HOMOGÉNKATALITIKUS AMINOKARBONILEZÉS: ÚJ MÓDSZER KIDOLGOZÁSA N-FERROCENOIL-AMINOSAV-ÉSZTEREK,
FERROCÉNGLIOXAMIDOK ÉS DISZUBSZTITUÁLT FERROCÉNSZÁRMAZÉKOK SZINTÉZISÉRE
Készítette:
Kuik Árpád okleveles vegyész
Témavezető:
Skodáné Dr. Földes Rita Egyetemi tanár
Pannon Egyetem Kémiai és Környezettudományok Doktori Iskola
Kémia Intézet
Szerves Kémia Intézeti Tanszék Veszprém
2010
Tartalomjegyzék
Tartalomjegyzék...1
Kivonat...3
Abstract ...5
Zusammenfassung...6
Bevezető...7
1. Irodalmi összefoglaló...9
1.1. A ferrocén 9 1.2. A ferrocénkarbonsavamidok tulajdonságai, felhasználásuk 11 1.2.1. Molekula- és ionreceptorok 12 1.2.2. Bioszenzorok 14 1.3. Ferrocén-amidok előállítása 15 1.4. N-Ferrocenoil-aminosavak és –peptidek előállítása 18 1.5. Palládium-katalizált reakciók 21 1.5.1. A jód-ferrocén és a dijód-ferrocén palládium-katalizált reakciói 23 1.5.2. Karbonilezési reakciók 32 2. Az eredmények bemutatása és értékelése...39
2.1. Célkitűzés 39 2.2. Jód-ferrocén aminokarbonilezése aminosav-észterek jelenlétében 39 2.3. 1,1’-dijód-ferrocén aminokarbonilezése különböző szekunder aminok jelenlétében 45 2.4. 1’-jód-ferrocénkarboxamid és 1’-jód-ferrocénglioxamid származékok karbonilezése és egyéb kapcsolási reakciói 50 2.5. Nem szimmetrikusan helyettesített 1,n’-ferrocén-dikarboxamidok előállítása 1,1’-dijód- ferrocénből egy lépésben 52 2.6. 1,n’-ferrocéndikarboxamidok szerkezetének vizsgálata 55 2.7. 1,1’-dijód-ferrocén aminokarbonilezése aminosav-észterek jelenlétében 62 3. Kísérleti rész...65 3.1. A kísérleti munka során felhasznált anyagok minősége 65 3.2. A kísérletek kivitelezése, az egyes származékok kinyerése 66
3.2.1. Jód-ferrocén előállítása 66
3.2.2. 1,1’-dijód-ferrocén előállítása 67
3.2.3. Aminosav-metil-észterek előállítása 68
3.2.4. Ferrocenoil-aminosav-metil-észterek előállítása jód-ferrocén aminokarbonilezésével
aminosav-metil-észterek jelenlétében 68
3.2.5. Ferrocenil-glioxil-aminosav-metil-észterek előállítása jód-ferrocén
aminokarbonilezésével aminosav-metil-észterek jelenlétében 71 3.2.6. 1,1’-dijód-ferrocén aminokarbonilezése aminok jelenlétében 74 3.2.7. 1’-jód-ferrocénkarboxamid és 1’-jód-ferrocénglioxamid Stille-reakciója 83
3.2.8. 1’-benzoil-N,N-dietil-ferrocénkarboxamid (15a) előállítása 85 3.2.9. Jódferrocén aminokarbonilezése aminok jelenlétében 86 3.2.10. 1,1’-dijód-ferrocén aminokarbonilezése aminosavészterek jelenlétében 87 3.3. Műszeres analitikai vizsgálati módszerek és készülékek 92 Összefoglalás...93 Rövidítések ...95 Irodalomjegyzék...96
Kivonat
A ferrocénszármazékok jó stabilitásuknak és kedvező elektrokémiai tulajdonságaiknak köszönhetően gyakorlati szempontból nagy jelentőséggel bírnak. Felhasználhatóak homogénkatalitikus szintézisekben ligandumként, redox aktív molekula- és ionreceptorként, redoxkapcsolóként, vagy bioszenzorként [1-8]. A ferrocénkarbonsav amid származékainak jelentőségét figyelembe véve fontos lehet egy új, egyszerű, ugyanakkor hatékony módszer kidolgozása az említett vegyületcsalád szintézisére.
