C H A P T E R 4
THE RHEOLOGY O F O R G A N I C GLASSES Rolf Buchdahl
I . I n t r o d u c t i o n 145 I I . A n e l a s t i c i t y and R e v e r s i b l e D e f o r m a t i o n s 148
I I I . Irreversible D e f o r m a t i o n s 152
1. V i s c o u s F l o w 152 2. Stress-Strain and U l t i m a t e - S t r e n g t h P r o p e r t i e s 153
I V . M a j o r E x p e r i m e n t a l T e c h n i q u e s 155 V . Effect of P h y s i c a l Variables o n A n e l a s t i c i t y 158
1. T e m p e r a t u r e and F r e q u e n c y 158 2. Pressure, Stress, and Orientation 162 V I . Effect of C h e m i c a l Variables o n A n e l a s t i c i t y and Irreversible D e f o r m a t i o n s . 165
1. M o l e c u l a r W e i g h t and M o l e c u l a r W e i g h t D i s t r i b u t i o n 165
2. M o n o m e r U n i t s 167 3. P l a s t i c i z a t i o n , C o p o l y m e r i z a t i o n , and P o l y m e r M i x t u r e s 175
N o m e n c l a t u r e 179 I. Introduction
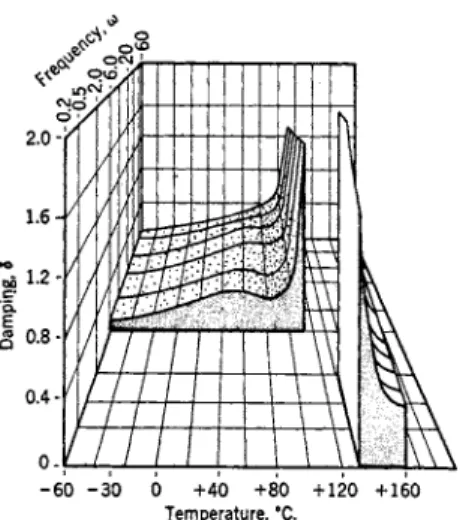
F r o m a mechano-rheological point of view, organic glasses can b e defined as a group of materials exhibiting (a) a high shear modulus G' of the order of 1 010 dynes / c m .2, (b) a considerable a m o u n t of mechanical damping δ as compared t o other materials of similar rigidity, and (c) a v e r y high resist- ance η t o purely viscous deformation. In the glassy state ( ? ' , δ, and η are slowly varying functions of the temperature. A s the material reaches the glass transformation region the mechanical properties undergo significant changes: T h e shear modulus and the (steady-state) viscosity decrease v e r y rapidly, whereas the mechanical damping increases as more and more of the energy of deformation is transformed into heat. A b o v e the glass trans- formation region the organic glasses are either viscous liquids or rubberlike materials. A rubber has a l o w shear modulus (G' 1 07 d y n e s / c m .2) and a low mechanical damping coefficient and, as a result, the d a m p i n g goes through a m a x i m u m in the glass transformation region. Viscous liquids, on the other hand, have a zero shear modulus and the mechanical d a m p i n g is v e r y large or infinite. In addition, G' and δ are also functions of the time or the frequency, whereas η is b y definition independent of time. T h e depend- ence of these quantities on frequency and temperature is shown best in a
145
146 ROLF BUCHDAHL
- 6 0 - 3 0 Ô + 4 0 + 8 0 + 1 2 0 + 1 6 0 Temperature, ° C .
FIG. 1. Damping of polymethylmethacndate as a function of temperature and frequency. [From K . Schmieder and K . Wolf, Kolloid-Z. 127, 65 (1952).]
three-dimensional diagram (see Fig. I ) .1 Under certain experimental con- ditions (?', δ, and η m a y also be stress (or strain) dependent and a c o m p l e t e phenomenological description then b e c o m e s very c o m p l e x . In the discussion to follow, stress or strain-dependent p h e n o m e n a will b e treated separately, whenever possible.
F r o m a molecular point of view, organic (like inorganic) glasses can b e defined as supercooled liquids d e v o i d of any long-range order. X - r a y measurements2 on polystyrene—a typical organic g l a s s2 a— s h o w clearly that (a) the diffraction pattern is characteristic of amorphous solids and (b) the position of the diffraction halos remains unchanged in going from the
" g l a s s y " t o the " r u b b e r y " state. There still exists, however, considerable uncertainty as t o the exact mechanism of the glass transformation and the nature of the glass s t a t e .3 ,4 T w o distinctly separate mechanisms can b e re- sponsible for the "freezing-in" of a liquid t o a glass: a relaxation mechanism or an equilibrium mechanism. On the basis of the relaxation mechanism, the glass transformation exists only because the system did not have sufficient time t o establish complete t h e r m o d y n a m i c equilibrium with its changed surroundings.5 O n the other hand, the equilibrium mechanism assumes that
1 T h e interdependence of temperature and frequency with respect t o G' and δ will b e discussed in Section V .
2 S. K r i m m and Α . V . T o b o l s k y , Textile Research J. 21, 805 (1951).
2 a This remark pertains t o polystrene made b y a free radical polymerization process and does n o t apply t o crystalline polystyrene obtained b y other initiators.
3 R . F . B o y e r and R . S. Spencer, Advances in Colloid Sei. 2 , 1 (1946).
4 W . Kauzmann, Chem. Revs. 43, 219 (1948).
6 F . Simon, Ergeh, exakt. Naturw. 9, 222 (1930).
RHEOLOGY O F ORGANIC GLASSES 147
the glass transformation arises from a structural change in the system. T h e fact that time (or frequency) has a marked effect n o t only on the tempera- ture region at which the glass transformation occurs, b u t also on the mag- nitude of the " d i s c o n t i n u i t y " has led m a n y investigators t o conclude that the glass transformation is a diffusion-controlled relaxation m e c h a n i s m6' 7 (for w o r k prior t o 1948 see ref. 4 ) . H o w e v e r , this interpretation ignores and leaves unanswered certain facts which lend support t o the equilibrium- mechanism interpretation:8 (a) F o r organic glasses it has not been possible to r e m o v e c o m p l e t e l y the discontinuities in the specific volume-tempera- ture c u r v e .9 (6) T h e relaxation mechanism is unable t o explain w h y , in a certain temperature range, the viscosity (or the relaxation times) suddenly increases and, as a result, it has been found necessary t o introduce a struc- tural c o n c e p t — a n undefined "degree of o r d e r " — t o represent the changing configurational state of the s y s t e m .10 (c) I n the study of the glass trans- formation b y x-ray diffraction2 and nuclear magnetic resonance11 changes are observed which are essentially independent of time and the correlation of the intensity of the diffraction pattern (as a function of temperature) with specific volume-temperature curves1 2—in the case of polystyrene—is of particular significance. I t appears that the c o m p l e t e elucidation of the nature of the glass transition will h a v e t o take into a c c o u n t b o t h mechan- isms mentioned a b o v e .1 2a
M a n y organic glasses undergo spontaneous crystallization in a time in- terval comparable t o or shorter than the time of the desired experiment.
H o w e v e r , the mechano-rheological properties of organic glasses have, with v e r y few exceptions, been studied on systems where spontaneous crystalli- zation is essentially nonexistent. T h e majority of materials falling in this category are substances of v e r y high molecular weight (polymers), such as polystyrene and p o l y m e t h y l m e t h a c r y l a t e ; these p o l y m e r i c organic glasses are characterized b y the fact that the intermolecular forces are relatively w e a k .13
6 E . Jenckel, Kolloid-Z. 120, 160 (1951); J. J. B e n b o w , Proc. Phys. Soc. (London) B67, 120 (1954).
7 R . O . Davies and G . O . Jones, Proc. Roy. Soc. (London) A217, 26 (1953).
8 R . Buchdahl and L . E . Nielsen, Appl. Phys. 21, 482 (1950).
9 R . S. Spencer and R . F . B o y e r , Appl. Phys. 17, 398 (1946).
1 0 J. Frenkel, " K i n e t i c T h e o r y of L i q u i d s . " Oxford Univ. Press, London, 1946.
