate constants appropriate to such a mechanism may be estimated by methods similar to those described by Slater.
69These constants are (26°, pH 5.4):
k
z= 2 Χ 10
91 X mole"
1X min."
1k
4= 4 Χ 10
5min.""
1k
b= 5 Χ 10
81 X mole"
1X min."
1/c
e= 0.93 Χ 10
2minr
1k
7= 6 Χ 10
3min."
19. C Y T O C H R O M E - I N D E P E N D E N T E L E C T R O N T R A N S P O R T 441
KS( D P N H ) =
i r
8( H
2o
2) =
= 1.2 X 10 5M , H202 present in excess
= 2 X 10~4 M, D P N H present in excess
C . D I S T R I B U T I O N O F F L A V O P R O T E I N P E R O X I D A S E S
D P N H peroxidase has, so far, been found in three streptococcal strains which had previously been shown to carry out the atypical peroxidations listed in Section I I I , A. T h e organisms are S. faecalis, strains 1 0 Ο 12 9·3 9 and B33a3 4 and S. mastitidis30 I t has been reported t h a t fresh, b u t not aged extracts of C perfringens exhibit a weak D P N H peroxidase activity.5 2 Balances for the D P N - d e p e n d e n t oxidation of acetaldehyde by extracts of C kluyveri50 indicate a four-electron reduction of 02. Since the D P N H oxidase of this organism catalyzes a two-electron reduction of oxygen,3 3* a D P N H peroxidase m a y be part of the over-all oxidase system. D P N H peroxidase is absent in L. mesenteroides, strain 39, and L. delbrueckii (un
published experiments of the a u t h o r ) .
Extensive surveys for the presence of D P N H peroxidase have not been carried out, since the catalase (and possibly cytochrome c peroxidase) present in cytochrome-containing organisms would offer serious inter
ference. (Mammalian cytochrome c, in t h e absence of a specific peroxidase, catalyzes a heat-stable peroxidation of D P N H ,7 0 however, the turnover is very low—2.5 moles D P N H oxidized per min per mole cytochrome c, at p H 5.4.) As mentioned in Section I I I , A, there are suggestions t h a t non- hematin peroxidases m a y be present even in organisms which use iron enzymes.
I t is possible t h a t there exist flavoprotein peroxidases for "noncoenzyme"
substrates. W i t h crude xanthine oxidase preparations, peroxide formed in the initial stages of hypoxanthine oxidation is later used as an oxidant.7 1 T h e properties of xanthine oxidase, however, indicate t h a t peroxide is not a physiological oxidant for the enzyme. I t has been suggested2 0 t h a t the peroxidase activity of crude xanthine oxidase preparations m a y be attrib
utable to a contaminating enzyme.
IV. Diaphorases
A. S I G N I F I C A N C E O F D I A P H O R A S E A C T I V I T Y
Diaphorases are enzymes t h a t couple the oxidation of reduced pyrid' nucleotide to the reduction of artificial or non physiological electron ceptors [equation (7)]. Although m a n y diaphorases have been shown t
flavoproteins, it m a y be premature to conclude t h a t diaphorase a c P N H + H+ + A —• PN+ + AH 2
442 Μ . I . D O L I N
per se always implies flavoprotein catalysis. Since diaphorases do not use physiological oxidants, t h e question always arises as to the significance of these enzymes. A diaphorase m a y represent (a) an enzyme whose physio
logical oxidant is unknown, or (b) an enzyme which, for various reasons, m a y have lost its ability to react with the true physiological electron acceptor.
With regard to t h e latter, it has been suggested t h a t t h e diaphorase of pig heart is derived from cytochrome c reductase.7 2 Recent work, however, in
dicates t h a t heart muscle diaphorase and cytochrome c reductase are, in fact, separate enzymes7 3 a which m a y both have been derived from a com
mon precursor (a lipoflavoprotein).7 4
T h e question of whether a given oxidant is artificial may, in itself, be difficult to decide. Substrate concentrations of free flavins, for instance, sometimes serve as electron acceptors. I t m a y not always be clear whether these reactions have a special significance or whether the flavins act merely as nonspecific oxidation-reduction dyes. T h e reduction of menadione (vitamin K3) is sometimes regarded as of no special interest since (a) quinones are known to be nonspecific oxidants for several flavoproteins (Chapter 6, Section IV, C), and (6) in cytochrome-containing organisms, reduction of exogenous menadione (or quinone) would bypass the cyto
chrome system and the a t t e n d a n t phosphorylations (Chapter 6, Section VI, A). These arguments lose their force when applied to lactic acid bac
teria. I n the latter, the establishment of any alternate p a t h w a y to oxygen is potentially useful (see Section I X ) . Menadione (or a physiological equiva
lent), b y virtue of its autoxidizability, m a y link the oxidation of reduced pyridine nucleotide to 02 r e d u c t i o n .4 8'7 5
B . D I A P H O R A S E S O F M I C R O O R G A N I S M S
M a n y diaphorase activities have been reported for crude extracts of bacteria. Since physiologically functional flavoproteins have alternate electron acceptors and since there m a y be present in extracts several flavo
proteins concerned with the oxidation of reduced pyridine nucleotide (e.g., five separate enzymes in S. faecalis), systematic examination of a given system is necessary before the relation between various diaphorase activi
ties can be determined. Table I I I summarizes the properties of t h e purified diaphorases. On reduction of Straub's soluble diaphorase with D P N H , t h e enzyme undergoes absorption changes7 7 very similar to those described for D P N H peroxidase (Section I I I , B ) . T h e behavior of t h e S. faecalis dia
phorase, on its reduction b y substrate, is described in Section IV, C.
