www.elsevier.com/locate/brainres Available online at www.sciencedirect.com
Research Report
Partial rescue of geldanamycin-induced TrkA depletion by a proteasome inhibitor in PC12 cells
Gergely Berta
a,b, Alexandra Harci
a,b, Okta´via Tarja´nyi
a,b, Mo´nika Vecsernye´s
a,b, Andra´s Balogh
a,b, Marianna Pap
a,b, Jo´zsef Szebere´nyi
a,b, Gyo¨rgy Se´ta´lo´ Jr.
a,b,naDepartment of Medical Biology, Medical School, University of Pécs, Pécs, Hungary H-7643, Pécs, Szigeti út 12., Hungary
bSignal Transduction Research Group, János Szentágothai Research Centre, Pécs, Hungary H-7624, Pécs, Ifjúság útja 20., Hungary
a r t i c l e i n f o
Article history:
Accepted 10 May 2013 Available online 20 May 2013
Keywords:
PC12 cell Geldanamycin MG-132 TrkA
Protein kinase Rat
a b s t r a c t
In this work we tried to identify mechanisms that could explain how chemical inhibition of heat-shock protein 90 reduces nerve growth factor signaling in rat pheochromocytoma PC12 cells. Geldanamycin is an antibiotic originally discovered based on its ability to bind heat-shock protein 90. This interaction can lead to the disruption of heat-shock protein 90- containing multimolecular complexes. It can also induce the inhibition or even degrada- tion of partner proteins dissociated from the 90 kDa chaperone and, eventually, can cause apoptosis, for instance, in PC12 cells. Before the onset of initial apoptotic events, however, a marked decrease in the activity of extracellular signal-regulated kinases ERK 1/2 and protein kinase B/Akt can be observed together with reduced expression of the high affinity nerve growth factor receptor, tropomyosine-related kinase, TrkA, in this cell type. The proteasome inhibitor MG-132 can effectively counteract the geldanamycin-induced reduc- tion of TrkA expression and it can render TrkA and ERK1/2 phosphorylation but not that of protein kinase B/Akt by nerve growth factor again inducible. We have found altered intracellular distribution of TrkA in geldanamycin-treated and proteasome-inhibited PC12 cells that may, at least from the viewpoint of protein localization explain why nerve growth factor remains without effect on protein kinase B/Akt. The lack of protein kinase B/Akt stimulation by nerve growth factor in turn reveals why nerve growth factor treatment cannot save PC12 cells from geldanamycin-induced programmed cell death.
0006-8993/$ - see front matter&2013 Elsevier B.V. All rights reserved.
http://dx.doi.org/10.1016/j.brainres.2013.05.015
Abbreviations: BDNF, brain-derived neurotrophic factor; Cy3, afluorophor cyanin dye; DMEM, Dulbecco's Modified Eagle's Medium;
DMSO, dimethyl-sulfoxyde; EDTA, ethylenediaminetetraacetic acid; EGTA, ethylene glycol tetraacetic acid; ErbB2, an oncogenic receptor tyrosine kinase, analogous to Her2/neu; ERK1/2, extracellular signal-regulated kinase 1 and 2; GA, geldanamycin, an inhibitor of Hsp90; Hsp90, a heatshock protein weighing 90 kDa; MAP Kinase, mitogen-activated protein kinase; MEK, mitogen- activated protein kinase/ERK kinase; MG-132, a peptidyl-aldehyde proteasome inhibitor, also known as Z-Leu-Leu-Leu-al; NGF, nerve growth factor; p62, a scaffold protein; PBS, phosphate buffered saline; PC12, rat pheochromocytoma cell line; PDGFRα, the alpha subunit of the platelet-derived growth factor receptor; PKB, protein kinase B, also known as Akt¼AKT8 virus oncogene cellular homolog; PMSF, phenylmethanesulfonylfluoride; PVDF, polyvinylidene difluoride; SDS, sodium dodecyl sulfate; TBS, Tris-buffered saline; TrkA and B, tropomyosin-related kinases A and B
nCorresponding author at: Department of Medical Biology, Medical School, University of Pécs, Pécs, Hungary H-7643, Pécs, Szigeti út 12., Hungary. Fax:+36 72 536 453.
E-mail addresses:gyorgy.setalo.jr@aok.pte.hu,setalogy@citromail.hu (G. Sétáló Jr.).
Our observations can help to better understand the mechanism of action of geldanamycin, a compound with strong human therapeutical potential.
&2013 Elsevier B.V. All rights reserved.
1. Introduction
Geldanamycin is a member of the ansamycin antibiotic family originally discovered in Streptomyces bacteria (DeBoer et al., 1970). Heat-shock protein 90 (Hsp90), an abundantly expressed, homodimeric, ATP-dependent molecular chaperone (Whitesell and Lindquist, 2005) was identified as a geldanamycin target by means of affinity chromatography (Whitesell et al., 1994).
As part of multimolecular complexes Hsp90 maintains a num- ber of proteins, including signaling kinases, in their functionally correct conformation (Pearl et al., 2008;Pratt and Toft, 2003).
Ansamycin antibiotics can efficiently compete with, and hence inhibit the ATP binding of 90 kDa chaperones (Grenert et al., 1997,1999;Jez et al., 2003;Onuoha et al., 2007;Prodromou et al., 1997;Rao et al., 2009). The resulting disruption of heat- shock protein function can cause the dissociation of Hsp90- containing complexes, or it can alter their intracellular locali- zation even compromising their stability. Binding of geldana- mycin and its derivatives to Hsp90 can shift it from a refolding chaperone to one that promotes the degradation of client proteins inside Hsp90-containing complexes (Schneider et al., 1996). Hsp90 is also capable of passive, ATP/ADP-independent chaperoning (Jakob et al., 1995;Scheibel et al., 1998;Shaknovich et al., 1992).