A munka célja N-ferrocenoil-aminosav-észterek, a ferrocénvázas vegyületek között újszerű ferrocénglioxamidok, valamint egyéb ferrocénszármazékok szintézise volt, a ferrocénszármazékok előállítása során eddigiekben nem alkalmazott palládium katalizált homogénkatalitikus karbonilezési eljárással.
A Pannon Egyetem Szerves Kémia Intézeti Tanszékén elkezdett jód-ferrocén karbonilezési kísérletek során új típusú ferrocénglioxamidokat szintetizáltak, ferrocénkarboxamid melléktermék keletkezése mellett.
A munka folytatásaként a szerző vizsgálta a jód-ferrocén palládium katalizált aminokarbonilezését aminosav-észterek mint nukleofil reakciópartnerek jelenlétében.
Megállapította, hogy a korábbi jód-ferrocén és szekunder aminok karbonilezési reakcióinál tapasztaltakkal ellentétben nem a hőmérséklettel, hanem a megfelelő bázis alkalmazásával lehet a reakciók szelektivitását eredményesen befolyásolni. A vizsgálatok eredményeiből meghatározta a ferrocénglioxamidok, illetve ferrocénkarboxamidok előállításához optimális reakciókörülményeket. Bizonyította, hogy karbonilezési körülmények között a bázisként alkalmazott DBU is acilezhető.
Az 1,1’-dijód-ferrocén aminokarbonilezésének vizsgálata rámutatott, hogy a megfelelő reakciókörülmények megválasztásával 1’-jód-ferrocénkarboxamidok és –glioxamidok közepes hozammal szintetizálhatóak, melyek további homogénkatalitikus reakciókba vihetők, változatos szerkezetű heterodiszubsztituált ferrocénszármazékokat eredményezve.
A részletes kutatómunka eredményeként egyszerű és hatékony eljárás született két különböző amidocsoportot tartalmazó 1,1’-ferrocéndikarboxamidok, illetve 1,n’-ferrocenoil-
aminosav-észtereket előállítására, melyekre rendkívül kevés példát találunk az irodalomban.
Abstract
Homogeneous catalytic aminocarbonylation: development of a new method to synthesize ferrocenoyl amino acid esters, ferroceneglioxamides and
disubstituted ferrocene derivaives
Since the discovery of ferrocene there is an increasing interest in the synthesis of its substituted derivatives that can be used in diverse fields of chemistry. Ferrocene-based systems are excellent ligands in asymmetric catalysis, but they can also be used as redox switches, molecular and ion receptors or as biosensors. The pharmacological aspects of the research concerning these compounds are also remarkable.
The main goal of this work was the synthesis of ferrocenoyl amino acid esters novel ferrocene glyoxamides and other ferrocene derivatives by homogeneous catalytic aminocarbonylation. A detailed investigation of the effects of the reaction conditions has been carried out in order to find the optimal conditions leading to the ferrocene products in good yield and with high selectivity.
In the experiments, iodoferrocene and 1,1’-diiodoferrocene were used as substrates. The nucleophilic reaction partners were primary and secondary amines and amino acid methyl esters.
Numerous new ferrocene compounds have been prepared which may have practical importance and may serve as building blocks for new electrochemical or biochemical sensors.
