β
β β
β-LAKTÁMOK KOBALT-KATALIZÁLT SZINTÉZISÉNEK ÉS KATALIZÁTOR KOMPLEXEINEK VIZSGÁLATA
Ph.D. DOKTORI ÉRTEKEZÉS
Készítette:
Fördıs Eszter Okleveles vegyész
Témavezetı: Dr. Ungváry Ferenc
Egyetemi tanár, a kémiai tudományok doktora
PANNON EGYETEM
KÉMIAI ÉS KÖRNYEZETTUDOMÁNYOK DOKTORI ISKOLA
VESZPRÉM
β β β
β-LAKTÁMOK KOBALT-KATALIZÁLT SZINTÉZISÉNEK ÉS KATALIZÁTOR KOMPLEXEINEK VIZSGÁLATA
Értekezés doktori (Ph.D.) fokozat elnyerése érdekében Írta:
Fördıs Eszter
Készült a Pannon Egyetem Kémiai Tudományok Doktori Iskolájának keretében.
Témavezetı: Dr. Ungváry Ferenc, egyetemi tanár
Elfogadásra javaslom (igen / nem) ……….
(aláírás)
A jelölt a doktori szigorlaton …... % -ot ért el.
Veszprém,……… ……….
a Szigorlati Bizottság elnöke Az értekezést bírálóként elfogadásra javaslom:
Bíráló neve: …... …... igen /nem
……….
(aláírás) Bíráló neve: …... …... igen /nem
……….
(aláírás)
A jelölt az értekezés nyilvános vitáján …...% - ot ért el.
Veszprém,……… ……….
a Bíráló Bizottság elnöke A doktori (Ph.D.) oklevél minısítése…...
………
az EDT elnöke
TARTALOMJEGYZÉK
BEVEZETÉS
11. IRODALMI ÁTTEKINTÉS
41.1. Diazoalkánok
41.2. Ketének
71.3. Iminek
91.4. Laktámok
111.5. Diazoalkánok reakciói átmenetifém-karbonilokkal
141.6. Karbén-hidas kétmagvú kobalt-karbonilok
161.7. Foszfán-szubsztituált karbén-hidas kétmagvú kobalt-karbonilok
191.8. Terminális karbén ligandummal rendelkez ı kétmagvú kobalt-
karbonilok
211.9. Dikobalt-karbonil komplexek és
13C illetve
14C izotóppal jelzett
szén-monoxid reakciója
221.10. Oktakarbonil-dikobalt és trifenil-foszfán reakciója
242. KÍSÉRLETI EREDMÉNYEK ÉS ÉRTÉKELÉSÜK
282.1. Etoxikarbonil-karbén-hidas dikobalt-karbonil komplexek kép-
z ı désének és kémiai tulajdonságainak vizsgálata
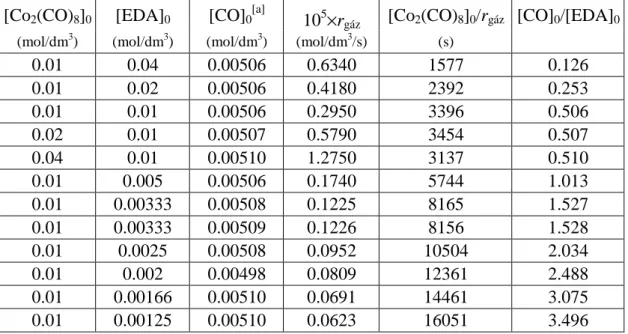
28 2.1.1. Az oktakarbonil-dikobalt és etil-diazoacetát reakciójának kinetikaivizsgálata 29
2.1.2. A Co2(CO)7(CHCO2Et) (1) komplex és etil-diazoacetát reakciójának
vizsgálata 35
2.1.3. A Co2(CO)7(CHCO2Et) (1) komplex és PPh3 reakciójának vizsgálata 39 2.1.4. A Co2(CO)5(CHCO2Et)(PPh3)2 (4) komplex elıállítása és vizsgálata 49 2.1.5. A Co2(CO)6(dppm) (5) és a Co2(CO)5(CHCO2Et)(dppm) (6)
komplexek (dppm = Ph2PCH2PPh2) szerkezetvizsgálata és kémiai
tulajdonságainak vizsgálata 61
2.2. β-Laktám szintézisek etil-diazoacetát iminek jelenlétében meg-
valósított kobalt-katalizált karbonilezésével
69 2.2.1. Az N-terc-butil-transz-α-etoxikarbonil-β-fenil-β-laktám katalitikusszintézise etil-diazoacetátból Co2(CO)8 katalizátor prekurzorral 70 2.2.2. Az N-terc-butil-transz-α-etoxikarbonil-β-fenil-β-laktám szintézise
etil-diazoacetátból trifenil-foszfánnal módosított kobalt katalizátor
jelenlétében 80
3. KÍSÉRLETI RÉSZ
823.1. Általános technika
823.2. Etoxikarbonil-karbén-hidas dikobalt-karbonil komplexek el ı -
állítása és kémiai tulajdonságainak vizsgálata
843.2.1. Co2(CO)7(CHCO2Et) (1) elıállítása 84
3.2.2. Co2(CO)6(CHCO2Et)2 (2) elıállítása 85
3.2.3. Az oktakarbonil-dikobalt és etil-diazoacetát reakciójának kinetikai
vizsgálata 86
3.2.4. A Co2(CO)7(CHCO2Et) (1) komplex és etil-diazoacetát reakciójának
kinetikai vizsgálata 87
3.2.5. Co2(CO)7(CHCO2Et) (1) és PPh3 reakciójának kinetikai vizsgálata 87 3.2.6. Co2(CO)6(CHCO2Et)(PPh3) (3) elıállítása 88 3.2.7. Co2(CO)5(CHCO2Et)(PPh3)2 (4) elıállítása 89
3.2.8. A 3 és 4 komplex és 13CO reakciója 91
3.2.9. A 3 komplex és 13CO reakciójának vizsgálata 92
3.2.10. A 4 komplex és 13CO reakciójának vizsgálata 93 3.2.11. A Co2(CO)5(CHCO2Et)(PPh3)2 (4) komplex és szén-monoxid reak-
ciójának kinetikai és egyensúlyi vizsgálata 94
3.2.12. A Co2(CO)6(CHCO2Et)(PPh3) (3) komplex szén-monoxiddal való
reakciójának vizsgálata etil-alkohol jelenlétében 94 3.2.13. Etil-diazoacetát és etanol reakciója CO nyomás alatt 2 mol% 3
komplex jelenlétében 95
3.2.14. Etil-diazoacetát és etanol reakciója CO nyomás alatt 2 mol%
Co2(CO)7(PPh3) komplex jelenlétében 95
3.2.15. Co2(CO)6(CHCO2Et)2 (2) és PPh3 reakciója etanol jelenlétében 96
3.2.16. A Co2(CO)5(CHCO2Et)(PPh3)2 (4) komplex szén-monoxiddal való
reakciójának vizsgálata etil-alkohol jelenlétében 96 3.2.17. Etil-diazoacetát és etanol reakciója CO nyomás alatt 2 mol% 4
komplex, Co2(CO)6(PPh3)2, valamint [Co(CO)3(PPh3)2][Co(CO)4]
komplex jelenlétében 96
3.2.18. Co2(CO)6(dppm) (5) elıállítása 98 3.2.19. Co2(CO)5(CHCO2Et)(dppm) (6) elıállítása 99
3.2.20. Az 5 és 6 komplex és 13CO reakciója 100
3.2.21. Etil-diazoacetát és etanol rakciója CO nyomás alatt 2 mol%
Co2(CO)6(dppm) (5) komplex jelenlétében 101
3.3. Kobalt-katalizált β-laktám szintézisek