The PC12 rat pheochromocytoma cell line is a popular model system to study nerve cell differentiation and survival in vitro (Greene and Tischler, 1976). In serum-containing culturing medium PC12 cells do not require nerve growth factor (NGF) for survival, but upon addition of it they develop a sympathetic neuron-like phenotype. After binding to its ligand, TrkA trans-/autophosphorylation and activation (Kaplan and Miller, 2000) elicits prolonged activation of the ERK-cascade in PC12 cells, leading to differentiation (Tischler and Greene, 1975;Vaudry et al., 2002). TrkA has been shown to interact with Hsp90 in human neuroblastoma (Farina et al., 2009), and also in acute leukaemic cells (Rao et al., 2010). After the withdrawal of trophic factors the machinery of pro- grammed cell death is activated in PC12 cells, whereas NGF stimulation of its cognate receptor tropomyosin-related kinase (TrkA) and its downstream indirect effector protein kinase B/Akt can inhibit apoptosis (Chao, 2003;Greene, 1978).
Depending on the examined cell type the result of gelda- namycin treatment can be differentiation or programmed cell death. Apoptosis of PC12 cells, for example, can be induced by geldanamycin as reported byLópez-Maderuelo et al. (2001).
Interestingly, they could still observe this phenomenon when NGF was present during geldanamycin treatment.
In this work we set out to identify mechanisms that could explain how geldanamycin treatment could interfere with NGF signaling via TrkA. We investigated the expression and intra- cellular distribution of TrkA, the high affinity NGF receptor in the presence of geldanamycin. Using immunological methods we detected the disruption of the TrkA-Hsp90 association with
reduced levels and the intracellular redistribution of TrkA in geldanamycin-treated PC12 cells. We hypothesize that from its new location TrkA may no longer be able to transmit signals towards protein kinaseB/Akt and ERK1/2 with equal efficiency.
The changed overall balance of signaling could, consequently, reduce the chances of cell survival.
2. Results
2.1. Preliminary studies
Pilot experiments repeatedly confirmed that dimethyl-sulfoxy de (DMSO), the vehicle of the compounds geldanamycin and MG-132, had no effect on the examined parameters of the ex- periments at the concentrations applied (0.025 v/v % for single treatments and 0.05 v/v % when geldanamycin and MG-132 were applied in combination), (data not shown).
2.2. The effect of geldanamycin on PC12 rat pheochromocytoma cells is time and dose dependent
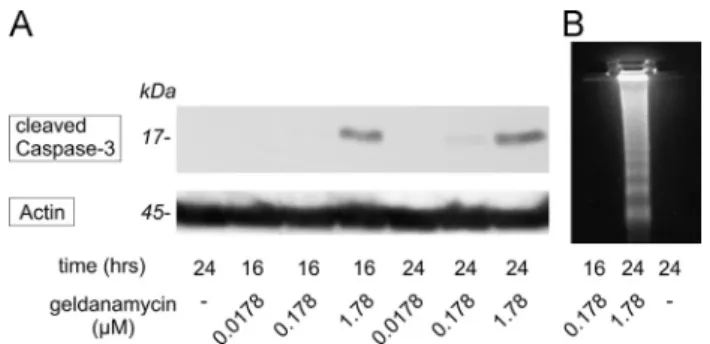
PC12 cells have responded to geldanamycin in a dose- and time-dependent manner. In keeping with recent results of Toyomura et al. (2012)if treated with the higher dose 1.78μM (1mg/ml) of the antibiotic the cells responded with apoptosis already after 16 h as shown by the activation of caspase-3 on our Western-blot (Fig. 1.A, upper panel). One tenth of this dose (0.178μM) had to be present in the culture for at least 24 h to produce initial signs of caspase-3 cleavage but proved
Fig. 1–Dose- and time-dependence of geldanamycin treatment in PC12 cells. Signs of apoptosis were detectable in the presence of 1.78μM (1μg/ml) geldanamycin after 16 h or 24 h but not with the 0.178μM dose for 16 h. (A) Western blot demonstrating the activation of effector caspase-3 (upper panel). Simultaneous probing of the membrane for actin verified equal loading of the lanes (lower panel). Shown is a representative of the series repeated four times with similar results. (B) Internucleosomal DNA fragmentation analysis of PC12 cells after geldanamycin treatments. The experiment has been performed four times. The presented photo is representative of the series producing similar results.
to be safe and was not followed by programmed cell death if used only for 16 h. These results have also been confirmed by DNA fragmentation analysis (Fig. 1B).
2.3. Geldanamycin reduces the level of TrkA
Next we examined the expression level of the TrkA NGF receptor under our above tested, non-apoptotic experimental settings with geldanamycin. The treatment unable to elicit apoptosis led to a markedly reduced expression of TrkA protein in PC12 cultures (Fig. 2, upper panel and graph, untreated control versus lane with 0,178μM geldanamycin (GA) alone for 16 h).
2.4. Inhibition of proteasome function partially restores TrkA protein in geldanamycin-treated PC12 cells
When the specific proteasome inhibitor compound MG-132 (Lee and Goldberg, 1998) was applied together with geldanamycin the
effect of the latter to reduce the amount of TrkA was substan- tially weaker (Fig. 2, upper panel and graph, lanes treated with both geldanamycin (GA) and MG-132).