Zusammenfassung
Homogenkatalytische Aminocarbonylierung: Entwicklung von neuen Methoden zur Synthese von Ferrocenoyl-Aminosäureester, Ferrocen- glyoxamide und zweifach substituirten Ferrocen-Derivaten
Seit der Entdeckung von Ferrocen gibt es ansteigende Interesse für die Synthese von ihren Derivaten, die in verschiedenen Einsätze verwendet werden können. Die Ferrocen-gegründete Systeme sind ausgezeichnete Liganden für asymmetrische Katalyse und sie können auch als redox Schalter, Rezeptoren für Moleküle and Ionen oder als Biosensoren angewendet werden. Die pharmakologische Verwendungen von diesen Verbindungen sind auch beachtenswert.
Das wichtigste Ziel dieser Arbeit war die Synthese von Ferrocenoyl-Aminosäureesters, von neuartigen Ferrocenyl-Glyoxamide und von anderen Ferrocen-Derivaten durch homogenkatalytische Carbonylierung. Der Einfluss der Reaktionsbedingungen wurde erforscht, um die Zielprodukte mit gutem Umsatz und hoche Selektivität darstellen zu können. Jodo- Ferrocen and 1,1’-Dijodo-Ferrocen wurden in den Versuchen als Substrate eingesetzt und primäre und sekundäre Amine und Aminosäure-Methyl-Ester wurden als nukleophile Reaktionspartner gewählt .
Zahlreiche neue Ferrocene Verbindungen wurden hergestellt, die über praktische Bedeutung verfügen und auch als Bausteine für neuartige elektrochemische oder biochemische Sensoren geeignet sein können.
Bevezető
A ferrocén felfedezésével [9], szerkezetének tisztázásával [10, 11] új korszak nyílt meg a fémorganikus kémia területén. Számos eljárást dolgoztak ki a legkülönbözőbb ferrocénszármazékok szintézisére, melyek gyakorlati szempontból rendkívül hasznos tulajdonságokkal rendelkeznek. Használják őket homogénkatalitikus szintézisekben ligandumként, redox aktív molekula- és ionreceptorként, redoxkapcsolóként, vagy bioszenzorként, de farmakológiai jelentősége - a ferroqin maláriaellenes hatása - is jól ismert [1- 8, 12].
Az említett vegyületek közül az N-ferrocenoil-aminosavszármazékok biokémiai és farmakológiai szempontból is igen nagy jelentőséggel bírnak. Jól alkalmazhatóak például élő szervezetekben lejátszódó elektrontranszfer folyamatok vizsgálatára, vagy különböző molekulákra érzékeny bioszenzorként.
A karbonilezési reakciók tárgykörén belül az aminok jelenlétében lejátszódó aminokarbonilezési reakciókra vonatkozóan igen részletes irodalmi anyag áll rendelkezésre.
A kiterjedt kutatások ellenére ferrocénszármazékok ily módon történő előállítását egyedül kutatócsoportunk vizsgálta.
Az irodalmi adatok azt mutatják, hogy az aminokarbonilezés eredménye rendkívül érzékeny a reakciókörülmények változásaira, legyen az akár a kiindulási anyagkánt használt szerves molekula, az alkalmazott bázis vagy a nukleofil reagensként használt amin, de a reakció hőmérséklete, a szén-monoxidnyomás, oldószer, katalizátor mind jelentősen befolyásolják a karbonilezés kimenetelét.
A ferrocénkarbonsav amidszármazékai gyakorlati jelentőségének okán fontos lehet egy, az eddigieknél hatékonyabb és ugyanakkor egyszerűbb eljárás kidolgozása. Erre kínál lehetőséget az előbb említett, különböző nukleofil reagensek jelenlétében végrehajtott aminokarbonilezési eljárás.
Kísérleteim kiindulási vegyületeként jód-ferrocént, illetve 1,1’-dijód-ferrocént, nukleofil reakciópartnerként primer és szekunder aminokat, illetve aminosav-metil-észtereket használtam.
A vizsgálataim főként arra irányultak, hogy új, hatékony eljárást dolgozzak ki
ferrocénkarboxamidok és ferrocénglioxamidok előállítására. Az előállított új vegyületek elkülönítésén és karakterizálásán túlmenően egy esetben a vegyület mélyreható szerkezetvizsgálatát is elvégeztem. Továbbá egyéb homogénkatalitikus eljárások kombinációjával új ferrocénszármazékok szintézisének lehetőségét is vizsgáltam.