1023.3.1. N-terc-butil-transz-α-etoxikarbonil-β-fenil-β-laktám elıállítása 102 3.3.2. N-benzil-transz-α-etoxikarbonil-β-fenil-β-laktám elıállítása 103 3.3.3. N-metil-transz-α-etoxikarbonil-β-fenil-β-laktám elıállítása 105 3.3.4. Etil-3-(difenil-metilén-amino)-3-oxo-propanoát elıállítása 106 3.3.5. Etil-diazoacetát és N-terc-butil-benzaldimin reakciójának kinetikai
vizsgálata Co2(CO)8 katalizátor prekurzor jelenlétében 107 3.3.6. Co2(CO)7(CHCO2Et) (1) komplex és N-terc-butil-benzaldimin reak-
ciójának kinetikai vizsgálata 108
3.4. Az N-terc-butil-transz-α-etoxikarbonil-β-fenil-β-laktám szinté- zise trifenil-foszfánnal módosított kobalt katalizátor jelenlété-
ben
1083.4.1. Polimerhez kötött kobalt-komplex elıállítása 108 3.4.2. Etil-diazoacetát foszfán-módosított kobalt katalizátorokkal végzett
karbonilezése N-terc-butil-benzaldimin jelenlétében 109
ÖSSZEFOGLALÁS
111IRODALOMJEGYZÉK
115KIVONAT
Etil-diazoacetát N-terc-butil-benzaldimin jelenlétében egy reakcióedényben végrehajtott kobalt-katalizált karbonilezésével 95%-os hozammal képzıdik az N-terc-butil-transz-α- etoxikarbonil-β-fenil-β-laktám, amelynek szerkezetét egykristály röntgendiffrakciós vizsgálattal jellemeztük. A módszer alkalmas N-metil- és N-benzil-szubsztituált β-laktám származékok elı- állítására is.
Reakciókinetikai és infravörös spektroszkópiai vizsgálatok eredményeire támaszkodva a karbonilezés mechanizmusát két egymással együttmőködı katalitikus ciklus feltételezésével írtuk le. A β-laktám képzésben az egyes ciklusok súlyát az alkalmazott szén-monoxid : etil- diazoacetát koncentráció arány szabályozza. Foszfánokkal módosított kobalt katalizátorok is alkalmasnak bizonyultak az etil-diazoacetát karbonilezésére.
ABSTRACT
The one-pot laboratory synthesis of N-tert-butyl-trans-α-ethoxycarbonyl-β-phenyl-β- lactam (TBL) with 95% yield has been achieved by the cobalt-catalyzed carbonylation of ethyl diazoacetate (EDA) in the presence of N-tert-butyl-benzaldimine (BTB). The structure of TBL was established by single crystal X-ray diffraction analysis. The method of synthesis was found to be applicable for the preparation of structurally similar other β-lactams as well.
Based on results of kinetic and IR spectroscopic investigations a cooperative two-cycle mechanism was suggested for the catalytic carbonylation of ethyl diazoacetate. The ratio of participation of the cycles in the TBL production depends on the concentration ratio of carbon monoxide and ethyl diazoacetate. Phosphane-modified carbonyl cobalt catalysts were found to be suitable for the carbonylation of ethyl diazoacetate as well.
AUSZUG
Ein Eintopf-Verfahren wurde entwickelt zur einfachen Herstellung von N-tert-butyl- trans-α-ethoxycarbonyl-β-phenyl-β-lactam (TBL) durch kobalt-katalysierte Carbonylierung von Diazoessigsäure-ethylester (EDA) in Gegenwart von N-tert-butyl-benzaldimin. Die Struktur des Produktes wurde durch Einkristall Röntgen-Diffraktion ermittelt. Das Verfahren ist geeignet zur Herstellung von struktur-ähnlichen N-methyl- und N-benzyl-β-laktam Derivate.
Auf Grund der Ergebnisse von kinetischen und IR-spektroskopischen Untersuchungen wurde ein kooperativer Zwei-Zyklus Mechanismus für die Carbonylierung von EDA vorgeschlagen. Der Anteil der Zyklen in der TBL-Produktion ist von dem Verhältnis der CO- und EDA-Konzentration abhängig. Phosphan-Modifizierte Kobalt Katalysatoren erwiesen sich aktiv in der Carbonylierungsreaktion von EDA.
BEVEZETÉS
A diazoalkánok diazo-csoportjának helyettesítése az izoelektronos szén-monoxiddal dinitrogén felszabadulását és ketén képzıdését eredményezheti.
R2C=N=N + CO R2C=C=O + Nδ 2
δ
A szakirodalomban, nagy számban találhatók e reakcióval kapcsolatban álló közvetett és közvetlen kísérleti bizonyítékok.[1] Ezek valószínősítik, hogy a ketén képzıdése több részlé- pésen keresztül történik. A szakmai közvélemény által elfogadott részlépésnek tekintik a karbén intermedierhez vezetı dinitrogén kiszakadást, valamint a karbén intermedier és a szén- monoxid keténné való összekapcsolódását. A reakció végeredményére hatással van az aktiválás módja és az átmenetifém-komplexek jelenléte.
A fenti reakció gyakorlat számára hasznos katalitikus megvalósításának kutatásában hat éve veszek részt. Munkám kiinduló pontja Tuba Róbert PhD értekezésében[2] összefoglalt meg- állapításai voltak, miszerint etil-diazoacetát és szén-monoxid etanol jelenlétében katalitikus mennyiségő oktakarbonil-dikobalt hatására lejátszódó reakciója 100%-os kitermelést megköze- lítıen dietil-malonáthoz vezet.