1 1 L . V . H o l r o y d , R . S. C o d i n g t o n , B . A . M r o w c a , and E . G u t h , J. Appl. Phys. 22, 696 (1951); H . S. G u t o w s k y and L . H . M e y e r s , J. Chem. Phys. 21, 2122 (1953).
12 T . G . F o x , Jr., and P . J. F l o r y , / . Appl. Phys. 21, 581 (1950).
l 2a J. H . G i b b s , / . Chem. Phys. 25, 185 (1956).
1 3 H . M a r k and Α . V . T o b o l s k y , "Physical Chemistry of High P o l y m e r i c S y s t e m s , "
p . 146. Interscience, N e w Y o r k , 1950.
148 R O L F B U C H D A H L
II. Anelasticity a n d R e v e r s i b l e D e f o r m a t i o n s
F o r an ideal or perfectly elastic b o d y the various moduli of elasticity completely specify the mechanical behavior at a given temperature. H o w - ever, organic glasses are not ideal elastic bodies, so that a modulus of rigidity Gf—which is of particular interest from the rheological point of v i e w — d o e s n o t suffice t o describe the behavior, and it is necessary t o in- troduce the c o n c e p t of a complex modulus G defined b y the well-known expression14
G =£= G' + iG" ( 1 )
where G" is the " i m a g i n a r y " out-of-phase-part of the rigidity modulus and measures the damping, or energy dissipated into heat, within the material.
T h e description of the mechanical behavior of anelastic b o d i e s1 4 , 1 4a in terms of complex variables has the advantage that n o reference need be made as t o the mechanism of damping, or t o a particular experimental m e t h o d . Anelasticity has often been expressed in terms of a " v i s c o s i t y " or internal friction; such terms i m p l y a certain mechanism for the dissipation of energy.
On the other hand, anelasticity is frequently described b y such terms as logarithmic decrement, attenuation factor, half-width of the resonance peak, and resilience. These terms imply a certain t y p e of experimental technique. It can easily be shown that all of the a b o v e w a y s of expressing anelasticity can b e expressed formally in terms of G' and G", or the ratio G"/G'.16
I n the following, a brief derivation will be given of the terms most fre- quently used during this discussion. These terms are a matter of choice and others could have been selected. F o r details, see V o l . I I , Chapter 1.
T h e equation of m o t i o n of a vibrating system can be derived b y con- sidering the forces acting on the system. F o r a mechanical system these forces m a y be (a) inertial force, Fi = —M d2y/dt2 where M is the mass of the vibrating system, y is the amplitude of the deformation at any time, t;
(b) restoring force, F2 = —G'y™ where G' is the spring constant; (c) fric-
14 C . M . Zener, " E l a s t i c i t y and Anelasticity of M e t a l s . " U n i v . of C h i c a g o Press, C h i c a g o , 1948; J. D . F e r r y , W . M . Sawyer, and J. N . A s h w o r t h , J. Polymer Sei. 2 , 593 (1944).
1 4a J. D . F e r r y , " R h e o l o g y : T h e o r y and A p p l i c a t i o n s / ' V o l . I I , C h a p t e r 11. A c a - demic Press, N e w Y o r k , 1957.
1 5 T h e author is i n d e b t e d t o D r . L . E . Nielsen for this treatment of anelasticity in terms of a c o m p l e x modulus ; the subsequent equations are given for one c o o r d i n a t e o n l y , b u t can easily b e extended t o more c o m p l i c a t e d cases.
1 6 T h i s definition assumes the v a l i d i t y of H o o k e ' s law and is justified for small d e f o r m a t i o n s ; for an extension t o " l a r g e " deformations see reference 17. T h e spring c o n s t a n t can be represented b y various kinds of m o d u l i , and might also contain different shape factors.
RHEOLOGY OF ORGANIC GLASSES 149
tional forces leading to damping, F3 = —iG"y\ (d) external forces Ft =
— F(t). For free vibrations (F4(0 = 0) and using equation (1) the general equation of m o t i o n18 becomes
M
È
+ Gy = 0 (2)The solution is of the form
—at iwt (iw — a) t / r > \
y = y0e e = y0eK (3)
where a is the attenuation factor and w is the frequency of vibration. Sep- arating this equation into real and imaginary parts results in
G' = M(w2 - a2)
(4) (?" = 2waM
T h e attenuation of a free vibration can be expressed in terms of the logarith- mic decrement δ defined as the natural logarithm of the ratio of t w o suc- cessive maxima F i and F2 which o c c u r at a time interval Ρ equal t o the period of vibration,
δ = In (Fx/72) = aP (5)
whereΡ = -
(6)
w
Substitution of (5) and (6) into (4) gives G' and G" in terms of experiment- ally observable quantities
pin
G1 = Mv? - (7a)
4:Mw2 v
1 7 T . Alfrey, " M e c h a n i c a l B e h a v i o r of H i g h P o l y m e r s , " Interscience, N e w Y o r k , 1948.
1 8 T h e s e equations are valid whenever the frequency of deformation is sufficiently l o w t o minimize inertial effects (~d2y/dx2). W h e n the p e r i o d of v i b r a t i o n b e c o m e s short e n o u g h t o a p p r o a c h the time required for the p r o p a g a t i o n of a w a v e t h r o u g h the solid, an equation of the f o l l o w i n g f o r m must b e used, in place of the a b o v e given equation of m o t i o n :
P dt* dx2
where ρ is the density of the material and χ is the direction in w h i c h the w a v e is travel- ing. A g a i n it can b e shown t h a t this e q u a t i o n can b e s o l v e d in a general w a y for inelas- tic b o d i e s if one replaces the real m o d u l u s b y a c o m p l e x m o d u l u s . ( K o l s k y1 8a has given an excellent review of the p r o p a g a t i o n of w a v e s t h r o u g h such solids.)
1 8a H . K o l s k y , "Stress W a v e s in S o l i d s . " Oxford U n i v . Press, L o n d o n , 1953.
150 ROLF B U C H D A H L
- - ρ Γ - '
δ- ^ Μ
( 7 b)I t is o f t e n m o r e desirable t o express results in terms of the total m o d u l u s (G = s/G2) and the ratio of the imaginary t o the real part of the modulus, G"/G'm, the latter quantity is frequently referred t o as the mechanical dis- sipation factor. F r o m previous equations it is easily s h o w n t h a t
/ / 2
G = g ( 4 / + ô2) =
W + ^ (8α)
G" 4*-δ G' 4τ γ2 - δ2
(86)
For small d a m p i n g (δ < 1.0) the second term in (8a) can b e neglected and the dissipation factor is given t o a g o o d approximation b y
G' 7 Γ
iwt-t
(Sc)
For forced vibrations [F4(0 = Ft* ] G' and G"/G' are given b y the fol- lowing expressions:
G' ~ w2 M, where wr is the resonance frequency
%~ = —^-= ——ττ^1, where — — is the half w i d t h of the reso-
G' 2 \ / 3 wr v>r
nance peak.
T h e fact that it is necessary t o introduce a c o m p l e x modulus (or a c o m - plex compliance = 1/modulus) t o describe the mechanical behavior of organic glasses means, as stated a b o v e , that their properties depend on time and n o t o n l y on temperature. A s a relaxation p r o c e s s ,14 anelasticity is thus denned as a measure of the dissipation of stored energy into heat b y whatever mechanism. Anelasticity, creep, and stress relaxation are therefore manifestations of the same process (or processes) which occur during the deformation of m o s t solid bodies. A l t h o u g h this was recognized in a qualitative w a y m a n y years a g o ,1 9 , 20 it has o n l y been quite recently that the interrelationship of these quantities has been clearly formulated in the m o s t general m a n n e r .2 1 , 22 F o r example, the relationship between the
1 9 W . V o i g t , Ann. Physik 47, 671 (1892).