2.5. Dissociation of TrkA and Hsp90 upon geldanamycin treatment
Having seen the reduced level of TrkA upon inhibiting Hsp90 by geldanamycin we decided to examine the intracellular association of the high affinity NGF receptor and the 90 kDa chaperone by means of co-immune precipitation. While TrkA and Hsp90 were found to co-precipitate in untreated PC12 cells, after geldanamycin treatment their interaction was no longer observable and it remained disrupted even if the proteasome inhibitor was added together with geldanamycin (Fig. 3, upper two panels). The marked reduction in the amount of the associated chaperone was not due to its reduced overall levels as shown by Western blot of the lysates (Fig. 3, lower panel).
2.6. Inhibition of the proteasome function can restore NGF-induced TrkA phosphorylation in geldanamycin-treated PC12 cells
In order to evaluate if the TrkA protein saved by MG-132 from degradation was still responsive to NGF we performed Western blotting with an antibody specific to the phosphory- lated form of TrkA following geldanamycin and MG-132 treat- ments. The NGF-inducibility of TrkA phosphorylation effectively blocked by geldanamycin could successfully be restored by the simultaneous inhibition of the proteasome (Fig. 4, upper panel and graph). In fact, NGF stimulation of TrkA phosphorylation appeared to be somewhat stronger in lysates derived from cells treated with both geldanamycin and MG132. This observation is in accordance with data reported bySong et al. (2009)andSong and Yoo (2011)about the effect of MG-132 on TrkA.
Fig. 2–Expression levels of TrkA (upper panel) before and after geldanamycin treatment without and with proteasome inhibition by MG-132 in PC12 cells. Simultaneous probing of the Western blot membrane for actin (lower panel) reflects the loading of the lanes. The graph demonstrates relative intensities of TrkA compared to the untreated control and normalized against the actin loading control. The values shown are mean7S.D. of four independent experiments.
n¼Significantly different from the untreated control (P¼0.0005). ¤¼Significantly different from the untreated control (P¼0.0007). #¼Significantly different from GA treatment (0.178lM) alone (P¼0.0021). §¼Significantly different from GA treatment (1.78lM) alone (P¼0.0016).
Fig. 3–The effect of geldanamycin treatment without and with proteasome inhibition by MG-132 on the association of TrkA and Hsp90 in PC12 cells. (IP, TrkA immuno- precipitation followed by Western blotting with anti-TrkA, or anti-Hsp90 to detect the co-precipitated chaperone;
W, Western blotting of the cell lysates with anti-Hsp90).
Shown is a representative of the series repeated three times with similar results.
2.7. Reduced ERK 1 and 2 phosphorylation following geldanamycin treatment
Next we examined if NGF activation of TrkA could success- fully be transmitted onto downstream members of protein kinase cascades in cells treated with both geldanamycin and MG-132. The phosphorylation of ERK 1 and 2 as an easy measure of detecting NGF's downstream effect revealed that
the lower dose and shorter duration of geldanamycin treat- ment not able to initiate apoptosis, could already weaken the level of ERK 1 and 2 phosphorylation induced by NGF (Fig. 5, upper panel and graphs, left four samples).
2.8. Inhibition of the proteasome function restores the NGF-inducible ERK1/2 phosphorylation in geldanamycin- treated PC12 cells
The simultaneous addition of the proteasome inhibitor MG-132 could restore the NGF-inducibility of ERK1/2 in geldanamycin- treated cultures (Fig. 5, upper panel and graphs, right four samples) when compared to treatments with geldanamycin and NGF only (Fig. 5, upper panel and graphs, left four lanes).
The somewhat elevated baseline phosphorylation of ERK upon inhibition of the proteasome can be attributed to the ERK phosphorylation inducing ability of MG-132 itself (Hashimoto et al., 2000). Reprobing the membrane for total ERK1/2 excluded the possibility of the observed changes in ERK1/2 phosphoryla- tion being due to loading differences of the lanes (Fig. 5, lower panel).
2.9. Phosphorylation of Akt is inhibited following geldanamycin treatment
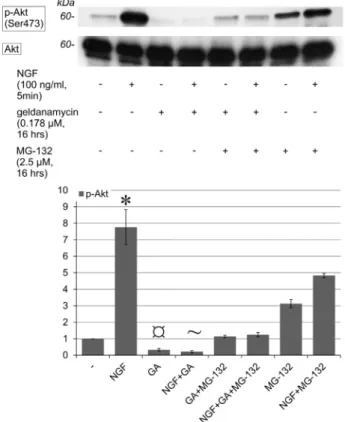
Not only the NGF activation of ERK1/2 but that of protein kinase B/Akt was also compromized in geldanamycin-treated PC12 cultures (Fig. 6, upper panel and graph, left four samples). The baseline phosphorylation of Akt was also reduced after geldanamycin treatment to barely detectable.
2.10. Inhibition of the proteasome function does not restore NGF-inducible protein kinase B/Akt phosphorylation in geldanamycin-treated PC12 cells
The proteasome inhibitor MG-132, when applied together with geldanamycin under the same conditions that could salvage the NGF activation of ERK1 and 2, could not restore activation of protein kinase B/Akt upon NGF treatment.
Baseline Akt phosphorylation, however, was elevated after pretreatment with the proteasome inhibitor as also reported earlier by Moises et al. (2009) (Fig. 6 and graph, right four samples).
2.11. Altered intracellular distribution of TrkA in geldanamycin- and MG-132-treated PC12 cells
The proteasome inhibitor MG-132, when applied together with geldanamycin, could efficiently save a substantial portion of TrkA protein as seen on Western blots (Fig. 2, upper panel and graph). The intracellular localization of salvaged TrkA, nevertheless, was dramatically different when compared to the TrkA immunoreactivity pattern of untreated PC12 cells.
In untreated cells a substantial portion of TrkA was in the immediate vicinity of the plasma membrane (Fig. 7, panel A).