EtO2CCH=N=N + CO + EtOH
1 mol% Co2(CO)8
25 °C; 50 bar EtO2CCH2CO2Et + N2
Etil-diazoacetát és oktakarbonil-dikobalt sztöchiometrikus reakciójából kétmagvú karbén-hidas komplexeket izoláltak, amelyekrıl kísérletileg bizonyították, hogy a dietil- malonát-képzıdés intermedierjei.[3] A dietil-malonát keletkezését a kobalthoz kötött etoxikarbonil-karbén és szén-monoxid kapcsolódásával feltételezett etoxikarbonil-ketén inter- medier és etanol reakciójával magyarázták.
Co2(CO)8 + EtO2CCHN2 Co2(CO)7(CHCO2Et) + EtO2CCHN2 Co2(CO)6(CHCO2Et)2
+ 2CO + EtOH 25 °C
1 bar + EtO2CCH2CO2Et
Co2(CO)6(CHCO2Et)2 Co2(CO)7(CHCO2Et)
+ 2CO + EtOH 25 °C
1 bar Co2(CO)8 + EtO2CCH2CO2Et Co2(CO)7(CHCO2Et)
Munkám elsı részében a vizsgálatokat kiterjesztettem az intermedier kobalt komplexek foszfán származékainak elıállítására és kémiai tulajdonságainak tanulmányozására. Azt tapasz- taltam, hogy a foszfán-szubsztituált kobalt komplexek is alkalmas prekurzorok a diazoalkánok katalitikus karbonilezésében.
A kutatás a továbbiakban arra irányult, hogy az etil-diazoacetát katalitikus karbonilezési reakcióját β-laktámok elıállítására alkalmazzam. A PhCH=NR típusú iminek jelenlétében vég- zett karbonilezéssel jó hozammal sikerült a β-laktám szerkezeti egységet tartalmazó N-alkil-α- etoxikarbonil-β-fenil-β-laktámok sztereoszelektív elıállítása.
EtO2CCH=N=N + CO + 5 mol% Co2(CO)8
25 °C; 50 bar + N2
PhCH=NR
N Ph
O EtO2C
R
A β-laktám szerkezeti egységet tartalmazó vegyületek széles spektrumú antibakteriális hatással rendelkeznek és számos egyéb fontos biológiai hatásuk van. A penicillineknél és a cefalosporinoknál hatásosabb enzim inhibítor β-laktám molekulák felfedezéséért és gazdaságos elıállításáért széleskörő kutatás folyik.
A fenti témakörben végzett munkám eredményérıl három Intézményi, egy Országos Tudományos Diákköri Konferencián,[4-7] egy nemzetközi tudományos konferencián[8], diplo- madolgozatomban[9] és szakfolyóiratokban megjelent tudományos közleményekben számoltam be.[10-15] A témával összefüggı további eredmények leírását tartalmazza még egy megjelenés alatt álló kézirat.[16]
A Bevezetésben hivatkozott munkák:
[1] Irodalmi összefoglalás: N. Ungvári, F. Ungváry, Carbonylation of diazoalkanes, in Modern Carbonylation Methods, Kollár, L. (Ed.), Wiley-VCH, Weinheim, Chapter 8 (2008) pp 199-221.
[2] Tuba Róbert PhD Disszertáció, Veszprém (2003)
[3] R. Tuba, F. Ungváry, J. Mol. Catal. A: Chem. 203 (2003) 59-67.
[4] Fördıs Eszter: Új módszer alkil-kobalt-karbonilok és acil-kobalt-karbonilok elıállítására, Tudományos Diákköri Dolgozat, Bemutatva: ITDK 2003. nov. 26. Veszprém
[5] Fördıs Eszter: Oktakarbonil-dikobalt és etil-diazoacetát reakciójának vizsgálata, Tudományos Diákköri Dolgozat, Bemutatva: ITDK 2004. nov. 24. Veszprém
[6] Fördıs Eszter: Trifenil-foszfán-szubsztituált dikobalt-karbén-karbonil komplexek elıállítása és kémiai tulajdonságainak vizsgálata, Tudományos Diákköri Dolgozat, Bemutatva: ITDK 2005 nov. 23. Veszprém [7] Fördıs Eszter: Oktakarbonil-dikobalt és etil-diazo-acetát reakciójának vizsgálata, Tudományos Diákköri
Dolgozat, Bemutatva: OTDK 2005. márc. 21-23. Budapest
[8] Fördıs Eszter: Reactions of ethoxycarbonylcarbene-bridged dicobalt carbonyl triphenylphosphane
complexes; International Symposium on Homogeneous Catalysis, ISHC-XVI. Firenze, 2008. júl. 6-11. Book of Abstracts pp. 220.
[9] Fördıs Eszter, Diplomamunka, Veszprém (2006)
[10] R. Tuba, E. Fördıs, F. Ungváry, J. Mol. Catal. A: Chem. 236 (2005) 113-118. Preparation of triphenylphosphane substituted ethoxycarbonylcarbene-bridged dicobalt carbonyl complexes and their application as catalyst precursors in carbonylation of ethyl diazoacetate into diethyl malonate
[11] R. Tuba, E. Fördıs, F. Ungváry, Inorg. Chim. Acta 358 (2005) 4081-4085. Kinetics and mechanism of the reaction of octacarbonyl dicobalt with ethyl diazoacetate
[12] E. Fördıs, N. Ungvári, T. Kégl, F. Ungváry, Eur. J. Inorg. Chem. (2006) 1875-1880. Reactions of 13CO with Ethoxycarbonylcarbene-Bridged Dicobalt Carbonyl Complexes:[µ2-{Ethoxycarbonyl(methylene)}- µ2-(carbonyl)-bis(tricarbonyl-cobalt)(Co-Co)] and [Di-µ2-{ethoxycarbonyl(methylene)}- bis(tricarbonylcobalt)-(Co-Co)]
[13] E. Fördıs, N. Ungvári, L. Párkányi, G. Szalontai, T. Kégl, F. Ungváry, Inorg. Chim. Acta 361 (2008) 1832-1842.
Structure of Co2(CO)6(dppm) and Co2(CO)5(CHCO2Et)(dppm) (dppm = Ph2PCH2PPh2) and reaction with 13CO and exchange with 13CO: an experimental and computational study
[14] N. Ungvári, E. Fördıs, T. Kégl, F. Ungváry, Inorg. Chim. Acta 362 (2009) 1333-1342.