2 0 Ε . Wiechert, Wied. Ann. Phys. Lp. 5, 335 (1893); Ann. Physik 40, 817 (1913);
Η . Leaderman, "Elastic and Creep Properties of Filamentous Materials and Other H i g h P o l y m e r s , " T h e Textile Foundation, Washington, D . C , 1943.
2 1 B . Gross, J. Appl. Phys. 18, 212 (1947) ; 19, 257 (1948).
2 2 F . Schwarzl, Physica 17, 830, 923 (1951).
R H E O L O G Y O F O R G A N I C GLASSES 151
complex shear modulus and the "creep s p e c t r u m " φ ( τ ) is of the following f o r m .
where w = 1/t and τ is a relaxation time which is a property of the material.
E q u a t i o n s of this t y p e , h o w e v e r , cannot b e applied directly t o the experi- mental data and it has been found necessary t o introduce approximations using Laplace or Fourier transforms,23 or mechanical models consisting of springs and d a s h p o t s .17 Based on such approximations which can b e carried t o various degrees of refinement,24 the following interrelationships between measurable quantities h a v e been obtained, (a) T h e p r o d u c t of the creep φ ( ί ) and "relaxation f u n c t i o n " ψ(ί) is approximately unity,
(b) A t equal times, t = 1/w, the real part of the c o m p l e x modulus is ap- proximately identical with the relaxation curve, G' (1/t) ~ G [1 — ψ(ί)].
given b y Z e n e r .14
Intimately associated with the p h e n o m e n o n of anelasticity is the the- oretical c o n c e p t of a retardation, or a relaxation time, r . T h e quantities, n o t t o b e confused with the experimentally obtainable relaxation times U , are characteristic constants of the materials determining stress r i or strain T2 d e c a y in a material. I t was soon r e c o g n i z e d20 that single or even multiple values of τ were insufficient t o describe the behavior of anelastic bodies, and it b e c a m e necessary t o introduce t w o continuous distribution functions, F(T\) and F(T2) for stress and strain relaxation, respectively. These func- tions, or spectra as they are sometimes called, are one more w a y of specify- ing the properties of viscoelastic bodies and, until it is possible t o derive t h e m with the aid of a molecular theory, their usefulness is rather limited.
S o m e rather interesting attempts h a v e been m a d e recently t o derive re- laxation spectra for dilute p o l y m e r s o l u t i o n s2 5 , 26 b u t for solids, including organic glasses, n o satisfactory solution t o this difficult p r o b l e m is ap- p a r e n t .2 6a
2 3 B . Gross, " M a t h e m a t i c a l Structure of the T h e o r i e s of V i s c o e l a s t i c i t y . ' ' H e r - mann, Paris, 1953.
2 4 J. D . F e r r y and N . L . Williams, / . Colloid Sei. 7 , 347 (1952).
F . Schwarzl and A . J. Staverman, Physica 18, 791 (1952).
2 6 J. G . K i r k w o o d , Ree. trav. chim. 68, 649 (1949).
2 6 P . E . R o u s e , Jr., J. Chem. Phys. 21, 1272 (1953).
2 6a A t t e m p t s t o e x t e n d consideration following R o u s e t o rubber-like solids have been quite successful. [See J. D . F e r r y , R . F . Landel and M . L . Williams, J. appl.
Phys. 26, 359 (1955)].
(Γ1 [1 + φ(ί) X G[l - φ(ί)] } ~ 1.
Tr Yd
In
ψ(ί)~2
L
dIn
t Î=7T/8Ù>. Other expressions similar to (c) have been
152 ROLF B U C H D A H L
ο"
r l O - 1 0 .
50X.
^—
20X.Creep
Recovery
J
500 1000 0 500 1000 Time, min.
F I G. 2. C r e e p and r e c o v e r y of an organic glass ( p o l y s t y r e n e ) at t w o temperatures.
[From E . K l e i n and E . Jenckel, Z. Naturforsch. 7a, 800 (1952).]
In order t o obtain the functions F(n) and F(72) from the experimental data, it is necessary t o m a k e use of certain a p p r o x i m a t i o n s2 1 , 22 as indicated a b o v e , and the error introduced b y these approximations depends o n the true relaxation spectrum (which is u n k n o w n ) and can therefore b e estimated only with difficulty, using progressively higher orders of a p p r o x i m a t i o n ; in general, the approximations are m o r e reliable when the spectrum is b r o a d . Within the limits of these approximations the following equations, relating these functions t o measurable quantities, are v a l i d :
F(r)
« -
j t G'(w)tF(r) « I \tG"(w)]
Similar expression can also b e given for F (τ).
III. Irreversible D e f o r m a t i o n s 1. V i s c o u s F L O W
I t is usually assumed that a certain fraction of a creep deformation below the glass transformation temperature is due t o viscous flow. T o establish the validity of this assumption, it is necessary t o follow the creep deforma- tion b y a recovery experiment (at the same or slightly higher temperature) as shown in Fig. 2 . O n l y that fraction of the deformation which does n o t recover is of a purely viscous nature. Quite frequently it is suggested that the linear portion of a creep curve is due t o viscous flow. T h i s argument assumes, however, that the viscosity coefficient itself is independent of time and also that the linear term can readily be separated from the re- maining portion of the creep curve b y graphical or algebraic m e t h o d s .
A m o n g organic glasses the viscosity of g l u c o s e7 has been studied more ex- tensively than a n y other material. I t is found that within, and slightly b e - low, the glass transformation region the v i s c o s i t y is v e r y high ( 1 012 t o 1 014 poises) and independent of shear stress; furthermore, within a narrow
R H E O L O G Y O F O R G A N I C G L A S S E S 153
temperature range (34 t o 28° C . ) the viscosity varies exponentially with temperature and has an activation energy of a b o u t 125 k c a l . / m o l e . Similar, b u t n o t as extensive, data for other low-molecular-weight organic glasses have been summarized b y Jenckel.6 D u r i n g the last few years several in- vestigators2 7 a bc h a v e studied the long-time creep behavior of high-molecu- lar-weight organic glasses, without specifying the viscosity coefficient in a n y detail. A c t u a l l y our k n o w l e d g e of this field is extremely limited. All that is k n o w n with reasonable certainty is that organic glasses can deform b y purely viscous flow and that the coefficient of viscosity is at least 1 010 poises or higher. W h e t h e r or n o t the viscosity coefficient is a function of t i m e ; whether or n o t it is always independent of stress, and if not, w h a t the functional relationship is; w h a t the temperature, dependence of the vis- cosity is b e l o w the glass transformation and t o w h a t extent the viscosity depends on the chemical composition—all these questions h a v e y e t t o b e answered.
2. S T R E S S - S T R A I N A N D U L T I M A T E - S T R E N G T H P R O P E R T I E S
I t is clear from the foregoing that the stress-strain behavior of an inelastic b o d y at a given rate of strain must b e a function of this rate. Indeed, the p h e n o m e n o n is observed every time the stress-strain relationship of organic glasses is investigated as a function of the rate of strain and a typical ex- ample is shown in Fig. 3. T o the extent that such deformations are reversible and conform t o the B o l t z m a n n superposition principle, it should b e possible t o predict the shape of such curves from the relaxation function of the stress d e c a y at equivalent t i m e s .17 A rather satisfactory agreement can b e o b - tained between calculated and measured stress-strain d a t a of organic glasses, as shown b y K n o w l e s and D i e t z28 in the case of plasticized and un- plasticized p o l y m e t h y l m e t h a c r y l a t e . T h e extension of this kind of analysis of stress-strain d a t a t o higher frequencies (or shorter times) is of particular value in clarifying the as y e t p o o r l y understood behavior of organic glasses under impactlike deformation. T h e p r o b l e m s connected with accurate stress-strain measurements at high frequencies and amplitudes are quite formidable,29 b u t in recent years s o m e of these techniques h a v e been applied t o p o l y m e r s and organic glasses with promising r e s u l t s .3 0 , 3 0a
A s the stress (or strain) increases, one begins t o approach the region where the material will break and the interpretation of experimental data
2 7a J. A . Sauer, J. M a r i n , and C . C . H s i a o , J. Appl. Phys. 20, 507 (1949).