Table IV lists some of t h e diaphorase activities t h a t have been described crude and partially purified systems. Where the information is available, identity between diaphorase activity and other D P N H- o x i d i z i n g activi-
is indicated. For simplicity, the menadione- and flavin-dependent
9. C Y T O C H R O M E - I N D E P E N D E N T E L E C T R O N T R A N S P O R T 443 TABLE III
PURIFIED DIAPHORASES
Enzyme Sub
strate Oxidant
Pros
thetic group
Molec
ular weight0
T O N6 Reference
Straub's soluble D P N H Methylene blue FAD 67,000 8500 76, 77
diaphorase (pig 2,6-Dichloro- — 76, 77
heart) phenol-indo-
phenol
New yellow en T P N H Methylene blue FAD 60,000 130* 78a zyme (yeast)
Streptococcus faeca D P N H Ferricyanide FMN 53,000 6,000 30, 39, 78b
lis diaphorase 2,6-Dichloro- 29,000 30, 39, 78b
phenol-indo- phenol
p-Benzoquinone 88,000 30, 39, 78b
α Minimum molecular weight, based on flavin content.
6 Moles reduced pyridine nucleotide oxidized per mole bound flavin per minute.
c Calculated from data given in reference 78a.
The diaphorases do not react with cytochrome c; however, it has been reported7 3* that Straub's soluble diaphorase will catalyze cytochrome c reduction in the presence of lipoic acid. S. faecalis diaphorase does not catalyze this reaction nor does it use peroxide or any of the acceptors with which D P N H peroxidase shows no activity.
reactions are listed along with the electron acceptors t h a t are clearly artificial. I n t h e absence of exogenous autoxidizable acceptors, crude ex
tracts of Leuconostoc mesenteroides, strain 39, and Lactobacillus delbrueckii do not use 02 as an electron acceptor for D P N H oxidation. T P N H is very slowly oxidized by extracts of these organisms in the presence of menadione or 2,6-dichlorophenol-indophenol (approximately }£o of the rate given by D P N H ) (unpublished experiments of the author). Other menadione reductases are listed in Chapter 6, Section I I I , A.
C. P U R I F I E D D I A P H O R A S E O F S. faecalis
1. I S O L A T I O N A N D P R O P E R T I E S
One of t h e DPNH-oxidizing enzymes of S. faecalis, strain 10C1, is a diaphorase t h a t has been obtained in high purity. T h e enzyme, purified over 600-fold from extracts of S faecalis, has t h e following properties.7 8 b
a. Prosthetic Group. Firmly bound F M N is the sole prosthetic flavin pres
ent. Based on t h e F M N content, the minimum molecular weight of the enzyme is approximately 53,000.
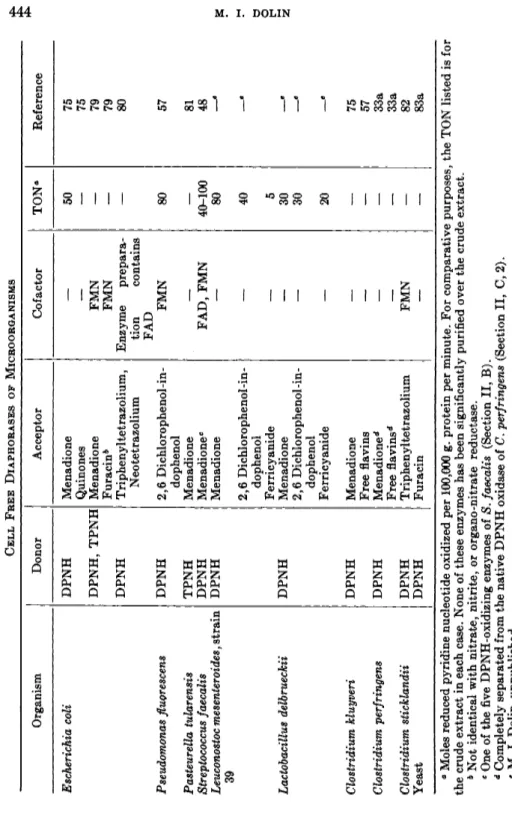
TABLE IV CELL FREE DIAPHORASES OF MICROORGANISMS Organism Donor Acceptor Cofactor TONe Reference Escherichia coli DPNH Menadione 50 75 Quinones — — 75 DPNH, TPNH Menadione FMN — 79 Furacin* FMN — 79 DPNH Triphenyltetrazolium, Enzyme prepara— 80 Neotetrazolium tion contains FAD Pseudomonas fluorescens DPNH 2,6 Dichlorophenol-in-FMN 80 57 dophenol Pasteurella tularensis TPNH Menadione — — 81 Streptococcus faecalis DPNH Menadione0 FAD, FMN 40-100 48 Leuconostoc mesenteroidesf strain QQ DPNH Menadione — 80
«
2,6 Dichlorophenol-in-— 40 e dophenol Ferricyanide — 5 Lactobacillus delbrueckii DPNH Menadione — 30 β 2,6 Dichlorophenol-in-— 30 β dophenol Ferricyanide — 20 β Clostridium kluyveri DPNH Menadione 75 Free flavins — — 57 Clostridium perfringens DPNH Menadione* — — 33a Free flavins'1 — — 33a Clostridium sticklandii DPNH Triphenyltetrazolium FMN — 82 Yeast DPNH Furacin — — 83a ° Moles reduced pyridine nucleotide oxidized per 100,000 g. protein per minute. For comparative purposes, the TON listed is for the crude extract in each case. None of these enzymes has been significantly purified over the crude extract. b Not identical with nitrate, nitrite, or organo-nitrate reductase. e One of the five DPNH-oxidizing enzymes of S. faecalis (Section II, B). d Completely separated from the native DPNH oxidase of C. perfringens (Section II, C, 2). * Μ. I. Dolin, unpublished.444 Μ. i. DOLIN