The TrkA signal of cells treated with the Hsp90 inhibitor geldanamycin was barely detectable at the level of individual cells (Fig. 7, panel B). In cultures treated with both geldana- mycin and the proteasome inhibitor MG-132 the TrkA signal regained in strength but was no longer shifted towards the Fig. 4–Stimulation of TrkA phosphorylation (p-TrkA, upper
panel) by NGF before and after geldanamycin treatment without and with proteasome inhibition by MG-132 in PC12 cells. The TrkA content of the samples is shown by the middle panel. Simultaneous probing of the Western blot membrane for actin (lower panel) served as loading control.
The graph demonstrates relative intensities of
phosphorylated TrkA compared to the untreated control and normalized against the corresponding TrkA signal following the normalization of both phosphorylated and
unphosphorylated TrkA against the actin loading control.
The values shown are mean7S.D. of four independent experiments.n¼significantly different from the untreated control (P¼0.0067). #¼Significantly different from NGF treatment alone (P¼0.0073). §¼Significantly different from GA treatment alone (P¼0.0033).‡¼Significantly different from GA+MG-132-treatment (P¼0.0178).
plasma membrane, instead it was evenly distributed through- out the entire cytoplasm. (Fig. 7, panel C). Proteasome inhibi- tion alone had no effect on the intracellular pattern of TrkA immunoreactivity when compared to untreated controls (not shown). (The intracellular localization of p-TrkA, unfortunately, could not be determined, because the antibody used for Western blot was not suitable for immunocytochemical detection of the
target protein as stated by the manufacturer and also supported by our repeatedly failed trials.)
2.12. Altered intracellular distribution of Hsp90 in geldanamycin- and MG-132-treated PC12 cells
Having seen the changes in the distribution of TrkA we decided to examine the intracellular localization of Hsp90, too. In untreated cells (Fig. 7, panel D) and upon geldanamy- cin treatment (Fig. 7, panel E) the Hsp90 immune signal had an even distribution predominantly over the cytoplasm. After combined treatment with geldanamycin and MG-132, how- ever, Hsp90 was markedly shifted towards the periphery of the cytoplasm (Fig. 7, panel F). Proteasome inhibition alone had no effect on the intracellular pattern of Hsp90 immunor- eactivity when compared to untreated controls (not shown).
The overall level of Hsp90 was not affected by MG-132 and/or geldanamycin treatments as shown by Western blot (Fig. 3, lower panel).
Fig. 5–ERK1/2 phosphorylation (p-ERK1 and 2) by NGF before and after geldanamycin treatment and its restoration by proteasome inhibition with MG-132 in PC12 cells by Western blotting (upper panel). Anti-ERK1/2 was used as loading control (lower panel). The graphs demonstrate relative intensities of p-ERK1 and 2 (upper and lower diagrams, respectively) compared to the untreated control and normalized against the loading control. The values shown are mean7S.D. offive independent experiments.
n¼Significantly different from the untreated control (Po0.0001 with both p-ERK1 and pERK2). #¼Significantly different from NGF treatment alone (Po0.0001 with both p-ERK1 and pERK2). §¼Significantly different from GA treatment alone (P¼0.0184 with p-ERK1, andP¼0.0314 with p-ERK2).‡¼Significantly different from GA+MG-132- treatment (Po0.0001 with both p-ERK1 and pERK2).
Fig. 6–(A) Stimulation of Akt phosphorylation (p-Akt) by NGF before and after geldanamycin treatment without and with proteasome inhibition by MG-132 in PC12 cells (upper panel).
Anti-Akt was used as loading control of the Western blot (lower panel). The graph demonstrates relative intensities of p-Akt compared to the untreated control and normalized against the loading control. The values shown are mean7S.D. offive independent experiments.n¼significantly different from the untreated control (P¼0.038). ¤¼Significantly different from the untreated control (P¼0.0153).¼Significantly different from the untreated control (P¼0.0072).
3. Discussion
In the present article we report that the treatment of wild type PC12 rat pheochromocytoma cells with geldanamycin leads to reduced levels of TrkA protein well before the onset of pro- grammed cell death. Drug sensitivity of cell lines including PC12 cells may vary due to clonal differences, passage number and culturing conditions (Romero et al., 2010). The dose of geldana- mycin used in our experiments was just a tenth of that indu- cing apoptosis in the experiments of others (López-Maderuelo et al., 2001). Furthermore, our treatment time was also reduced by about one third. Despite of the milder treatment, the NGF stimulation of ERK1/2 and protein kinase B/Akt were both severely compromized under such conditions.
The simultaneous treatment with the proteasome inhibi- tor MG-132 helped to keep the level of TrkA higher than that seen in the cultures treated with geldanamycin alone and it also rendered the phosphorylation of the receptor upon NGF
stimulation again possible. NGF activation of TrkA in cultures treated with both geldanamycin and the proteasome inhibi- tor was in fact somewhat stronger than that seen in PC12 cells without pretreatment. The ability of the proteasome inhibitor MG-132 to potentiate the effect of NGF has been reported by Song et al. (2009) and Song and Yoo (2011).
Nevertheless, the activating effect of NGF on TrkA could only be further transmitted onto ERK1/2 but not towards protein kinase B/Akt in cultures pretreated with both geldanamycin and MG-132.