2 7b W . N . F i n d l e y , J. Appl. Phys. 21, 258 (1950).
2 7c F. M a r i n , Y o h - H a n P a o , and G . Cuff, Trans. ASME 73, 705 (1951).
2 8 J. K . K n o w l e s and A . G . H . D i e t z , Trans. ASME 77, 177 (1955).
2 9 G . I. T a y l o r , J. last. Civil. Engrs. 26, 486 (1946).
3 0 H . K o l s k y , Proc. Phys. Soc. (London) B62 , 676 (1949).
3 0a J. C . Smith, J. Research 57, 83 (1956).
154 ROLF BUCHDAHL
0 0.02 0.04 0.06
True strain, in./in. 0.0Ö
FIG. 3. Effect of rate of strain on the ultimate strength properties of an organic glass (polymethylmethacrylate Mw — 3.2 Χ 1 06) .
[From F. K . K n o w l e s and A . G . H . D i e t z , Trans. ASME 77, 177 (1955).]
b e c o m e s increasingly difficult. I n the first place, the superposition principle will n o longer b e applicable. S e c o n d l y , organic glasses frequently d e v e l o p small but visible crack marks, a p h e n o m e n o n k n o w n as " c r a z i n g " ;3 1 - 33 these small fracture surfaces or flaws not o n l y g r o w in size under an applied stress b u t also p r o d u c e v e r y large stress concentrations which, in turn, lead t o brittle fracture34 and l o w ultimate-strength properties.35 H o w e v e r , the same material—polystyrene, for example—which exhibits brittle fracture under tension will behave as a ductile material under c o m p r e s s i o n .36 D u c t i l -
31 B . Maxwell and L . F . R a h m , Ind. Eng. Chem. 41, 1988 (1949).
3 2 C . C . Hsiao and J. A . Sauer, J. Appl. Phys. 21, 1071 (1950) ; 24, 957 (1953).
3 3 M . A . Sherman and B . M . A x i l r o d , ASTM Bull. No. 191, 65 (1953).
3 4 P . W . B r i d g m a n [/. Appl. Phys. 18, 246 (1947)] has p o i n t e d o u t t h a t fractures are always brittle, b u t that the essential differences o c c u r prior t o actual rupturing of the material.
3 6 R . N . H a w a r d , " T h e Strength of Plastics and G l a s s . " Interscience, N e w Y o r k , 1948.
3 6 T . S. Carswell and H . K . N a s o n , " S y m p o s i u m on P l a s t i c s . ' ' A m . S o c . T e s t i n g Materials, Philadelphia, 1944; see also H . K o l s k y [Nature 166, 235 (1950)] for the d e - pendence of birefringence on strain in the glassy state.
Crosshead rate, in./in.
A Β C D
0.02 0.08 0.32 1.28
RHEOLOGY O F ORGANIC GLASSES 155
ity—and the related phenomena of yielding and "cold flow"—is generally associated with the flow of a solid under applied stress and is therefore of particular interest from a rheological point of view. For crystalline bodies clear evidence about the nature of the process of plastic deformation—
resulting from slip along certain crystal planes—has been provided b y experiments on single crystals.37 Slip takes place along a given slip plane and direction when the shear stress acting along them reaches a critical value. Because it is believed that slip spreads consecutively and not si- multaneously over a slip plane, there is usually not enough energy available t o produce a change (such as melting3 8) in a slip plane. It appears unlikely, although not altogether impossible, that this same mechanism is responsible for the ductility in amorphous glasses. Müller and J ä c k e l3 9 , 3 9a have sug- gested that "cold flow" of organic glasses can be interpreted as a successive melting or softening process and recently demonstrated experimentally a significant temperature rise in the draw zone during a stress-strain experi- m e n t .40 These experiments lend strong support t o the assumption that the ultimate-strength properties of organic glasses are intimately connected with the temperature dependence of the elastic and inelastic properties at small deformations or amplitudes. It is also interesting to note that the tem- perature dependence of the ultimate-strength properties41 show many simi- larities t o the temperature dependence of (?' and G"/G', and B o y e r42 has drawn attention t o the relationship between tensile strength and the glass transformation temperature.3 9a
IV. M a j o r Experimental Techniques
T h e experimental techniques which have been used to study the rheo- logical behavior of organic glasses are not unique t o this class of materials.
In fact, almost all of them have been employed, at some time or another, to study the mechanical behavior of metals, glasses, and other solid bodies.
T h e various methods can be subdivided into certain groups, (a) transient techniques which include stress relaxation, creep, free (damped) vibrations, and what is c o m m o n l y described as "stress-strain measurements";43 (b) forced vibration techniques which include measurements at resonance fre- quencies, direct measurements of stress and strain, wave propagation (of
37 A. H . Cottrell, "Dislocations and Plastic Flow in Crystals," Oxford Univ.
Press, London, 1953.
3 8 R . Fürth, Proc. Roy. Soc. (London) A177, 217 (1941).
3 9 F. H . Müller and K . Jäckel, Kolloid-Z. 129, 145 (1952); 137, 130 (1954).
3 9a I. J. Gruntfest, / . Polymer Set. 20, 491 (1956).
4 0 F. H . Müller and P. Brauer, Kolloid-Z. 135, 1 (1954).
4 1 H . K . Nason, T . S. Carswell, and C . H . Adams, A ST M Spec. Tech. Publ. No. 78 (1949).
42 R . F. Boyer, J. Appl. Phys. 22, 723 (1951).
4 3 A . S . T . M . Standards on Plastics, Part 6 (1952).
156 ROLF BUCHDAHL
low and high amplitudes) and transducer measurements of shear to strain ratio. A critical review of many of these methods can be found in another chapter of this b o o k44 and in ref. 18, whereas a description of the more con- ventional engineering-type techniques is given in ref. 48 and discussed by Freuden thai.45
For organic glasses the majority of the experimental work has been done using the following techniques: creep, stress relaxation, free vibration, resonance vibration, and conventional stress-strain measurements; some of the other techniques, referred to above, have been used in isolated cases.
In order t o obtain accurate creep (or recovery) data of rigid materials it is necessary to measure small deformations very precisely and, if possible, continuously; these requirements can be met either b y optical or electrical methods using strain gages, differential transformers, transducer tubes, e t c .46 T h e nonrecoverable or purely viscous deformation is usually obtained from a measurement of the rate of creep in tension and the calculation of the viscosity coefficient assumes the validity of Trouton's rule, a relation- ship which, as Davies and Jones7 point out, has never been verified experi- mentally. In stress-relaxation measurements the continuous recording of the stress decay is equally desirable and can be achieved b y various means (see, for example, Stein and T o b o l s k y ) .47 For transparent substances creep and stress-relaxation measurements in tension can readily be combined with birefrigence measurements to follow changes in configurational or bond orientation.
There exist numerous ways to start free vibrations in a specimen and to record their decay, but it appears that the method of the torsion pendulum is the preferred one; it was first applied to organic polymers b y Kuhn and Künzle48 and the design of Nielsen49 using electronic means to measure and to record the vibration has greatly extended the range and precision of this method. In the vibrating-reed method one end of the specimen is attached to a ' ' d r i v e / ' of variable frequency, and the resonance frequency and half- width of the resonance peek is measured directly or recorded photographi- cally; this method has been used b y Sack et al.,b0 Nielsen,51 Horio and
4 4 J. D . Ferry, "Rheology : Theory and Applications," Vol. I I , Chapter 3. Academic Press, New York, 1957.