1. Irodalmi összefoglaló
Ebben a fejezetben szeretném összefoglalni munkám előzményeit, irodalmi hátterét.
Igyekszem bemutatni a dolgozat alapját képező ferrocénszármazékok tulajdonságait, felhasználási lehetőségeit, lehetséges előállítási módjait. Röviden áttekintem az általam használt jód-ferrocén és 1,1’-dijód-ferrocén palládium-katalizált homogénkatalitikus reakcióit.
Részletesebben tárgyalom az aminokarbonilezési reakciók általános jellemzőit.
1.1. A ferrocén
A ferrocén felfedezése, szerkezetének megismerése, karakterizálása az 1950-es évek elején a fémorganikus kémia robbanásszerű fejlődéséhez vezetett, és a vegyületet a mai napig töretlen tudományos érdeklődés övezi.
Először Kaely és Pauson számolt be egy új típusú vas-organikus vegyületről a „Nature”
hasábjain 1951-ben [9]. Ciklopentadienil-magnézium-bromidot reagáltattak vas(III)-kloriddal, azzal a céllal, hogy fulvalént állítsanak elő (1.1 egyenlet). Ehelyett egy meglepően stabil, narancssárga színű port kaptak. A stabilitást a ciklopentadienil-anion aromás jellegének tudták be.
FeCl3 + 3CpMgBr FeCp2 +1/2C10H10 + 3MgBrCl (1.1) Tőlük függetlenül Miller és munkatársai szintén „diciklopentadienil-vas”-at szintetizáltak (1.2 egyenlet).
FeCl3 + 2C5H6 500°C FeCp2 + H2
(1.2) A bisz(η5-ciklopentadienil)vas valódi szerkezetét ők még nem ismerték fel. Robert Burns Woodward és Goeffrey Wilkinson következtetett először a szendvics szerkezetre [10]. Tőlük függetlenül Ernst Otto Fischer szintén erre a megállapításra jutott [11]. Feltételezésük helyességét NMR és röntgen adatokkal igazolták.
A ligandum-fém kötés a ferrocénben többféleképp közelíthető meg. Az egyik lehetőség az, hogy a molekulát úgy tekintjük mint Fe(II)-nek két ciklopentadienidionnal (C5H5-
) alkotott komplexét. Egy másik megközelítés szerint két semleges C5H5-gyűrű koordinálódik a Fe(0)-hoz.
A helyzet ennél jóval bonyolultabb és különféle fém-ligandum kölcsönhatásokon alapul. A fém-
ligandum kötés a központi vasatom és a ciklopentadienilgyűrűk környezetében lokalizált, megfelelő szimmetriájú és energiájú pályák kölcsönhatásával írható le. A C5H5-csoport molekulapályái öt C(2p)-pálya kombinációjával származtathatók. A 6 -elektron a --és
3-pályán helyezkedik el, a 4- 5-pálya üres. A két Cp-csoportból és a vasatomból felépített ferrocén molekulában a -pályák szimmetrikus és antiszimmetrikus kombinációjával rendre a fématom dz2
- és pz-pályái lépnek kölcsönhatásba. Hasonlóképpen a --és 3-pályák kombinációi a dxz, dyz, px és py atomi pályákkal való kölcsönhatás révén stabilizálódnak. A kölcsönhatás részletei összetettek, de végeredményben a ligandum-fém kötés L → M koordinációs és M → L viszontkoordinációs komponensre bontható.
Kémiai tulajdonságait tekintve a ferrocén — aromás jellegénél fogva — számos aromás szénhidrogénekre jellemző reakcióba vihető. A ferrocén reakciókészségét alapvetően meghatározza az a tény, hogy a ciklopentadienil-gyűrű parciális negatív töltést hordoz, ezért a benzolszármazékokhoz képest elektrofil reagensekkel szemben nagyobb reakciókészséget mutat.