The intracellular distribution of the TrkA immune signal in untreated PC12 cells was cytoplasmic, as expected, with a substantial portion of the immunoreactivity being strongly shifted towards the periphery of the cytoplasm into the immediate vicinity of the plasma membrane. According to the Western blot results the TrkA immune signal has become much weaker in the presence of geldanamycin but has regained strength if the proteasome inhibitor MG-132 was Fig. 7–(A–C). The effect of geldanamycin treatment on the level and intracellular distribution of TrkA (red) in PC12 cells without and with proteasome inhibition by MG-132 [nuclei were counterstained with Hoechst 33342 (green), the zooming factor of the microscope was set at 4]. (A) Peripheral, membrane-associated dominance of TrkA immunoreactivity in untreated PC12 cells.
(B) Barely detectable cytoplasmic TrkA signal after geldanamycin treatment. (C) Partial restoration of TrkA throughout the cytoplasm upon inhibition of the proteasome by MG-132 in geldanamycin-treated PC12 cells.
(D–F) The effect of geldanamycin treatment on the intracellular distribution of Hsp90 (red) in PC12 cells without and with proteasome inhibition by MG-132 [nuclei were counterstained with Hoechst 33342 (green), (the zooming factor of the microscope was set at 3]. (D) Hsp90 immunoreactivity in untreated PC12 cells. (E) Unaltered Hsp90 signal after geldanamycin treatment.
(F) Peripheral accumulation of the Hsp90 immune reactivity under the plasma membrane following inhibition of the proteasome with MG-132 in geldanamycin-treated PC12 cells.
The microscopic experiments have been performed three times. The presented photos are representative of series with similar results.
also present during treatment with the Hsp90 antagonist.
Despite its regained intensity the original, membrane- associated fraction of the TrkA signal was not restored by MG-132 in geldanamycin-treated cells. This newly acquired intracellular position of TrkA raises the possibility that it might also have altered downstream signaling connections.
Interestingly, the Hsp90 immunoreactivity showed a shift into the opposite direction, towards the membrane in cells treated with both geldanamycin and MG-132. Currently we can only speculate about the significance of this latter observation. One possibility could be that MG-132, being a rather hydrophobic compound reaches preferentially the peripheral regions of the cytoplasm by diffusion. Here, it could lead to the accumulation of Hsp90's protein partners compromized by geldanamycin but saved by MG-132 from the proteasome. The accumulation of these proteins might result in a compensatory shift of Hsp90 capacity in the same direction.
The phenomenon will be worth further investigating.
Pre-organized molecular complexes are known to increase the efficiency of intracellular signal transduction markedly.
The altered intracellular localization of TrkA in cells treated with both geldanamycin and MG-132 raises the possibility that a substantial portion of TrkA may no longer be close enough to the plasma membrane to effectively stimulate all of its downstream cascades.Jullien et al. (2003)reported that gp140 TrkA is the only form of TrkA readily detectable at the PC12 cell surface. In their studies treatment by NGF produced a clearing of more than 70% of the receptor from the cell surface within 15 min of treatment. With similar results Grimes et al. (1996) have found that 66% of the receptor initially labeled at the PC12 cell surface was found inside the cells after 20 min of NGF treatment. Currently we cannot tell exactly why the TrkA receptors saved by protea- some inhibition in geldanamycin-treated PC12 cells did not return to their membrane-associated original location. There are several points at which inhibition of the proteasome could interfere with the regular and quite dynamic trafficking of TrkA. In experiments ofSommerfeld et al. (2000)the brain- derived neurotrophic factor (BDNF) receptor TrkB was down- regulated following the binding of BDNF. This could be prevented by the inhibition of the proteasome. TrkA is rapidly and transiently ubiquitinated upon addition of NGF and it has been shown that TrkA is deubiquitinated by proteasomes prior to degradation by lysosomes whereby the scaffold protein p62 acts as a shuttling protein in the internalization and transport of TrkA (Geetha et al., 2008). The kinetics of receptor trafficking post-internalization occurs as a sequel from early into late endosome, then into multivesicular bodies and proteasomes,finally degradation in the lysosomes or, alternatively, TrkA can also return to the membrane from early endosomes through recycling endosomes (Geetha and Wooten, 2008).
According to a study by Zhang et al. (2000) neuronal differentiation of PC12 cells is promoted by catalytically active Trk-s within endosomes in the cell interior. In contrast, survival responses are initiated by activated receptors at the cell surface where they orchestrate prolonged activation of protein kinase B/Akt. Thus, interaction between TrkA and intracellular signaling molecules are dictated by both phos- photyrosine residues of the activated receptors and by the
intracellular location of the latter. The activated receptor subunits with their phosphorylated tyrosines build scaffolding for the assembly of a multi-enzyme signaling complex (Lemmon and Schlessinger, 1994). The altered intracellular distribution of TrkA could explain, at least from the viewpoint of protein localization, why its activation by NGF could not lead to subsequent phosphorylation of protein kinase B/Akt under such conditions.
We believe that our observations may help to better understand the mechanisms of action of geldanamycin and MG-132, two compounds with strong human therapeutic potential.
4. Experimental procedures
4.1. ReagentsAll chemicals used were from Sigma-Aldrich (Budapest, Hungary) unless stated otherwise. NGF was purchased from Alomone Labs (Jerusalem, Israel).
4.2. Tissue culture
PC12 rat pheochromocytoma cells (kindly provided by G.M. Cooper) passage number 5 to 15) were plated onto plastic petri dishes or Thermanox (Nalgene Nunc Interna- tional, Rochester, NY, USA) coverslips (at 50% or 30% con- fluence, respectively) and maintained in DMEM comple- mented with 10% horse and 5% fetal bovine serum, for 24 h to achieve sufficient adhesion. The cultures were kept at 371C in a humidified atmosphere containing 5% CO2. Next day the media were switched to 0.5% horse serum-containing DMEM for 24 h to silence growth factor-stimulated pathways before and during treatments.