4 5 A. M . Freudenthal, "The Inelastic Behavior of Engineering Material and Struc- tures." Wiley, New York, 1950.
4 6 D . Telfair, T . S. Carswell, and Η . K . Nason, Modern Plastics, February (1944);
R . Stein and H . Schaevitz, Rev. Sei. Instr. 19, 835 (1948) ; E . Baker, Rev. Set. Instr. 22, 373 (1951).
4 7 R . S. Stein and Α . V . T o b o l s k y , Textile Research J. 18, 302 (1948).
4 8 W . K u h n and O. Künzle, Helv. Chim. Acta 30, 839 (1947).
4 9 L . E . Nielsen, Rev. Set. Instr. 22, 690 (1951).
5 0 H . S. Sack, J. M o t z , H . L . Raub, and R . N . Work, J. Appl. Phys. 18, 450 (1947).
5 1 L . E . Nielsen, A ST M Bull. 165, 48 (1950).
R H E O L O G Y O F O R G A N I C G L A S S E S 157
Onogi, and Ballou and S m i t h .53 T w o m e t h o d s based on the forced oscillations have been used primarily t o study the stress dependence of the internal friction : in the m e t h o d first introduced b y K i m b a l l54 the specimen is in the form of a cylindrical rod which is being rotated b y a suitable de- v i c e ; at the free end of the rod different loads can b e applied which deflect the rod vertically. T h e magnitude of the horizontal deflection during ro- tation is a measure of the phase lag between stress and strain. I n another method, first d e v e l o p e d b y L a z a n55 and e m p l o y e d b y other investigators,56 the specimen, in the form of a solid cylinder, is rigidly attached t o t w o large masses which are supported o n frictionless steel rollers. A mechanical oscillator, driven b y a synchronous m o t o r , is attached t o one of the masses and can generate alternate cycles of tension and compression in the speci- men. T h e large masses are varied until the system is operating at reso- nance and the d a m p i n g capacity is obtained from the measured value of the displacement v e c t o r from the oscillator force. (Reference has already been m a d e t o m e t h o d s which extend the conventional stress-strain mea- surements43 t o v e r y m u c h higher rates of l o a d i n g .2 9'3 0)
W h i c h m e t h o d should b e e m p l o y e d in a study of the rheological behavior of organic glasses depends, t o a v e r y large extent, on the particular func- tional relationship one wishes t o obtain between dependent and independent variables. T h i s is particularly true for d y n a m i c measurements (forced or free vibration and w a v e p r o p o g a t i o n ) , whereas the choice is v e r y limited for creep, stress-relaxation, and viscosity measurements. If one desires t o study the anelastic properties ((?', ( ? " , or G"/Gf) primarily as a function of the frequency it is usual t o c o m b i n e several different m e t h o d s in order t o cover a sufficiently large frequency range, as was d o n e , for example, b y N o l l e57 w h o e m p l o y e d altogether five different m e t h o d s t o cover a frequency range of 1 06 decades. E v e n then such a procedure contains certain inherent diffi- culties: (a) T h e frequency dependence usually is measured in selected regions because m o s t m e t h o d s perform satisfactorily o n l y within a rather narrow frequency range (one and one-half t o t w o d e c a d e s ) . (b) T h e various methods should b e checked against each other t o eliminate systematic er- rors (this can b e c o m e an experimental p r o b l e m of considerable m a g n i t u d e ) , (c) T h e moduli obtained d o not always refer t o the same deformation process, hence their interrelation requires a knowledge of Poisson's ratio, which is frequently assumed rather than determined for the specific experi-
5 2 M . H o r i o and S. O n o g i , Appl. Phys. 22, 977 (1951).
5 3 J. W . B a l l o u and J. C . Smith, Appl. Phys. 20, 493 (1949).
5 4 A . L . K i m b a l l , J. Appl. Mechanics 8, A37 and A135 (1949).
5 5 B . J. L a z a n , Trans. ASME 65, 87 (1943).
5 6 F . M . R o b e r t s o n and A . Y o r g i a d i s , Trans. ASME 68, Am (1946); J. A . Sauer and W . F . Oliphant, Am. Soc. Testing Materials, Proc. 49, 1119 (1949).
5 7 A . W . N o l l e , J. Appl. Phys. 19, 753 (1948).
158 R O L F B U C H D A H L
mental conditions. I t is therefore n o t surprising t o find that various efforts h a v e been m a d e t o obtain the frequency dependence b y other procedures.
T h e theoretical approach is based o n the assumption of equivalence, under certain conditions, of temperature and frequency. T o the extent that this procedure is valid, which is discussed in the following section, it is possible t o study the frequency dependence over an extremely wide range (up t o 1 4 or 1 5 decades) b y making measurements at various temperatures. T h e pre- ferred experimental approach consists in studying the mechanical proper- ties over as wide a frequency as possible with a single instrument; using a m e t h o d based on the measurement of the hysteresis l o o p , Philippoff58 has been able t o cover six decades of frequencies b e l o w 1 0 c.p.s.
A l t h o u g h in principle, a n y instrument can b e put at the desired tempera- ture, certain m e t h o d s are m u c h t o b e preferred over others for various experimental reasons, such as the size of temperature bath, effect of tempera- ture on supports and measuring devices. Creep, stress relaxation, the tor- sion-pendulum, free-vibration and reed resonance instruments are particu- larly well suited t o m a k e measurements over a w i d e temperature range, because the specimen size can b e quite small and the measuring devices can b e readily separated from the specimen itself. I n creep and stress-relaxation measurements one obtains only one quantity (the modulus or c o m p l i a n c e ) , whereas the torsion pendulum and the vibrating-reed give modulus and the logarithmic decrement or loss factor. I t is o b v i o u s that creep or stress relaxation measurements can cover a v e r y large frequency range p r o v i d e d one is patient enough, b u t in actual practice the range seldom extends b e y o n d 1 03 t o 1 04 decades, from 1 c.p.s. (t = 1/w) d o w n w a r d . F o r the torsion pendulum and the vibrating-reed instrument the frequency range is m o r e restricted, between one and t w o decades extending from a b o u t 0 . 0 5 t o 3 c.p.s. and 1 0 t o 5 0 0 c.p.s., respectively. T h e vibrating-reed instrument can give more precise measurements of the modulus and the loss factor at small losses, whereas the torsion pendulum does n o t possess quite the same pre- cision b u t can cover a m u c h wider range of anelastic properties.
V . Effect o f Physical V a r i a b l e s o n Anelasticity 1. T E M P E R A T U R E A N D F R E Q U E N C Y
Before reviewing the experimental material it seems appropriate t o dis- cuss briefly the interdependence of these t w o variables in their effect on the anelastic properties. If anelasticity is essentially a relaxation p h e n o m e n o n , it is immediately obvious—as has been already pointed out in Section I—that these properties must b e b o t h time and temperature dependent. Further- more, as relaxation times generally decrease with increasing temperature
6 8 W . Philippoff, Appl. Phys. 24, 685 (1953).