Reactions of triphenylphosphane-substituted ethoxycarbonylcarbene-bridged dicobalt carbonyl complexes with carbon monoxide or 13CO: An experimental and theoretical study
[15] E. Fördıs, R. Tuba, L. Párkányi, T. Kégl, F. Ungváry, Eur. J. Org. Chem. (2009) 1994-2002.
Application of the octacarbonyl dicobalt-catalyzed carbonylation of ethyl diazoacetate for the one-pot synthesis of N-tert-butyl-trans-α-ethoxycarbonyl-β-phenyl-β-lactam
[16] N. Ungvári, E. Fördıs, T. Kégl, F. Ungváry, Inorg. Chim. Acta, közlésre elfogadva 2009. szept. 11.
Mechanism of the cobalt-catalyzed carbonylation of ethyl diazoacetate
1. IRODALMI ÁTTEKINTÉS
A diazoalkánok karbonilezésére legalkalmasabbnak látszó karbonil-kobalt komplexek megismerését, és e komplexek iminek jelenlétében β-laktám katalitikus szintézisét célzó kísér- leteim megalapozásához a legfontosabbnak vélt részeket foglalom össze röviden az alábbiak- ban.
1.1. Diazoalkánok
A diazoalkánok rendkívül sokoldalú kémiájában a tájékozódásom kiindulópontja a Houben-Weyl megfelelı kötetei [1,2], valamint egy diazo-kémiáról szóló speciális monográfia volt [3].
Az általánosságban R2CN2 képlettel leírható diazoalkánok molekulán belüli polarizált- sága mezomer határszerkezetekkel szemléltethetı (1.1 séma). Ezekre, valamint elméleti kémi- ai számítások eredményeire gyakran hivatkoznak a diazoalkánok különféle kémiai reakcióinak értelmezésekor.
R2C=N=N R2C-N==N R2C-N=N R2C-N=N
ahol R leggyakrabban: H, alkil, alkenil, aril, alkilOC(=O), alkilC(=O) különféle párosításban
1.1 séma
A dipoláris tulajdonsággal rendelkezı diazoalkánok α-szénatomja, mint nukleofil cent- rum elektrofil reagensek, mint például a proton és karbonilvegyületek számára támadható és dinitrogén felszabadulással különféle termékekhez lehet jutni. Így például sósavval alkil- kloridhoz, karbonsavval karbonsavészterhez, fenollal fenoléterhez, aldehiddel és ketonnal lánchosszabbított származékhoz lehet jutni. Savhalogenidekkel acil-diazoalkánokat lehet elıál- lítani. Olefinekkel és acetilénekkel 1,3-dipoladdiciós termékek képzıdnek [1].
Diazoalkánokat, mint karbén prekurzorokat széles körben alkalmaznak kiindulási anyagként a szintetikus kémiában [4]. Különösen átmenetifém komplexekkel katalizált reakci- ókban kedvelt reagensek a különféle diazoalkánok [5]. E szintetikus kémiában rendkívül hasz- nos vegyületek a megfelelı primer amin diazotálásával csak olyan esetekben állíthatók elı,
ahol az elsıdlegesen képzıdı diazónium-vegyület diazoalkánná alakulását kísérı protonvesz- tést a molekulában lévı elektronszívó csoport elısegíti. Például glicin-etilészter- hidrogénklorid diazotálásával így etil-diazoacetát képzıdik (1.1 reakció):
EtO2CCH2NH2 HCl + NaNO2 EtO2CCHN2 + 2 H2O + NaCl (1.1)
A példában szereplı etil-diazoacetátban az etoxi-karbonil-csoport a közönséges diazoalkánokhoz képest további töltés-delokalizációt tesz lehetıvé, amellyel lényegesen meg- növeli a molekula termodinamikai stabilitását (1.2 séma).
O C EtO
CH N=N
O C EtO
CH N N
O C EtO
CH N=N O
C EtO
CH N N
1.2 séma
Ilyen stabilizáló csoport hiányában a primer amin diazotálásakor keletkezı diazóniumion spontán dinitrogénvesztéssel karbéniumionná alakul és csak annak további át- alakulási termékeihez lehet jutni. Ilyen esetekben a diazovegyület elıállítására a megfelelı primer amin diazotálása helyett más szintézisutat kell választani. A leggyakrabban alkalmazott klasszikus eset például a diazometánnál az, hogy a metil-amin valamilyen acilezett származé- kát vetik alá diazotálásnak (1.2 reakció), és a képzıdıtt nitrozovegyületet lúggal elbontják [6]
(1.3 reakció):
CH3NH3Cl + H2NC(=O)NH2 CH3N(NO)C(=O)NH2
HNO2
CH3NHC(=O)NH2
- NH4Cl (1.2)
CH3N(NO)C(=O)NH2 + KOH CH2N2 + KOCN + 2 H2O (1.3)
Trimetilszilil-diazometán esetében a megfelelı Grignard reagenst viszik reakcióba al- kalmas diazocsoport-átvivı reagenssel, mint például difenil-foszforil-aziddal [2] (1.4 reakció).
Me3SiCH2Cl + Mg Me3SiCH2MgCl (PhO)2P(=O)N3 Me3SiCHN2 (1.4)
A diazoalkánok hı, fény, átmenetifémek és átmenetifém-vegyületek hatására dinitrogén kilépésével karbéneket, illetve átmenetifém-komplexek esetében ún. karbenoidokat - karbénként reagáló vegyületeket - eredményeznek (1.5 reakció).
∆∆∆∆, hνννν, M vagy [M]
R2CN2 R2C + N2
ahol M = átmenetifém
(vagy karbenoid) (1.5)
Az R-csoport minısége szerint nagy különbségek vannak a diazoalkánok termikus sta- bilitásában. A diazometán (CH2N2) vagy a diazoetán (CH3CHN2) rendkívül bomlékony, és csak közvetlenül a felhasználás elıtt híg oldatban szokás elıállítani. Ezzel szemben, alfa- helyen karbonil-csoportot tartalmazó diazoalkánok viszonylag stabilis molekulák. Az etil- diazoacetát (EtO2CCHN2) például desztillálással tisztítható kereskedelemi termék. A diazo- malonésztert (EtO2CC(N2)CO2Et) pedig a legstabilisabb diazoalkánok közé sorolják. Ugyan- csak kereskedelemben vásárolható termék a trimetilszilil-diazometán (Me3SiCHN2) is 2 mol/dm3 koncentrációjú oldat formájában, amely hőtıszekrényben hosszú ideig tárolható.
A diazoalkánt a megfelelı alkil-diazónium-ion konjugált bázisának tekintik (1.6 reakció).