9. C Y T O C H R O M E - I N D E P E N D E N T E L E C T R O N T R A N S P O R T 445
6. Substrate Specificity. T P N H cannot replace D P N H as t h e electron donor. Members of all t h e known classes of flavoprotein oxidant have been tested as potential electron acceptors; however, t h e only oxidants found were ferricyanide, 2,6-dichlorophenol-indophenol, and a series of quinones and naphthoquinones. Quinones in general are very efficient oxidants of the reduced flavoprotein. W i t h p-benzoquinone, for instance, t h e KB for quinone is 5 X 10~6 Af, t h e turnover ( T O N ) is 8.8 Χ 104 moles of D P N H oxidized per min. per mole of enzyme-bound F M N , and k> t h e second-order rate constant for reoxidation of reduced diaphorase b y p-quinone is 1.0 X 101 0 M~l X m i n .- 1. T h e latter value exceeds t h a t found for t h e decomposi
tion of peroxide b y catalase. I t is of interest t h a t 2,3-dimethoxy-5-methyl- p-benzoquinone (coenzyme Q without t h e isoprenoid side chain) is also an active oxidant (TON, with 10~4 Μ quinone, = 2.7 Χ 104 moles D P N H oxidized per min. per mole bound F M N , and k = 2.7 Χ ΙΟ8 M~l X min.""1).
T h e values quoted apply a t p H 7.0, 24° C , in phosphate buffer. T h e insolu
ble quinones, coenzyme Qio, and vitamin Ki are inactive.
c. pH Optimum. T h e pH-activity curve is complex, with a peak a t p H 4.5, a plateau between p H 7.5 and 8.5, and a minimum between p H 5.0 and 6.5.
d. Inhibition of the Enzyme. Like D P N H peroxidase, the diaphorase is not appreciably affected b y hydrogen peroxide or b y other — S H inhibitors.
These findings are consistent with t h e fact t h a t S. faecalis carries out a physiologically useful flavoprotein respiration.
2. E N Z Y M E - S U B S T R A T E C O M P L E X F O R M A T I O N
T h e diaphorase has a typical flavoprotein spectrum, with peaks a t 270, 370, and 450 πΐμ. On t h e addition of D P N H , t h e yellow color of t h e enzyme is bleached and there is a 9 0 % decrease in absorbance of t h e 450-πΐμ band.
T h e enzyme does not form t h e enzyme-substrate complex described for the D P N H peroxidase system (Section I I I , B ) ; however, t h e diaphorase does form a complex with D P N H somewhat analogous to t h e one demon
strated for microsomal cytochrome reductase.8 3 b Examination of t h e ultra
violet difference spectrum of diaphorase reduced with one equivalent of D P N H (absorbance of D P N H - r e d u c e d enzyme minus absorbance of oxi
dized enzyme) reveals t h e presence of an intermediate (presumably D P N - bound to reduced flavoprotein) which absorbs a t approximately 320 ιημ.
When t h e alcohol dehydrogenase system is used t o generate D P N H , t h e diaphorase can be reduced in t h e presence of catalytic concentrations of D P N , without formation of t h e 320-ιημ band. I t is difficult therefore t o de
cide whether t h e kinetically active form of t h e diaphorase is free, reduced flavoprotein, or reduced flavoprotein complexed with D P N . With quinones as oxidants, kinetic d a t a suggest t h a t t h e active form of t h e reduced en
zyme is t h e ternary complex of reduced flavoprotein, D P N , a n d quinone.