R H E O L O G Y O F O R G A N I C G L A S S E S 159
one should expect a certain correlation between measurements c o n d u c t e d at increasing frequencies and decreasing temperatures, and vice versa, and indeed this has been observed m a n y times. T h e question then arises whether or n o t it is possible t o obtain a quantitative relationship which c o m b i n e s the effect of b o t h variables on the mechanical behavior. I t is o b v i o u s that a fundamentally valid relationship of this kind w o u l d be of great usefulness from a theoretical and experimental p o i n t of v i e w . A l f r e y17 was p r o b a b l y the first one t o suggest a quantitative relationship, n a m e l y , that a change in temperature from T2 t o Τι (T2 < Ti) shifts the distribution function F(\n r ) o n a logarithmic time scale b y an a m o u n t
without changing the shape of the distribution function, where Ea is an activation energy and R is the gas constant. T h i s idea has been extended and further d e v e l o p e d b y F e r r y59 and T o b o l s k y and A n d r e w s60 and has led t o the d e v e l o p m e n t of the " m e t h o d of reduced variables," w h i c h is based on the following assumptions: (a) T h e m o d u l i Gi defined b y the equation G'{w) = XG%w2n2/(l + W2T?) are proportional t o the absolute temperature T, and t o the density, p, of the material, (b) All the relaxation times, τ » , h a v e the same temperature dependence, that is, when the temperature is changed from a reference temperature, T0, t o another temperature, T, every η is multiplied b y the same factor at. (c) T h e multiplication factor, at y can b e determined independently from the temperature dependence of the steady-state viscosity, 77. Schwarzl and S t a v e r m a n ,61 investigating the conditions a material must fulfill in order t o justify the procedure of replacing any change of temperature b y a shift o n the logarithmic time scale [which is identical with assumption ( 6 ) ] , c o n c l u d e that the functions describing the temperature dependence of the various processes must b e the same, or t h a t the apparent activation energies of all molecular rate processes must b e identical. T h e fact that for a few p o l y m e r i c systems—
and particularly for p o l y i s o b u t y l e n e6 2— g o o d agreement has been obtained between at values determined from steady-state viscosity and d y n a m i c mechanical measurements lends strong support t o the assumptions under- lying the m e t h o d of reduced variables. Nevertheless, the validity and the usefulness of this procedure, when extended t o the glass state, is still rather questionable for the following reasons: (a) Extensive steady-state or non-
6 9 J. D . F e r r y , J. Am. Chem. Soc. 72, 3746 (1950).
6 0 Α . V . T o b o l s k y and R . D . A n d r e w s , J. Chem. Phys. 13, 3 (1945).
6 1 F . Schwarzl and A . J. Staverman, J. Appl. Phys. 23, 838 (1952).
6 2 E . R . Fitzgerald, L . D . G r a n d i n e , Jr., and J. D . F e r r y , Appl. Phys. 24, 650 (1953).
160 ROLF B U C H D A H L
F I G . 4. Approximate frequency and temperature dependence of E' and E"/E'.
[From A . W . N o l l e , J. Polymer Sei. 5, 1 (1950).]
recoverable viscosity data at the low temperature side of the glass transi- tion and in the glassy region itself are practically nonexistent, which makes it impossible t o check the validity of the theory and—under these circum- stances—reduces the m e t h o d t o a curve-fitting procedure. (b) It is doubtful whether the first assumption is valid for the glassy state as it is based on the kinetic theory of rubber elasticity, (c) D i r e c t63 and i n d i r e c t64 evidence has been accumulated which shows that assumption 2 is n o t always appli- cable in the glassy state : the relaxation processes d o n o t necessarily h a v e the same temperature dependence. I t w o u l d appear, therefore, t o b e m o r e desirable t o represent the dependence of such data in a three-dimensional diagram (as shown, for example, in Fig. 1) in order t o bring out clearly the detailed mechanical behavior of the glass state.
Because of the difficulties associated with measurements covering a v e r y broad frequency range at a fixed temperature, relatively few detailed in- vestigations have been carried out up t o n o w and N o l l e ' s w o r k65 on several amorphous c o p o l y m e r s (isobutylene-isoprene, butadiene-acrylonitrile, styrene-butadiene) is still one of the most comprehensive studies in this field. His data show (Fig. 4 ) that G' and G"/G' are only v e r y slightly de- pendent on frequency for sufficiently high G' values and l o w internal fric- tion; recent w o r k on un vulcanized GR-S r u b b e r66 (a c o p o l y m e r of s t y r e n e -
6 3 S. Iwayanagi and T . Hideshima, / . Phys. Soc. Japan 8, 368 (1953) ; A. B . T h o m p - son and D . W . W o o d s , Trans. Faraday Soc. 52, 1383 (1956).
6 4 R . M . Fuoss, J. A C . S. 63, 378 (1941); A . D y s o n , Polymer Sei. 7, 133 (1951).
6 5 A . W . N o l l e , J. Polymer Sei. 5, 1 (1949); see also M a x w e l l .72
6 6 L . J. Zapas, S. L . Shufler, and T . W . D e W i t t , Rubber Age 72, 763 (1953).
R H E O L O G Y O F O R G A N I C G L A S S E S 161
butadiene) confirms the essential frequency independence of Gf and G"/G' at l o w temperatures o v e r four frequency decades. I n order t o establish ex- perimentally the existence of a frequency dispersion (at constant tempera- ture) it would b e necessary t o extend a creep or stress-relaxation measure- ment t o time values which are, for all intents and purposes, impossible t o reach. Under these circumstances various investigators continue the ex- perimental investigations at a higher temperature and, using the previously referred t o procedure of curve fitting, obtain the well-known dispersion curves as a function of the reduced frequency. T h e inherent insensitivity of this m e t h o d t o detect the fine s t r u c t u r e67 of anelasticity in the glassy state can easily lead t o an oversimplification of the experimental facts as has been shown in the case of p o l y m e t h y l m e t h a c r y l a t e ; c o m b i n i n g stress-re- laxation data obtained at various temperatures, T o b o l s k y and L o u g h l i n68 obtained a reduced stress-relaxation function which is decreasing in a m o n o - tone fashion toward the dispersion region whereas several other investiga- t o r s6 3, 6 9 - 72 using different techniques h a v e s h o w n that, within the same temperature region, the behavior is m o r e c o m p l e x (see Fig. 1 ) .
T h e study of G' and G"/G' as a function of temperature, at essentially constant frequency, is easier from an experimental point of v i e w . B o t h quantities appear t o reach a constant value as the temperature decreases although there exist n o experimental data for organic glasses b e l o w — 200° C . A s the temperature is increased, the shear modulus and the internal friction will decrease and increase, respectively. T h e material under investigation m a y exhibit o n l y a single dispersion region (see Fig. 4 ) or it might possess multiple (or secondary) dispersion regions, as shown in Fig. 1. I t appears that measurements of the internal friction as a function of temperature—
preferably at a few frequencies—are best suited t o detect this fine structure of the anelastic properties whose existence has o n l y been recognized during the last few years (for example, A l f r e y ' s b o o k1 7 does n o t discuss it at all) and w h i c h seems t o b e a m u c h m o r e c o m m o n p h e n o m e n o n than had been at first anticipated. A s the material approaches the dispersion region w h i c h is due t o the transition from the glassy t o the rubbery or liquid state, the general features of the frequency and temperature dependence of G' and G"/Gf are always the s a m e : T h e shear modulus decreases b y t w o t o three decades and the internal friction goes through a m a x i m u m . H o w e v e r , the width of this dispersion region varies significantly a m o n g different high
6 7 F. Schwarzl and A . J. Staverman, Appl. Sei. Research A4, 127 (1953).
6» Α . V . T o b o l s k y and F. R . Laughlin, J. Polymer Sei. 8, 543 (1952).
™ K . Schmieder and K . Wolf, Kolloid-Z. 127, 65 (1952).
7 0 J. H e y b o e r , Kolloid-Z. 148, 36 (1956).
7 1 E . A . W . Hoff, Κ . D e u t s c h , and W . R e d d i s h , J. Polymer Sei. 13, 565 (1954);
E . A . W . Hoff, D . W . R o b i n s o n , and A . H . W i l l b o u r n , 18, 161 (1955).