R2CH-N2 R2C=N2 + H (1.6)
Az alkil-diazónium-ion pKa értékei között nagy különbségeket állapítottak meg az R- szubsztituens minıségétıl függıen. Így például a metán-diazónium-ion (mindkét R = H) ese- tében a pKa érték 10. Ezzel szemben a diazo-észtereknek és a diazo-ketonoknak megfelelı diazónium-ionok (legalább az egyik R = alkoxi-karbonil, illetve acil) pKa értékei −5, illetve −2 értékőek. A diazónium-ionok pKa értékei között megállapított 12-15 nagyságrend különbségre szokás visszavezetni a diazometán és más alkil-diazo-vegyület, valamint a különféle diazo- karbonil-vegyület kémiai viselkedésben tapasztalható nagy eltéréseket.
A diazo-alkánok veszélyességével foglalkozó irodalom és a diazo-alkánokkal való munkánál betartandó munkavédelmi elıírások tömör [2,7] és részletesebb [1,2] leírásaiból tájékozódtam.
1.2. Ketének
A ketének kumulált kötésrendszerő, igen reakcióképes, spontán dimerizálódásra hajla- mos karbonilvegyületek, felfedezésük Hermann Staudinger nevéhez főzıdik [8]. Fontos szere- pet játszanak mint intermedierek a szerves szintézisekben. A ketének szobahımérsékleten álta- lában nem tárolhatóak, ezért közvetlenül felhasználásuk elıtt szokták azokat elıállítani. Az általánosságban R2C=C=O képlettel leírható ketének molekulán belüli polarizáltsága mezomer határszerkezettel szemléltethetı [9] (1.3 séma), melyre gyakran hivatkoznak a ketének külön- féle kémiai reakcióinak értelmezésekor.
R C R
C O
R C R
C O
1.3 séma
A karbonil csoport melletti szénatomon jelentıs negatív töltés van, összhangban e szén- atom megjelenési helyével a ketének 13C NMR spektrumában. Az általánosságban R2C2=C1=O képlettel leírható keténekre jellemzı 13C NMR kémiai eltolódás-tartomány: 178-203 ppm a karbonil C1 szénatom esetében, és 33-48 ppm a C2 szénatom esetében a hozzá kapcsolódó R csoportok minıségétıl függıen [10]. A ν(CO) rezgés az infravörös spektrumban a hármaskötés tartományában, 2100 és 2200 cm−1 között jelentkezik.
A keténeket általában a megfelelı karbonsavból vagy annak származékaiból szokták elıállítani [11]. A csoport legfontosabb képviselıje az alapvegyület, a ketén: CH2=C=O, melynek nagyipari elıállítása ecetsav pirolízisével történik [12] (1.7 reakció).
(1.7)
CH3COOH ∆∆∆∆ H2C=C=O + H2O
A ketének nem adják a karbonilcsoportra jellemzı kondenzációs reakciókat. Két leg- fontosabb reakciótípusuk a HX típusú molekulák acilezése [13,14] és a dimerizáció [15]. A HX típusú molekulák acilezése a karbonilcsoportra jellemzı nukleofil addícióval indul, a C=O kettıs kötés azonban ezt követıen – enol-keto átalakulás révén – regenerálódik. Keténnel ezen az úton tehát acetilcsoport vihetı be a molekulába (1.8 és 1.9 reakciók). Példák:
H2C=C=O + EtOH CH3COOEt (1.8)
(1.9)
Ezzel a reakcióval – a HX molekulától függıen – nagyon sokféle acetilszármazék állít- ható elı, így HX = alkohol esetén ecetsavészter, HX = amin esetén nitrogénen helyettesített acetamid, stb. Legfontosabb ezek közül a ketén reakciója ecetsavval, amely az ecetsav- anhidrid elıállításának ipari módszere (1.10 reakció).
H2C=C=O + CH3COOH (CH3CO)2O (1.10)
A ketén szobahımérsékleten spontán dimerizál és diketénné alakul át (1.11 reakció).
Amennyiben a ketént nem közvetlenül elıállítása után használják fel, úgy az diketén formájá- ban tárolható, mert a dimerizáció reverzibilis. A diketént 500°C-ra melegítve depolimerizálódik [15].
2 H2C=C=O H2C
C O
C O
H2C
(1.11)
A négytagú β-lakton győrős szerkezet miatt a diketén maga is reakcióképes, és mint acilezıszer, az acetoacetil csoport [CH3C(O)CH2C(O)-] bevitelére alkalmas. Diketén és etil- alkohol reakciója a szerves szintézisekben gyakran használt acetecetészter egyik elıállítási módja.
A nagy reakciókészségő ketének közül az alkoxikarbonil-keténeket eddig nem sikerült izolálni. Intermedier vegyületként való képzıdésére például a metoxikarbonil-ketén esetében az alkohol jelenlétében nyert észter alapján következtettek [16]. Mások alkohol távollétében a metoxikarbonil-ketén spontán dimerizációját feltételezték, de elkülönítési kísérleteik nem jár- tak sikerrel, mert csak egy vöröses polimerként jellemzett anyagot kaptak [17]. Nagy mólfeleslegő szén-szuboxid és etanol reakcióelegyében még alacsony hımérsékleten is csak a végtermék dietil-malonátot sikerült kimutatni a közbensı termékként feltételezhetı etoxikarbonil-ketén helyett [18] (1.12 reakció). Valószínőleg az etoxikarbonil-ketén reakciója etanollal sokkal gyorsabb, mint a képzıdésének sebessége.
O=C=C=C=O + EtOH [EtO2CCH=C=O] EtOH
EtO2CCH2CO2Et (1.12)
1.3. Iminek
Az iminek C=N kettıs kötést tartalmazó reakcióképes vegyületek [19], amelyek szá- mos kémiai átalakítás építıelemei. A reakciókészség alapja a C=N kettıs kötés 1.4 sémán lát- ható polarizációja.
R1 C R2
Nδ- R3
δ+
1.4 séma
A karbonilvegyületek primer aminokkal a félacetálokhoz hasonló félaminálokon át, vízvesztéssel iminekké, régebbi nevükön Schiff-bázisokká alakulnak át (1.13 reakció). Az aromás karbonilvegyületek iminszármazékai delokalizált elektronszerkezetük folytán jóval stabilisabbak, mint alifás analógjaik, amelyek csak inert körülmények közt tárolhatók, hiszen oxigén vagy víz jelenlétében könnyen hidrolizálnak vagy gyorsan oligomerizálódnak.