4.3. Immunoprecipitation and Western blotting
After treatment the cultures were collected in ice-cold lysis buffer (50 mM Tris-base, pH 7.4, 150 mM NaCl, 10% glycerol, 1mM EGTA, 1mM Na-orthovanadate, 5μM ZnCl2, 100 mM NaF, 10μg/ml aprotinin, 1μg/ml leupeptin, 1mM PMSF, 1% Triton X-100) and frozen at−201C. Thawed samples were homoge- nized by vortexing for 20 s. The homogenate was centrifuged at 40,000gat 41C for 30 min and the protein concentration of the supernatant was determined (Lowry's method, Detergent Compatible Protein Assay Kit, Bio-Rad, Hercules, CA, USA). For caspase-3 activity measurements cells were lysed in Chaps cell extract buffer and processed as suggested by the antibody supplier (Cell Signaling Technology, Beverly, MA, USA).
For immunoprecipitation equal amounts of the samples (0.5 mg) were brought to 0.5μg/μl by the addition of lysis buffer and immunoprecipitated with 1μg of anti-TrkA anti- body (R&D Systems, Minneapolis, MN, USA) under gentle rotation for 2 h. Next, the precipitates were collected with Protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and washed according to the manufacturer's instructions. Conditions to minimize nonspecific Hsp90
contamination of the precipitates were adjusted as described in our previously published work (Sétáló et al., 2002).
Samples prepared from equal amounts of protein (see immunoprecipitates above or for Western blotting 50μg in case of TrkA and 25μg for the rest) were mixed with Laemmli buffer (the 4 stock contained 25 ml 1 M Tris-HCl, pH 6.8, 40 ml glycerol, 8 g SDS, 10 ml 100 mM EDTA, 10 ml 100 mM EGTA and 1 ml 1% bromophenol blue brought up to 100 ml with distilled water) and denatured by boiling. Subsequently, they were loaded onto 10% (or 7.5% for TrkA and 18% for cleaved caspase-3) SDS-containing polyacrylamide gels and separated based on molecular size. The gels were electro- blotted onto PVDF membranes (Hybond-P, GE Healthcare, United Kingdom).
Immunological detection of the protein of interest was carried out by first blocking the membrane in 3% nonfat dry milk in TBS-Tween (10 mM Tris-base, 150 mM NaCl, 0.2% Tween-20, pH 8.0), followed by addition of the primary antibody [phospho-p44/42 MAP kinase to detect phospho- ERK1/2, anti-Akt and phospho-Akt (Ser 473) beta-Actin and phospho-TrkA (Tyr490), cleaved caspase-3 (Asp175), (Cell Signaling Technology), ERK-1 (C-16), or ERK-2 (C-14), Trk (C- 14), Hsp90 (Santa Cruz Biotechnology)] diluted in the blocking solution and incubated overnight. Antisera were diluted 1:1000. At this dilution rate methodical control samples prepared from antigen-deficient (nnr5) cells' lysates (for anti-TrkA and anti-phospho-TrkA), or by the omission of the primary antibodies (in all cases) produced no specific immune signal expected at the right molecular weight range in pilot experiments. Excess antibody was removed by five washes of TBS-Tween. Membranes were incubated with a horseradish-peroxidase(HRP)-conjugated goat anti-rabbit sec- ondary antibody (Pierce, Thermo Fischer Scientific, Rockford, IL, USA) diluted 1:10,000 in blocking solution. Five washes in TBS-Tween were followed by detection of the enhanced chemiluminescent signal (Immobilon Western, Millipore Cor- poration, Billerica, MA, USA). Densitometry was carried out using the ImageJ software (National Institutes of Health, USA).
4.4. Data presentation of Western blots
The presented photos are representative of series with similar results. Densitometric values are the mean7S.D. for the indi- cated number of independent experiments. Significance of differences was determined using ANOVA testing applying Bonferroni corrections for multiple samples.Pvalues˂0.05 were considered to be significant. Significant differences considered as relevant to major findings are marked in the graphs and their correspondingPvalues are indicated in thefigure legend.
4.5. DNA fragmentation analysis
Adherent andfloating/dead cells alike were scraped into their culturing media and pelleted by centrifugation at 41C in 15 ml plastic tubes. After the removal of the supernatant the pellet was lysed in 0.5 ml ice-cold lysis buffer (5mM Tris- HCl, 5 mM EDTA, 0.5% Triton-X 100, pH 7.4) for 20 min on ice.
The detergent-insoluble fraction was removed by centrifuga- tion (at 40,000g for 30 min at 41C) and the lysate was
deproteinized by two rounds of phenol and one round of chloroform extraction. DNA was precipitated by the addition of 1 ml absolute ethanol and 50μl 3 M Na-acetate to the 0.5 ml protein-free lysate and incubation at −201C overnight.
The precipitated nucleic acid was collected by centrifugation (at 100,000g for 30 min at 41C). The pellet was washed in 0.5 ml of 70% ethanol, then vacuum-dried. The dry material was solubilized in 20μl of Tris-EDTA (pH 7.4) buffer and the contaminating RNA was digested with RNase A treatment.
The remaining DNA fragments were resolved by electrophor- esis in a 1.8% agarose gel and visualized for imaging by the addition of 100-fold diluted SYBR Gold nucleic acid dye (Molecular Probes, Eugene, OR, USA).
4.6. Fluorescence microscopy
Treatments were stopped by a quick rinse in 371C PBS (1.37 mM NaCl, 0.27 mM KCl, 0.43 mM Na2HPO47H2O, 0.14 mM KH2PO4, pH 7.4) followed byfixation in 4% parafor- maldehyde in PBS, pH 7.4 for 1 h at room temperature. Excess fixative was washed out in three changes of PBS, then three changes of TBS (50 mM Tris-HCl, pH 7.4, 150 mM NaCl).