7 2 Β , M a x w e l l , J. Polymer Sei. 20, 551 (1956).
1 6 2 R O L F B U C H D A H L
polymeric organic glasses. These variations appear t o b e a function of the molecular and colloidal structure of the system (see Section V I ) .
2 . P R E S S U R E , S T R E S S , A N D O R I E N T A T I O N
It is well k n o w n that pressure has a significant effect on the irreversible viscous d e f o r m a t i o n73 and it w o u l d therefore b e of interest t o investigate t o what extent this quantity affects the anelastic properties of an organic glass.
Hughes et al.u are apparently the only ones w h o h a v e studied this p r o b l e m experimentally. I t is found that, for polystyrene, the rigidity modulus in- creases b y approximately 6 % for an increase in pressure of 1 5 , 0 0 0 p.s.i.
whereas the bulk modulus increases over the same range b y a b o u t 3 0 % . Corresponding t o this rise in bulk modulus Poisson's ratio increases uni- formly. F o r p o l y m e t h y l m e t h a c r y l a t e the changes with pressure are m o r e complex.
T h e behavior of organic glasses at large stresses (or strains) has been t o u c h e d u p o n in a previous section in c o n n e c t i o n with a discussion of the ultimate-strength properties. F o r m a n y engineering applications the energy dissipation during cyclic stress application (at various stress values) is of considerable importance. A l t h o u g h there exist some questions2 7 15 whether or n o t the data which h a v e been obtained so far are completely free of sys- tematic errors, the major conclusions arrived at b y several investiga-
tors2 7 a' 7 5' 76 are as follows: (a) T h e damping capacity increases approxi-
mately with the second or third p o w e r of the applied stress. (6) T h e d a m p i n g capacity is—within the investigated range—independent of fre- q u e n c y , this being in general agreement with the fact that, at the tempera- tures where these measurements are usually made, one would h a v e t o g o t o extremely l o w frequency t o find the dispersion region, (c) T h e dependence of the damping capacity on stress and strain amplitude appears t o undergo a significant change as the fatigue strength v a l u e77 of the material is ap- proached.
Until n o w the effect of molecular orientation has not been taken into account, and it was assumed the configuration of the p o l y m e r molecules was completely r a n d o m . H o w e v e r , it is well k n o w n that chain segments of or- ganic glasses can b e aligned or orientated preferentially along a given direc-
7 3 S. Glasstone, Κ . J. Laidler, and H . E y r i n g , " T h e T h e o r y of R a t e P r o c e s s e s , "
M c G r a w - H i l l , N e w Y o r k , 1941.
7 4 D . S. Hughes, Ε . B . Blankenship, and R . Minis, J . Appl. Phys. 21, 294 (1950).
7 5 F. M . R o b e r t s o n and A . Y o r g i a d i s , Trans. ASME 68, A173 (1946); A . Y o r g i a d i s , Product Eng. 25, N o . 11, 164 (1954).
7 6 A . L . K i m b a l l and D . E . L o w e l l , Phys. Rev. 30, (1927) ; B . F . Lazan and A . Y o r g i - adis. A . S . T . M . S y m p o s i u m on Plastics p . 66 (1944).
7 7 B . J. Lazan, F . P o d n i e k s , and R . J o h n s o n , S o c i e t y of R h e o l o g y M e e t i n g , N e w Y o r k , N o v e m b e r , 1953.
R H E O L O G Y O F O R G A N I C GLASSES 163
0 400 800 1200 Hot stretch. %
F I G. 5. Effect of a m o u n t of e l o n g a t i o n , at various temperatures in the r u b b e r y state, on the birefringence orientation b e l o w the glass transformation temperature.
[ F r o m K . J. Cleereman, H . J. K a r a m , and J. L . Williams, Modern Plastics, 30, N o . 9, 119 (1953).]
tion and this partial ordering can readily b e detected b y birefringence measurements, p r o v i d e d the polarizabilities of the b a c k b o n e chain and side groups of the p o l y m e r are sufficiently different. ( F o r a detailed discussion of the relationship between orientation and birefringence, see T r e l o a r .7 8) B y stretching a p o l y m e r in the rubbery state, followed b y rapid quenching b e l o w the glass transformation temperature, it is possible t o incorporate into the material various amounts of orientation. T h e extent of orientation depends essentially on the relative magnitude of the viscoelastic t o the purely viscous responses of the p o l y m e r which, in turn, are a function of temperature, a m o u n t and rate deformation (or stretch), rate of quenching, and molecular w e i g h t ;7 9 , 80 Figure 5 shows, as a specific example, the de- pendence of birefringence orientation as a function of a m o u n t of stretch at several temperatures. A s a result of this partial ordering the anelasticity of the glass is changed: the damping and modulus b o t h increase and the transition region shifts t o lower temperatures (see Fig. 6 and T a b l e I ) . T h e shift in the glass transformation region t o lower temperatures is inter- pretated t o b e the result of a m o r e unstable configuration in the glass state which requires less thermal energy t o destroy, but the causes for the in-
7 8 L . R . G . T r e l o a r , " P h y s i c s of R u b b e r E l a s t i c i t y , " Oxford U n i v . Press, L o n d o n , 1949.
7 9 L . E . Nielsen and R . B u c h d a h l , J. Colloid Set. 5, 282 (1950).
8 0 K . J. Cleereman, H . J. K a r a m , and J. L . Williams, Modern Plastics, 30, N o . 9, 119 (1953).
164 R O L F B U C H D A H L
1 ,5 TO Q.
C TO Ο
<u
J: O.IO
ο -C
>
•a 0.05 ε Û TO
0
20 30 40 50 60 70 80 90 100 Temperature, X .
FI G . 6. Effect of orientation on damping. [From L. E . Nielsen and R . Buchdahl, J. Appl. Phys. 21, 488 (1950).]
T A B L E I
EF F E C T O F OR I E N T A T I ON O N T H E MO D U L US A N D DA M P I NG O F PO L Y S T Y R E NE I N T H E GL A S SY ST A T E8 5
Birefringence, A./mil. Dynamic Young1 s Modulus, dynes/cm.2
Half Width of Resonance Peak
0 (annealed) 3.24 X 1 010 0.046
0 (worked) 3.18 X 1 010 0.049
1580 3.46 X 1 010 0.050
2090 3.39 X 1 010 0.051
4500 4.04 X 1 010 0.054
crease in the damping at a temperature considerably below the glass trans- formation region are not as readily understood at this time.81 A s a uni- axially oriented glass strives t o reach a more stable configuration, it will contract in the direction of stretch. T h e rate of this internal stress relaxa- tion or annealing process 8 0 »8 2 , 83 is a function of time and temperature (Fig.
7) in the same w a y as the external stress relaxation is dependent on these
81 It should be pointed out that the anelasticity can be changed b y i' w o r k i n g "79
the glass above the transition region without leaving residual strains in the material as shown in Fig. 6; similar observations were made b y T o b o l s k y and McLaughlin for polymethyl methacrylate.82
8 2 J. R . McLoughlin and Α . V . T o b o l s k y , J. Polymer Sei. 7, 658 (1951).
8 3 F . H . Müller, Kolloid-Z. 95, 138, 306 (1941); Ε . F . Gurnee, L . T . Patterson, and R . D . A n d r e w s , Jr., Appl. Phys. 26, 1106 (1955).
RHEOLOGY O F ORGANIC GLASSES 165
0 . 7 0 0 . 6 0 . £ 0 . 5 0
~ 0 . 4 0
Ζο 0 . 3 0 to 2 0 . 2 0
0 . 1 0
1
) 0 Χf /
' g C-
I
ν* eC ^ 8 j ° C .- \
- 8 0= — 4 - 7 5 ° C . υϋ . I 11 1 0 1 0 0 1 0 0 0 1 0 , 0 0 0
T i m e , m i n .