PhCHO + H2NR Ph-CH OH
NHR
- H2O
Ph-CH=NR (1.13)
félaminál
Aszimmetrikus karbonilvegyületek esetén a C=N kötés az imineknél geometriai izome- rek képzıdését teszi lehetıvé, amelyek azonban általában igen könnyen izomerizálódnak. A C=C és C=N kettıs kötés körüli geometriai izoméria között lényeges különbség van annak következtében, hogy a nitrogénatomhoz nem két atom vagy atomcsoport kapcsolódik, hanem az egyik „szubsztituens” a nitrogénatom magános elektronpárja. Ez azzal a következménnyel jár, hogy a C=N kettıs kötésnél a két izomer átalakulása egymásba lényegesen könnyebben játszódik le, mint az alkének esetében.
Iminek esetében az átalakulás olyan gyors, hogy a két izomer nem különíthetı el, csak -100°C alatti hımérsékleten mutatható ki NMR-spektroszkópiával. Az izomerizációhoz a π- kötésnek ugyanis nem kell felszakadnia, mert az elektronpár és az alkilcsoport a nitrogénatom
„áthibridizálódása” segítségével is át tud kerülni a nitrogénatom másik oldalára. Ezt úgy kell elképzelni, hogy az sp2 nitrogénatom az átmeneti állapotban sp hibridállapotúvá alakul át, a C,N,Q-hármas egy egyenes mentén helyezkedik el és a magános pár a nemkötı p-pályára ke- rül. Ebbıl a helyzetbıl pedig a molekula egyaránt „visszaeshet” mindkét konfigurációba (1.5 séma).
C=N R R'
Q
C=N _ Q R
R' Q
C=N R R'
1.5 séma
Szekunder aminból és karbonilvegyületbıl nem képzıdhet C=N kettıskötéses vegyü- let. Ilyenkor, amennyiben a karbonilcsoporttal szomszédos α szénatomon van hidrogén, C=C kettıskötéses vegyület, enamin keletkezik. Az iminek és az enaminok – megfelelı molekula- szerkezet esetén – a keto-enol tautomériával analóg tautomer viszonyban állhatnak egymással.
Imineket elı lehet állítani a már említett módon aminok és karbonilvegyületek nukleofil addíciós reakciójában képzıdı félaminálok dehidratálásával [20], karbonsavak nitrozo-vegyületekkel alkotott kondenzációs reakciójában, Stieglitz-átrendezıdéssel [21,22], valamint Houben-Hoesch reakcióval [23,24].
Az iminek katalitikus hidrogénezéssel könnyen szekunder aminokká alakíthatók, ezen- kívül könnyen hidrolizálnak a megfelelı amin és karbonilvegyület keletkezése közben.
1.4. Laktámok
A laktámok tulajdonképpen győrős monopeptidek, az acilezett primer aminok egyik különleges típusát képviselik. A győrős belsı észterek, a laktonok nitrogéntartalmú analógjai.
Aminocsoportot megfelelı helyzetben tartalmazó amino-karbonsavakból keletkezhetnek. Álta- lában a γ-, δ- és ε-laktámok stabilisak, ezek az 5, 6, ill. 7 tagú győrők. Legfontosabb képvise- lıjük az ε-amino-hexánsavból levezethetı ε-kaprolaktám, melyet nagyipari méretekben állíta- nak elı, mint a Nylon-6 típusú szintetikus poliamidok (pl: a Perlon) monomerjét.
Laktámokat elı lehet állítani győrős oximok sav-katatalizált Beckmann- átrendezıdésével [25-27], Schmidt-reakcióval győrős ketonok és hidrogén-azid (HN3) reakció- jában [28,29], a már említett amino-karbonsavak győrőzáródási reakciójával, valamint Kinugasa reakcióval alkinek és nitronok (R1R2C=N+R3O-)Cu(I)-katalizált cikloaddíciós reak- ciójában, amely az in situ elıállított Cu(I)-acetilid közbensı terméken keresztül játszódik le [30,31].
A β-laktámok nitrogén heteroatomot tartalmazó négytagú győrős amidok, amelyek nagy biológiai jelentıséggel bírnak, mivel számos antibakteriális hatású vegyület fontos építı- elemei. Az elsı β-laktámot Herrmann Staudinger állította elı 1907-ben [32] az anilin és ben- zaldehid reakciójából levezethetı Schiff-bázis, az N-benzilidén-anilin és a difenil-ketén [2+2]
cikloaddíciós reakciójával (1.14 reakció).
N Ph
O Ph
Ph Ph
C=C=O Ph
Ph H
N-fenil-α-difenil-β-fenil-β-laktám
+ (1.14)
PhCH=N-Ph
Számos β-laktám szintézis ismert, de az egyik legegyszerőbb, széles körben alkalma- zott módszer a Staudinger reakció, amely ketének és iminek [2+2] cikloaddíciós reakciója (1.6 séma). A reakció lejátszódását egy ikerionos intermedieren keresztül két lépésben képzelik el [33-35]. Az elsı lépésben az imin-nitrogén magános elektronpárja gyors reakcióban megtá- madja a ketén sp hibridizációjú szén-atomját.
A második sebességmeghatározó lépésben egy elektrociklikus konrotációval lejátszódó győrőzáródás következik be.
C O C
R' R
N X C R'' H +
R' N H
O X
R R''
O N
R H
R''
X R' gyors
1.6 séma
Elektronküldı szubsztituens az imin nitrogén atomon és elektronszívó szubsztituens a keténben kedvez a β-laktám képzıdésnek. Apoláros oldószerekben szoros ion-párként ismert [Cp2Co]+[Co(CO)4]− (ahol Cp = η5-C5H5) [36] vagy Na+[Co(CO)4]− [37] hatására bekövetkezı katalizált Staudinger reakciót írtak le a difenil-ketén és az elektron-szegény Tos-N=CH- COOEt (ahol Tos = p-toluolszulfonil) benzotrifluorid oldószerben szobahımérsékleten végzett reakciójánál. Katalizátor nélkül nem észleltek reakciót. Katalizátor jelenlétében azonban 5 perc alatt 85%-os hozammal észlelték a megfelelı β-laktám képzıdését. A katalízist [Cp2Co]+[Co(CO)4]− esetében az 1.7 séma szerint képzelik el. Ehhez képest a Na+[Co(CO)4]− katalizátor esetében a különbség csupán annyi, hogy a [Cp2Co]+ szerepét a Na+ veszi át [38,39].
C O C
Ph Ph
[Cp2Co]+[Co(CO)4]-
(CO)4Co Ph OCoCp2
Ph
Tos N C
COOEt
H N
Tos
Ph EtOOC Ph
O
1.7 séma
A ketének közül az etoxikarbonil-ketén közvetlen alkalmazását a Staudinger reakció- ban megakadályozza az a tény, hogy ezt a ketént nagy reakciókészsége miatt még nem sikerült izolálni.