Blocking of nonspecific binding sites was carried out by incubation of the samples in 5% nonfat dry milk in TBS at 41C for 1 h under gentle rocking. The primary antibodies TrkA (R&D Systems) and Hsp90 (Santa Cruz Biotechnology) were diluted 1:500 in the blocking solution and incubated with the specimens overnight at 41C under gentle rocking.
Five washes in TBS followed. The fluorescent signal was generated by the addition of a Cy3-conjugated donkey-anti- rabbit antibody (Jackson Immuno Research, Cambridgeshire, UK) diluted in the blocking solution 1:500. At this dilution rate the methodical control samples prepared from antigen- deficient (nnr5) cells (for anti-TrkA) or by the omission of the primary antibodies (for all samples) produced no immune signal using the applied microscope settings. Nuclei were counterstained with Hoechst 33342 (Calbiochem, La Jolla, CA, USA).
4.7. Data presentation of confocal micrographs
Confocal images were generated using an Olympus FV-1000 laser scanning confocal system. Single optical sections from areas of interest were taken using a 40, long distance phase objective at zoom setting 4 for TrkA and setting 3 for Hsp90 immunefluorescence images.
Acknowledgments
This work was supported by grants from the Hungarian National Research Fund (OTKA T037528), from SROP-4.2.2/B-10/1-2010- 0029 Supporting Scientific Training of Talented Youth at the University of Pécs and from the University of Pécs (PTE ÁOK-KA- 2013/24). The purchase of the Olympus FV-1000 laser scanning confocal system was supported by grant GVOP-3.2.1-2004-04- 0172/3.0 to Pécs University. The sponsors/funding sources who providedfinancial support for the conduct of the research and preparation of the article had no involvement in study design; in the collection, analysis and interpretation of data; in the writing
of the report; and in the decision to submit the article for publication. The work described in this article has been carried out following the Uniform Requirements for manuscripts sub- mitted to Biomedical Journals.
We are indebted to Miss Anikó Kiss for her excellent technical assistance throughout the experiments.
r e f e r e n c e s
Chao, M.V., 2003. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev.
Neurosci. 4, 299–309.
DeBoer, C., Meulman, P.A., Wnuk, R.J., Peterson, D.H., 1970.
Geldanamycin, a new antibiotic. J. Antibiot. 23, 442–447.
Farina, A.R., Tacconelli, A., Cappabianca, L., Cea, G., Chioda, A., Romanelli, A., Pensato, S., Pedone, C., Gulino, A., Mackay, A.R., 2009. The neuroblastoma tumour-suppressor TrkAI and its oncogenic alternative TrkAIII splice variant exhibit geldanamycin-sensitive interactions with Hsp90 in human neuroblastoma cells. Oncogene 28, 4075–4094.
Geetha, T., Seibenhener, M.L., Chen, L., Madura, K., Wooten, M.W., 2008. p62 serves as a shuttling factor for TrkA interaction with the proteasome. Biochem. Biophys. Res. Commun. 374, 33–37.
Geetha, T., Wooten, M.W., 2008. TrkA receptor endolysosomal degradation is both ubiquitin and proteasome dependent.
Traffic 9, 1146–1156.
Greene, L.A., Tischler, A.S., 1976. Establishment of a
noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Nat. Acad.
Sci. U.S.A 73, 2424–2428.
Greene, L.A., 1978. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J. Cell Biol 78, 747–755.
Grenert, J.P., Sullivan, W.P., Fadden, P., Haystead, T.A., Clark, J., Mimnaugh, E., Krutzsch, H., Ochel, H.J., Schulte, T.W., Sausville, E., Neckers, L.M., Toft, D.O., 1997. The amino- terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J. Biol. Chem. 272, 23843–23850.
Grenert, J.P., Johnson, B.D., Toft, D.O., 1999. The importance of ATP binding and hydrolysis by hsp90 in formation and function of protein heterocomplexes. J. Biol. Chem. 274, 17525–17533.
Grimes, M.L., Zhou, J., Beattie, E.C., Yuen, E.C., Hall, D.E., Valletta, J.S., Topp, K.S., LaVail, J.H., Bunnett, N.W., Mobley, W.C., 1996.
Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J. Neurosci. 16, 7950–7964.
Hashimoto, K., Guroff, G., Katagiri, Y., 2000. Delayed and sustained activation of p42/p44 mitogen-activated protein kinase induced by proteasome inhibitors through p21(ras) in PC12 cells. J. Neurochem 74, 92–98.
Jakob, U., Lilie, H., Meyer, I., Buchner, J., 1995. Transient interaction of Hsp90 with early unfolding intermediates of citrate synthase. Implications for heat shock in vivo. J. Biol.
Chem. 270, 7288–7294.
Jez, J.M., Chen, J.C., Rastelli, G., Stroud, R.M., Santi, D.V., 2003.
Crystal structure and molecular modeling of 17-DMAG in complex with human Hsp90. Chem. Biol. 10, 361–368.
Jullien, J., Guili, V., Derrington, E.A., Darlix, J.L., Reichardt, L.F., Rudkin, B.B., 2003. Trafficking of TrkA-greenfluorescent protein chimerae during nerve growth factor-induced differentiation. J. Biol. Chem. 278, 8706–8716.
Kaplan, D.R., Miller, F.D., 2000. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 10, 381–391.
Lee, D.H., Goldberg, A.L., 1998. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403.
Lemmon, M.A., Schlessinger, J., 1994. Regulation of signal transduction and signal diversity by receptor oligomerization.
Trends Biochem. Sci. 19, 459–463.