Fi g . 7. Retractions of an oriented organic glass as a function of time and tempera- ture. (Temperature at which retraction is measured is indicated on graph.) [From K . J. Cleereman, H . J. K a r a m , and J. L . Williams, Modern Plastics, 30, N o . 9, 119
(1953).]
variables. Calculations of an apparent activation energy for the process b y the method of curve-shifting along the logarithmic time scale leads t o values of the order of 1 0 0 t o 2 0 0 kcal./mole, characteristic of deformation processes occurring in the transition range. Orientation has also a very marked effect on the stress-strain and ultimate-strength p r o p e r t i e s ,8 0 , 8 4*85
which is of importance in the technology of fibers and thin films. A sig- nificant fact is the development of a yield point and "cold flow" leading to materials of higher strength.
V I . Effect o f Chemical V a r i a b l e s on Anelasticity a n d N o n r e c o v e r a b l e Deformations 1. M O L E C U L A R W E I G H T A N D M O L E C U L A R - W E I G H T D I S T R I B U T I O N
Investigations of M e r z et αΖ.86 showed that Y o u n g ' s modulus and the dissipation factor of polystyrene in the glassy state are independent of molecular weight (or chain length) a b o v e a certain minimum molecular- weight value, as shown in Fig. 8 . On the basis of mechanical investigations it is impossible t o establish this minimum molecular-weight value because below a certain molecular weight ( ^ 1 3 0 , 0 0 0 for polystyrene) the specimen breaks into small fragments. However, it is possible t o obtain some in- formation on this point from dilatometric studies of the glass transition temperature and the specific volume as a function of molecular weight.
Fox and F l o r y12 showed that (a) the transition temperature for polystyrene is practically independent of molecular weight above 2 5 , 0 0 0 and (b) the molecular weight dependence of the specific volume and of the glass transi- tion temperature are closely related. T h e y interpreted the linear increase of the specific volume with 1/Mn as being the result of changes in the
8 4 F. Bailey, India Rubber World 118, 25 (1948).
8 5 L. E . Nielsen and R . Buchdahl, J. Appl. Phys. 21, 488 (1950).
8 6 Ε . H . M e r z , L. E . Nielsen, and R . Buchdahl, Ind. Eng. Chem. 43, 1396 (1951).
166 R O L F B U C H D A H L
4.0 -
Ε υ
21 - o / f r- T - » I I 1 1 1 1 1 20 30 40 50 60 70 80 90 100 110
T e m p e r a t u r e , ° C . Fi g . 86
Fi g . 8. M o d u l u s (8α) and dissipation factor (86) of p o l y s t y r e n e in the glass state as a function of molecular weight. M o l e c u l a r weight (M„) of f r a c t i o n s : Ο = 1.36 Χ 106
€ = 9.92 Χ 1 06; 3 = 7.65 Χ 1 05; W = 2.31 Χ 1 05; © = 1.46 Χ 1 0δ; X = M i x t u r e w i t h l o w molecular weight p o l y s t y r e n e : Mn — 5.12 Χ 103. [ F r o m Ε . H . M e r z , L . E . Nielsen, and R . B u c h d a h l , Ind. Eng. Chem. 43, 1953 (1951).]
average density of packing with the concentration of end groups; Alten- b u r g87 has expanded this idea along more quantitative lines and obtained reasonably g o o d agreement between the measured88 and calculated specific v o l u m e expansion coefficients for polystyrene of different molecular weight.
8 7 K . A l t e n b u r g , Ζ. physik. Chem. 201, 75 (1952).
8 8 K . Überreiter and K . Kanig, Z. Naturforsch. 6a, 551 (1951).
R H E O L O G Y O F O R G A N I C G L A S S E S 167
A p p l y i n g these considerations t o the mechanical behavior, the following conclusions can b e reached: (a) Because the dilatometric glass transition temperature and the mechanical glass transformation are different measure- ments of the same p h e n o m e n o n , the dependence on molecular weight should b e the same and experiments on mixtures (using a v e r y low-molecu- lar-weight polystyrene with a high-molecular-weight fraction (see F i g . 8 ) lend strong support t o this c o n c l u s i o n .8 8a (6) T o what extent different end groups affect the modulus and damping b e l o w the transition region has n o t been t o o clearly established, although Buchdahl and Nielsen89 h a v e inter- preted certain secondary maxima in Polyvinylchloride as being due t o the effect of specific end groups. T h e stress-relaxation function of polyisobutyl- ene fractions also is independent of molecular weight (between 300,000 and 800,000) in the glassy state and the transition region, as was shown b y A n d r e w s and T o b o l s k y .90
I t appears justified, on the basis of the results which have been obtained for polystyrene and polyisobutylene, t o generalize that the mechanical behavior of organic glasses (at small stresses or strains) is independent of molecular weight and that the apparent molecular-weight dependence, at low molecular weights, is n o t due t o the molecular weight itself, b u t is the result of the introduction of different chemical units in the form of end groups. ( T h e ultimate-strength properties are affected in the same manner b y changes in molecular w e i g h t .8 6 , 9 1) Extending these conclusions t o sys- tems containing a distribution of molecular weights (whole or unfractioned polymers) should not introduce any new effects in the mechanical behavior;
and, indeed, stress-relaxation68 and torsion-pendulum92 measurements on unfractionated polyisobutylene of different average molecular weight con- firm this c o n c l u s i o n93 (Figs. 9 and 10). I t is important t o note that in the rubbery region—long relaxation times—molecular weight has a marked effect on the rheological properties.
2 . M O N O M E R U N I T S
A discussion of the effects of the building b l o c k on the mechanical be- havior can conveniently b e separated into t w o aspects, (a) the properties b e l o w the glass transformation region (secondary dispersion regions), and (b) the effect of the m o n o m e r unit on the value of the glass transformation temperature.
8 8* T . G. F o x , Jr., and S. L o s h a e k , J. Polymer Sei. 15, 371, 391 (1955).
8 9 R . B u c h d a h l and L . E . Nielsen, J. Polymer Sei. 15, 1 (1954).
9 0 R . D . A n d r e w s and Α . V . T o b o l s k y , J. Polymer Sei. 7, 221 (1951).
9 1 A . G . H . D i e t z , P M M A R e p o r t s , M a s s . Inst, of T e c h n o l . , 1952.
9 2 K . Schmieder and K . W o l f , Kolloid-Z. 134, 149 (1954).
9 3 T h e v e r y limited facts k n o w n c o n c e r n i n g the effect of chain branching on the mechanical b e h a v i o r of organic g l a s s e s89 w o u l d indicate t h a t m o d e r a t e amounts of branching leave these properties u n c h a n g e d .
168 R O L F B U C H D A H L
ι ι ι ι ι • ι ι ι t ι I » t I I I 1 I
L o g t i m e , h o u r s
Fi g. 9. Stress-relaxation function of p o l y i s o b u t y l e n e s of different molecular weights in the glass and transition region. V i s c o s i t y average molecular w e i g h t s : 1-1.36 X 1 06; 2-2.80 X 1 06; 3-6.60 X 106. [From Α . V . T o b o l s k y and J. R . M c L o u g h l i n , J. Polymer Sei. 8, 543 (1952).]
ί ο9·
1 08-
1 07-
^1
-:
-
: -
:
1
1 2J
\ \ 1 2 3 3 1 5 0 1 0 0 5 0 0T e m p e r a t u r e . ° C . 5 0 1 0 0
4 . 0
3 . 5
2 . 5
2 . 0 a
1 . 5
1 . 0
0 . 5
Fi g. 10. Shear moduli and damping of p o l y i s o b u t y l e n e s of different molecular weights as a function of temperature. M o l e c u l a r w e i g h t s : I—50,000; II—100,000;
111—200,000. [From K . Schmieder and K . Wolf, Kolloid-Z. 134, 149 (1954).]