Az irodalomban két közleményt találtam, amelyekben Cr, Mo és W karbén- komplexeibıl [40], illetve Cr karbén-komplexekbıl [41] kiindulva iminek jelenlétében β- laktámokhoz jutottak. Az elsı esetben a karbenoidok és N-metil-benzaldimin reakcióelegyét két napon át 80-100°C-on 50 bar szén-monoxid nyomás alatt tartották, majd a reakcióelegyben képzıdött β-laktámot 90%-os kitermeléssel izolálták [40] (1.15 reakció). A második esetben pentakarbonil-króm-karbén komplexeket iminek jelenlétében besugározva nyertek β- laktámokat [41] (1.16 reakció). Valószínő, hogy ezeknél a sztöchiometrikus reakcióknál is közvetetten a Staudinger reakcióról van szó, ugyanis az alkalmazott reakciókörülmények kö- zött az elsıdleges termék a karbén ligandum és szén-monoxid összekapcsolódásából egy ketén, amely a reakció körülményei között átalakul a jelenlévı iminnel β-laktámmá.
(CO)5M C OEt SiPh3
+ CO + PhCH=NMe
N Ph3Si
EtO
O Me
Ph
50 bar 80-100 °C, 2 nap
90% izolált kitermelés (M = Cr, Mo, W)
(1.15)
(CO)5Cr C X R
(X = OR, NR2; R = alkil)
hνννν
(CO)4Cr X
C R C O
N R
X
O N
(1.16)
A fent idézett irodalmakban leírt eredmények azt igazolják, hogy nem szükséges feltét- lenül izolált ketént felkínálni az imin reakciópartnereként.
Karbén ligandumokat tartalmazó karbonil-kobalt komplexek hasonló β-laktámhoz ve- zetı átalakulását eddig még nem vizsgálták annak ellenére, hogy nagy számban ismertek ilyen komplexek. Az ismertté vált és érdeklıdésemet felkeltı karbén ligandumokat tartalmazó karbonil-kobalt komplexeket egy késıbbi alfejezetben részletesen bemutatom.
1.5. Diazoalkánok reakciói átmenetifém-karbonilokkal
A diazoalkánok és átmenetifém-komplexek reakciója néhány esetben koordinált diazoalkán komplexhez vezet, de általában a diazoalkán dinitrogénjének vesztésével jár, amely bomlást dediazotálásnak is nevezik [42].A dinitrogén-vesztés után a diazoalkánból megmara- dó rész egy karbén, amely sok esetben az átmenetifémhez koordinálódik és izolálható karbén- komplexhez vezet. Többek között az oktakarbonil-dikobaltról azt állították, hogy még 60°C- on sem hatásos katalizátora a diazoalkánok dediazotálásának [43].
Ezt az állítást cáfolta Tuba és Ungváry megfigyelése, miszerint az oktakarbonil- dikobalt az etil-diazoacetátot dinitrogén felszabadulás közben szobahımérsékleten vagy még alacsonyabb hımérsékleten is hatásosan elbontja. A dinitrogén felszabadulása mellett jó ki- termeléssel izolálható etoxikarbonil-karbén ligandumot tartalmazó kobalt-komplexek képzıd- nek, amelyek etanol jelenlétében katalizálják az etil-diazoacetát karbonilezését dietil- malonáttá [44,45].
E megfigyelések is alátámasztják, hogy a diazoalkán és átmenetifém-komplex reakció- jában képzıdı karbén-komplex - az un. karbenoid - a szabad karbénhez hasonló további átala- kulásokra képes. A diazoalkánok és átmenetifém-komplexek reakciója így két lépésre bontha- tó, melyek részei lehetnek az 1.8 séma szerint ábrázolt katalitikus ciklusnak is [46].
LnM LnM=CR2
LnM-CR2 N2
R2C=N2 N2
SCR2 S
S = I-, C=C , CO, C C , CR2
1.8 séma
A szabad koordinációs hellyel rendelkezı átmenetifém az LnM-típusú komplexben, mint elektrofil Lewis-sav centrum koordinálni képes a diazoalkánt, amely dinitrogén vesztésen keresztül a fémhez koordinált karbént, az ún. karbenoidot eredményez.
A karbén ligandumot az átmenetifém centrum képes átadni egy alkalmas elektrondús S: molekulának egy SCR2 típusú termék képzıdése közben és ezzel regenerálódik a diazoalkán koordinációjára képes LnM és kezdıdhet egy új ciklus. Ha az általános sémában szereplı S: reagens molekula helyére szén-monoxidot képzelünk, akkor a szén-monoxid és az LnM=CR2 karbenoid reakciójából levezethetı SCR2 típusú termék-molekula egy ketén.
Különbözı átmenetifémekhez kötött karbén és szén-monoxid keténhez vezetı reakció- jára több példa található az irodalomban, amelynek összefoglalása egy nemrég megjelent mo- nográfiában olvasható [47]. A legtöbb esetben csak feltételezik a ketén fenti úton való képzı- dését, mivel csupán olyan végsı reakciótermékeket izoláltak, amelyek a közbensı keténbıl vezethetık le. A trimetilszilil-diazometán és oktakarbonil-dikobalt reakciója esetében azonban a trimetilszilil-ketén preparatív elıállítására alkalmas módszer is ismeretes [48].
Az idézett eredmények azt is igazolják, hogy nem csak sztöchiometrikus reakciókat le- het karbonil-kobalt komplexekkel megvalósítani, hanem azok katalitikus ciklusok részei is lehetnek. Ezért megvizsgáltam, hogy mit írtak le eddig a potenciális katalizátor prekurzor karbén-hidas kétmagvú kobalt-karbonilokról.
1.6. Karbén-hidas kétmagvú kobalt-karbonilok
Az elsı karbén-hidas kétmagvú kobalt-karbonil komplexet (A) oktakarbonil-dikobalt, acetilén és szén-monoxid 190 bar nyomáson és 70°C-on 12 óra alatt végrehajtott reakciójával állították elı 1959-ben [49]. Szerkezetét röntgendiffrakcióval egyértelmően igazolták [50].