Az elektrofil szubsztitúciós reakcióban az elektrofil reakciópartner nem lehet oxidálószer, mert a Fe(II) → Fe(III) oxidációban keletkező ferricíniumion ( [Fe(Cp)2]+ ) elektrofilekkel nem támadható.
A Cp gyűrű nagy reakciókészségét mutatja, hogy Friedel-Crafts-acilezés során minden esetben termékelegyet kapunk. Például az alábbi reakcióban a monoacetil-ferrocén mellett megfigyelhető az 1,1’-diacetil-ferrocén keletkezése is (1.3 egyenlet) [13].
Fe
CH3 O
Fe
CH3 O
CH3
O CH3COCl +
AlCl3 Fe
(1.3) A központi vas szerepe a ferrocén elektrofil szubsztitúciós reakcióiban nem teljesen tisztázott. Az elektrofil ágens támadása révén elvben exo (A) vagy endo (B) típusú kationos ciklopentadién intermedier komplex alakulhat ki (1.4 egyenlet). Erős elektrofilek támadásakor, mint amilyen a fenti példában van, a ciklopentadienil gyűrű direkt megtámadásával az exo típusú intermedier képződik. Ha az intermedier keletkezése a központi vas megtámadásával kezdődik és
a szubsztituens innen vándorol a ciklopentadienil gyűrűre, akkor az endo komplexhez jutunk.
Ilyen intermedierek kialakulását a H+-nál gyengébb elektrofilek esetén figyelték meg [14].
Fe E
Fe
Fe E+
H
E
+ Fe H
E
+
+
Fe H + E
-H+
Fe E E=RCOCl/AlCl3, H+...
exo
endo E=Hg(II), Tl(III)...
A
B
(1.4) A fenti reakció mechanizmusának értelmezése ennél valamivel bonyolultabb, de annyi bizonyos, hogy az elektrofil reaktivitása mellett fontos szerep jut a ferrocénszármazékban lévő központi vas bázicitásának is.
1.2. A ferrocénkarbonsavamidok tulajdonságai, felhasználásuk
A ferrocénkarbonsav amidszármazékai széles körben alkalmazott építőkövek a szupramolekuláris kémiában. Használják őket redox aktív molekula- és ionreceptorként, redox kapcsolóként, vagy bioszenzorként [1-8]. Mindez a ferrocénvegyületek különleges szerkezeti és elektrokémiai tulajdonságainak köszönhető.
A ferrocénnek jól karakterizálható egy elektronos oxidációja, valamint mind a ferrocén, mind pedig a ferricínium kation stabilitása teszi a ferrocén származékait különösen alkalmassá arra, hogy elektrokémiai szondaként számos területen alkalmazhatóak legyenek. A ferrocénszármazékok redox sajátságát a szubsztituensek erősen befolyásolják. Ha ezekhez valamilyen vendég (guest) molekula, vagy ion kapcsolódik, az változást idéz elő a ferrocén egység redox viselkedésében, ami például ciklikus-voltammetria segítségével nyomon követhető.
A dolgozat korlátait figyelembe véve csak néhány konkrét példát említek a számos felhasználási lehetőség közül.
1.2.1. Molekula- és ionreceptorok
A redox érzékeny receptorok olyan molekulák, amelyek képesek egy vendég (guest) molekula vagy ion szelektív megkötésére, amely azután nemkötő kölcsönhatásokon, és/vagy különböző kötéseken keresztül hatást fejt ki a redox központra. A hatás elektrokémiai módszerekkel (általában ciklikus voltammetriával) mérhető.
Az amidszármazékok között találhatunk kationra, anionra, vagy neutrális molekulákra érzékeny receptorokat is.
Az első csoportra kiváló példát szolgáltatnak a ferrocéndikarbonsav N,N-dialkil-amid származékai, melyek az alkáli-fémionok közül szelektíven csak a lítiumiont kötik meg az 1.5 egyenletben feltüntetett komplexek képződése révén. A komplexek kialakulását különböző analitikai módszerekkel (13C-NMR, CV, UV-Vis) igazolták [1].