López-Maderuelo, M.D., Fernández-Renart, M., Moratilla, C., Renart, J., 2001. Opposite effects of the Hsp90 inhibitor geldanamycin: induction of apoptosis in PC12, and differentiation in N2A cells. FEBS Lett. 490, 23–27.
Moises, T., Wüller, S., Saxena, S., Senderek, J., Weis, J., Krüttgen, A., 2009. Proteasomal inhibition alters the trafficking of the neurotrophin receptor TrkA. Biochem. Biophys. Res. Commun.
387, 360–364.
Onuoha, S.C., Mukund, S.R., Coulstock, E.T., Sengerovà, B., Shaw, J., McLaughlin, S.H., Jackson, S.E., 2007. Mechanistic studies on Hsp90 inhibition by ansamycin derivatives. J. Mol. Biol. 372, 287–297.
Pearl, L.H., Prodromou, C., Workman, P., 2008. The Hsp90 molecular chaperone: an open and shut case for treatment.
Biochem. J. 410, 439–453.
Pratt, W.B., Toft, D.O., 2003. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. 228, 111–133.
Prodromou, C., Roe, S.M., O'Brien, R., Ladbury, J.E., Piper, P.W., Pearl, L.H., 1997. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular
chaperone. Cell 90, 65–75.
Rao, R., Lee, P., Fiskus, W., Yang, Y., Joshi, R., Wang, Y., Buckley, K., Balusu, R., Chen, J., Koul, S., Joshi, A., Upadhyay, S., Tao, J., Sotomayor, E., Bhalla, K.N., 2009. Co-treatment with heat shock protein 90 inhibitor 17-dimethylaminoethylamino-17- demethoxygeldanamycin (DMAG) and vorinostat: a highly active combination against human mantle cell lymphoma (MCL) cells. Cancer Biol. Ther. 8, 1273–1280.
Rao, R., Nalluri, S., Fiskus, W., Balusu, R., Joshi, A., Mudunuru, U., Buckley, K.M., Robbins, K., Ustun, C., Reuther, G.W., Bhalla, K.N., 2010. Heat shock protein 90 inhibition depletes TrkA levels and signaling in human acute leukemia cells. Mol. Cancer Ther. 9, 2232–2242.
Romero, C., Benedí, J., Villar, A., Martín-Aragón, S., 2010.
Involvement of Hsp70, a stress protein, in the resistance of long-term culture of PC12 cells against sodium nitroprusside (SNP)-induced cell death. Arch. Toxicol 84, 699–708.
Scheibel, T., Weikl, T., Buchner, J., 1998. Two chaperone sites in Hsp90 differing in substrate specificity and ATP dependence.
Proc. Nat. Acad. Sci. U.S.A 95, 1495–1499.
Schneider, C., Sepp-Lorenzino, L., Nimmesgern, E., Ouerfelli, O., Danishefsky, S., Rosen, N., Hartl, F.U., 1996. Pharmacologic shifting of a balance between protein refolding and
degradation mediated by Hsp90. Proc. Nat. Acad. Sci. U.S.A 93, 14536–14541.
Sétáló Jr., G., Singh, M., Guan, X.P., Toran-Allerand, C.D., 2002.
Estradiol-induced phosphorylation of ERK1/2 in explants of the mouse cerebral cortex: the roles of heat shock protein 90 and MEK2. J. Neurobiol 50, 1–12.
Shaknovich, R., Shue, G., Kohtz, D.S., 1992. Conformational activation of a basic helix-loop-helix protein (MyoD1) by the C-terminal region of murine HSP90 (HSP84). Mol. Cell. Biol 12, 5059–5068.
Sommerfeld, M.T., Schweigreiter, R., Barde, Y.A., Hoppe, E., 2000.
Down-regulation of the neurotrophin receptor TrkB following ligand binding. Evidence for an involvement of the
proteasome and differential regulation of TrkA and TrkB.
J. Biol. Chem 275, 8982–8990.
Song, E.J., Hong, H.M., Yoo, Y.S., 2009. Proteasome inhibition induces neurite outgrowth through posttranslational modification of TrkA receptor. Int. J. Biochem. Cell. Biol. 41, 539–545.
Song, E.J., Yoo, Y.S., 2011. Nerve Growth Factor-induced Neurite Outgrowth is Potentiated by Stabilization of TrkA Receptors.
44; 182–186 B.M.B. Reports.
Tischler, A.S., Greene, L.A., 1975. Nerve growth factor-induced process formation by cultured rat pheochromocytoma cells.
Nature 258, 341–342.
Toyomura, K., Saito, T., Emori, S., Matsumoto, I., Kato, E., Kaneko, M., Okuma, Y., Nakamura, H., Murayama, T., 2012. Effects of Hsp90 inhibitors, geldanamycin and its analog, on ceramide
metabolism and cytotoxicity in PC12 cells. J. Toxicol. Sci. 37, 1049–1057.
Vaudry, D., Stork, P.J., Lazarovici, P., Eiden, L.E., 2002. Signaling pathways for PC12 cell differentiation: making the right connections. Science 296, 1648–1649.
Whitesell, L., Mimnaugh, E.G., De Costa, B., Myers, C.E., Neckers, L.M., 1994. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins:
essential role for stress proteins in oncogenic transformation.
Proc. Nat. Acad. Sci. U.S.A. 91, 8324–8328.
Whitesell, L., Lindquist, S.L., 2005. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772.
Zhang, Y., Moheban, D.B., Conway, B.R., Bhattacharyya, A., Segal, R.A., 2000. Cell surface Trk receptors mediate NGF- induced survival while internalized receptors regulate NGF- induced differentiation. J. Neurosci. 20, 5671–5678.