Co(CO)3 (CO)3Co
C C
O C C C
O H O
H
A
Co(CO)3 (CO)3Co
C C
CF3 F
O
B
Co(CO)3 (CO)3Co
C C
CF3 F3C
O
C
Co(CO)3 (CO)3Co
C C
F F
D
F F
Az azóta eltelt idıben változatos úton különféle további karbén-hidas kétmagvú kobalt- karbonilt állítottak elı. A (CO)4CoCF2CF2Co(CO)4 komplex vákuumban való melegítésével nyerték a B [51,52], az oktakarbonil-dikobalt és bisz-trifluorometil-diazometán szobahıfokon 28 napig tartó reakciójával kapták a C [53,54], a Co(CO)3NO komplex és Br2F2C oldószerben való fotolízisével [55-57], valamint Hg[Co(CO)4]2 és ClF2CCO2Cs 110°C-on megvalósított reakciójával jutottak D-hez [58]. Az egy difluoro-karbén ligandumot tartalmazó komplexet (E) Na[Co(CO)4] és F2C(COCl)2 reakcióval állították elı [59]. Szubsztituált butenolid-karbént és szubsztituált vinilidén-karbént is tartalmazó hexakarbonil-dikobalt komplexek (F) elıállítását [60] és szerkezetét [61] Pályi és munkatársai írták le. F volt az elsı olyan hexakarbonil- dikobalt-komplex, amely két különbözı karbén ligandumot tartalmazott.
Co(CO)3 (CO)3Co
C C
O C C C
C R2 O
R1
F
Co(CO)3 (CO)3Co
C C F F
O
E
R3 R4
Tuba és Ungváry oktakarbonil-dikobalt és etil-diazoacetát reakciójával (1.17 reakció) egy, illetve két hídhelyzető etoxikarbonil-karbén ligandumot tartalmazó dikobalt-karbonil komplexeket (G, illetve H) állítottak elı [44,45].
Co2(CO)8 + EtO2CCHN2
- CO, - N2 - CO, - N2
+ EtO2CCHN2
(OC)3Co Co(CO)3 C
O
G
(OC)3Co Co(CO)3
H
(1.17)
CO2Et
CO2Et CH
CO2Et CH
CH
Mindkét komplex szén-monoxid és etanol jelenlétében dietil-malonátot eredményez (1.18 és 1.19 reakciók), amelynek képzıdését a kobalthoz kötött etoxikarbonil-karbén és CO kapcsolódásával feltételezett etoxikarbonil-ketén intermedier és etanol reakciójával magyaráz- ták.
+ 2CO + EtOH 25 °C
1 bar
+ EtO2CCH2CO2Et
Co2(CO)6(CHCO2Et)2 Co2(CO)7(CHCO2Et) (1.18)
+ 2CO + EtOH 25 °C
1 bar Co2(CO)8 + EtO2CCH2CO2Et
Co2(CO)7(CHCO2Et) (1.19)
A szerzık demonstrálták, hogy az etoxikarbonil-karbén ligandumok nem csak sztöchiometrikus reakcióban építhetık be a malonsav-etilészter származékba, hanem az elıál- lításukhoz használt oktakarbonil-dikobalt a malonsav-etilészter származékok katalitikus elıál- lításához (1.20 reakció) prekurzorként szolgálhat.
EtO2CCHN2 + CO + EtOH
1 mol% Co2(CO)8
25 °C; 50 bar EtO2CCH2CO2Et + N2 (1.20)
A feltételezett etoxikarbonil-ketén intermedier in situ átalakítására nem csak etanol, hanem egyéb OH csoportot tartalmazó vegyület, mint például metanol, terc-butanol, ciklohexanol, fenol [44,45] és mentol [47], vagy NH csoportot tartalmazó vegyület, mint pél-
EtO2CCHN2 + CO + HN O
25 °C, 50 bar, 24h 1 mol% Co2(CO)8
O N
EtO2CCH2C O
(1.21)
A ketén oktakarbonil-dikobalt katalizált képzıdését trimetilszilil-diazometán karbonilezésénél igazolták [48] (1.22 reakció). Ebben az esetben azonban nem sikerült a ketén képzıdésben feltételezhetıen intermedier szerepet játszó trimetilszilil-karbén komplex (Co2(CO)7(CHSiMe3) kimutatása.
Me3SiCHN2 + CO
10 mol% Co2(CO)8
25 °C; 1 bar Me3SiCH=C=O + N2 (1.22)
Acil-kobalt-tetrakarbonilokból kiindulva nyerték az I [62] és a J [63] komplexeket. A J komplex különlegessége az, hogy a szokásos hat karbonil ligandum közül egyet a karbén ligandumon lévı karbonil-oxigén koordinációja helyettesít. Formailag hasonló koordinációt írtak le már korábban Adams és munkatársai a K komplex esetében [64], amelyet tetrakarbonil-kobaltát-ion és difenil-imidoil-klorid reakciójával nyertek.
Co(CO)3 (CO)3Co
C C
OSiPh3 R
O
I
R = Me, MeOCH2
Co(CO)2 (CO)3Co
C C R O
O
J
R = Me, iPr, tBu
C O
R
Co(CO)2 (CO)3Co
C C R N
O
K
C N Ph R
Ph
A felsoroltakon kívül még számos olyan µ-karbén ligandumot tartalmazó dikobalt- karbonil komplexet állítottak elı, amelyekben a kobaltokhoz egy-egy π-ciklopentadienil ligandum is kapcsolódik [65-71].
1.7. Foszfán-szubsztituált karbén-hidas kétmagvú kobalt-karbo- nilok
Az elsı karbén-hidas kétmagvú kobalt-karbonil komplex (A) 1959-ben leírt [49] elıál- lítását követıen, néhány származékának elıállították a monotrifenil-foszfit- (L) és a monotributil-foszfán-szubsztituált (M) változatát [72].
L M
Co(CO)2[P(OPh)3] (CO)3Co
C C
O C C C
O
n O Pr H
Co(CO)2(PnBu3) (CO)3Co
C C
O C C C
O
nPr O H
Az A komplex 3-fenil-2-butén-4-olid-4-ilidén változatának ismeretes két különbözı foszfánnal diszubsztituált származéka (N és O) [73].
Co(CO)2(PnBu3) (nBu3P)(CO)2Co
C C
O C C C
O Ph O H
N
Co(CO)2(PRPh2) (Ph2RP)(CO)2Co
C C
O C C C
O Ph O H
O
R = izopentil
A B komplex két különbözı biszfoszfán-szubsztituált származéka (P és Q) is ismeretes [51,52].

![2.3 ábra. A kezdeti gázfejl ı dés sebessége (r 1 gáz ), valamint az 1 komplex kezdeti koncentráció csökkenésének sebessége (r 1 −[1] ) az argon atmoszférában, metilén-kloridos oldatban az 1](https://thumb-eu.123doks.com/thumbv2/9dokorg/872929.46952/43.892.205.698.703.1057/gázfejl-sebessége-koncentráció-csökkenésének-sebessége-atmoszférában-metilén-kloridos.webp)