Fe
NEt2 O NEt2
O
Fe
NEt2
O NEt2
O Li
Li Fe
Et2N O
Et2N O
2 Fe
NEt2 O NEt2
O Li
2 Li
(1.5) LiBF4 fokozatos adagolása az N,N-dietil-ferrocénkarboxamid oldatához a karbonil szénatom eltolódásának folyamatos növekedéséhez vezetett a 13C-NMR spektrumban, mivel a karbonil-oxigénhez koordinálódó Li+ növeli az oxigénatom elektronszívó hatását. A ciklikus- voltammogramon a LiBF4 adagolása a csúcspotenciál pozitív irányú eltolódását, illetve a csúcsáramok intenzitásának csökkenését, valamint nagyobb Li+ felesleg (>1ekv.) — mintegy 300mV-al nagyobb potenciálértéknél — egy újabb, a ferrocén-dikarboxamid/lítium=1/1 komplexhez tartozó potenciálcsúcs megjelenését eredményezte. Ugyanakkor Na+ és K+ ionok hozzáadása nem okozott detektálható változást a fenti vizsgálatok során.
Az 1.6 egyenletben látható amidot kloridionokkal reagáltatták. Az anion hasítja a receptor molekula intramolekuláris hidrogénkötését, és egyidejűleg koordinálódik mindkét amidocsoporthoz hidrogénkötésen keresztül. A kloridion koordinációja megváltoztatja a két ciklopentadienil gyűrű elfordulásának szögét, valamint a ferrocén/ferricínium redox pár redoxpotenciálját, így a kloridkoncentráció mérhető [2,3].
Fe N
O N H
CH3
O
H Fc
Fe N
O N
H CH3
O
H Fc
Cl Fe
N H CH3 O
O N
Fc H
Cl
(1.6) Az Astruc és munkatársai által kifejlesztett, amidoferrocenil-alkiltiolát ligandummal módosított arany elektród (1.1 ábra) szelektíven ismeri fel a dihidrogén-foszfát aniont. A H2PO4-
anion — hidrogénkötés-akceptor negatív töltésű oxigénje, illetve hidrogénkötés-donor savas OH csoportja miatt — két hidrogénkötésen keresztül is képes koordinálódni az ugyancsak kettős tulajdonságú amidocsoporttal rendelkező receptor molekulához (1.1 ábra). Más szervetlen anionokkal végzett vizsgálatok (pl. HSO4-, NO3-, Cl-, és Br-) nem okoztak jelentős változást a ciklikus voltammogramon [4].
S S S S
Fe
N O
H
Fe
N O
H
Fe
N O
H
Fe
N O
H
Fe
O N R
H
P O O
O H OH
1.1 ábra Amidoferrocenil-alkiltioláttal módosított arany elektród és a H2PO4- anionnal kialakított hidrogénkötések
Ferrocéntartalmú amidopiridil receptorok 1,1’- és 1,3- regioizomerjei heterociklusos molekulákkal képeznek komplexeket komplementer hidrogénhidakon keresztül (1.2 ábra). A redoxaktív ferrocenilcsoport jelenlétének köszönhetően a komplexképződés folyamata elektrokémiai módszerekkel szintén nyomon követhető [5,6].
O
N
N
N O
N
N
N
Et E t
O H H O
H H
Fe
N N
O
O O
H H
N N
O N
N
R
R O
H H
HN H N O
1.2 ábra Ferrocéntartalmú amidopiridil receptorok heterociklusos vegyületekkel alkotott komplexei
1.2.2. Bioszenzorok
A bioszenzorok olyan mikró kivitelű elektrokémiai, optikai, vagy egyéb érzékelők, melyek felületére biológiailag aktív anyagot kötnek. A detektált jel függvénykapcsolatban van egy mérendő anyag mennyiségével.
A géndiagnosztika egyik kulcskérdése a DNS molekula érzékeny és egyszerű detektálásának lehetősége. A legáltalánosabban alkalmazott vizsgálati módszer, hogy egy próba DNS-t, mely a célpont DNS komplementer szekvenciájával rendelkezik, radioaktív izotóppal, például 32P-al jelölnek meg és a két DNS molekula között kialakult kapcsolat miatt detektálhatóvá válik a keresett DNS szakasz. Habár a radioizotópos nyomjelzés a jó detektálhatóság miatt széles körben elterjedt, veszélyessége, illetve a jelző izotóp rövid élettartama miatt más alternatív megoldások válhatnak jelentőssé. Az egyik lehetőség az elektrokémiai módszerrel történő detektálás. Habár a DNS közönséges körülmények között redox-inaktív, különböző módszerekkel, például ferrocénkarbonsav származékok hozzákapcsolásával aktívvá tehető (1.3 ábra). Az így kapott próba DNS által keresett DNS nemcsak hogy elektrokémiai úton jól detektálható, de mennyiségileg is meghatározható[7].
Fe O
N H
O P O
O O
NNNN...NNN
1.3 ábra Ferrocén tartalmú próba DNS
Heller és Degani glükóz-oxidáz enzimek aminosav oldalláncaihoz kapcsoltak ferrocén egységeket peptid kötéssel (1.4 ábra). Ezáltal közvetlen elektronikus kapcsolat jöhet létre egy elektród és az enzim FAD/FADH központja között, ami pusztán az enzim és az elektród között nem tud kialakulni. Azáltal, hogy a ferrocén/ferricínium pár képes elektronokat közvetíteni az enzim és az elektród között, lehetőség nyílik az 1.4 ábrán látható enzimkatalizált reakcióban keletkező redukált állapotú FADH direkt reoxidálására. Ha az elektródra feszültséget kapcsolunk elektronvándorlás indul meg, a mérhető áram erőssége (I) függvénykapcsolatban van a glükóz koncentrációval [8].
D-glükóz
D-glükonsav- lakton
FAD
FADH2
2 Fc ferrocén + 2 H+
2 Fc+
ferrocínium ion 2 e- glükóz-oxidáz
Fe O
NH Lys-(glükóz-oxidáz)
1.4 ábra Ferrocénnel módosított glükóz oxidáz enzim működése
1.3. Ferrocén-amidok előállítása
A ferrocén felfedezését, karakterizálását követően számos kutatócsoport irányította figyelmét ferrocénszármazékok, köztük a ferrocénkarboxamidok szintézisére. Az 1950-es évek második felére már több módszer is létezett az amidok előállítására.
Weliky és Gould jó néhány, különböző ferrocénszármazékokhoz vezető reakció utat dolgozott ki benzoil-ferrocénen keresztül. A benzoil-ferrocén oximjának Beckman- átendeződésével jutottak el mindössze 8 %-os hozammal a ferrocénkarboxanilidhez is (1.7 egyenlet) [15].
Fe PhCCl O AlCl3
Fe
P h O
NH2OH
Fe
P h N
TosCl Fe
NH O HO
Ph
(1.7) Rausch és munkatársai ferrocénből kiindulva több N-szubsztituált ferrocénkarboxamidot állítottak elő. A ferrocént kis feleslegben alkalmazott alumínium-klorid jelenlétében a megfelelő izocianáttal reagáltatva, közepes hozammal nyerték a kívánt karboxamidokat (1.8 egyenlet) [16].
Fe RN=C=O , AlCl3 Fe
NH O
R
(1.8) Az izocianát segítségével történő amidszintézis másik érdekes lehetősége, amikor nem ferrocénnel reagáltatják a megfelelő izocianátot, hanem lítium-ciklopentadieniddel. Ezután a monokarbamoil-szubsztituált ciklopentadienid vas(II)-kloriddal lejátszódó reakciójából kapják meg a monokarbamoil-, illetve dikarbamoil-ferrocént (1.9 egyenlet) [17].
F e
NHR O Li
Li O
N H
R
O
H N R Li
F e
NHR O
O NHR FeCl2 Li
+ R-N=C=O
(1.9) Gyakran alkalmazott módszer a ferrocénkarbonsav kloridjával történő acilezés (1.10 egyenlet). Némi hátrányt jelent azonban a savklorid nagy reakcióképessége, ami megnehezíti a vegyület előállítását, tisztítását, tárolását. A módszer által elért hozam: 20-40% [18].
Fe Fe
Cl O
Fe
NR2 O
OH O
PC l5 benzol
RNH2,vagy R2NH piridin
(1.10) Ezt a hátrányt Rotello és munkatársai azzal igyekeztek kiküszöbölni, hogy a szintézis intermediereként a megfelelő savfluoridot használták (1.11 egyenlet), amely megfelelően reaktív az amid képzéshez, ugyanakkor elég stabilis ahhoz, hogy tisztítása (átkristályosítás, szublimáció, kromatográfia) könnyen megoldható legyen, emellett szobahőmérsékleten bomlás nélkül eltartható.
Fe Fe
F O
Fe
NR2 O
OH O
N
N N F
F F
R2NH
80% 90- 99%
(1.11) A savklorid és az amin kapcsolása általában bázis hozzáadását igényli, hogy az megvédje a támadó nukleofil reakciópartnert a protonálódástól. Meglepő, hogy savfluorid esetében erre nincs szükség, ami további előnyöket jelent használatakor. Először is, bázisra érzékeny szubsztrátum jelenlétében nem játszódik le mellékreakció, másodszor, nincs szükség vizes feldolgozásra a reakció végén [19].
Speiser azt vizsgálta, hogy különböző karbodiimidek hogyan képesek aktiválni a ferrocénkarbonsavat, hogy az erős nukleofil reagenssel támadható legyen. A legtöbb vizsgált N,N’-diszubsztituált karbodiimid a ferrocénkarbonsavval igen stabil N,N’-diszubsztituált N- ferrocén-karbamid származékot eredményezett, ellentétben az 1-etil-3-(3-dimetil-amino-propil)- karbodiimid-hidrokloriddal (EDC). Éppen ezért alkalmas ez utóbbi karbodiimid a ferrocén- karbonsav aktiválására. Ferrocénkarbonsav, EDC és 3-(trietoxi-szilil)-propil-amin reakciójában közepes hozammal szintetizálták a megfelelő ferrocénkarboxamidot [20].
Kraatz hidroxi-benzotriazolt használt a ferrocénkarbonsav aktiválására. A keletkező ferrocenoil-benzotriazol-észter diklórmetános oldatát reagáltatta imidazollal (1.12 egyenlet). Az elért hozam 66% [21].
Fe Fe O
OBt O
N CH2Cl2, Et3N N
sz obahõm ., 16h hozm : 66%
H N
N +
(1.12) A leírt módszerek — 1,1’-ferrocén-dikarbonsavból kiindulva — természetesen alkalmasak 1,1’-ferrocéndikarboxamidok szintézisére is. Az 1,1’-heterodiszubsztituált ferrocéndikarboxamid származékok előállítása azonban némileg bonyolult, hosszadalmas, többlépéses szintézist igényel azáltal, hogy a legtöbb esetben az egyik karboxilcsoportot védeni, míg a másikat aktiválni kell. A védőcsoport eltávolítását újabb aktiválási lépés követi és ezek után alakulhat ki a nem szimmetrikus ferrocéndikarboxamid származék. Ez lehet az egyik oka, hogy ilyen vegyületek szintézisére csak elvétve találunk példát az irodalomban. Az egyik példa Sekine és munkatársai nevéhez fűződik. Ferrocéndikarbonsav-metil-észtert reagáltattak N-(t-butoxi-karbonil)-1,3- propándiaminnal 2-klór-1-metil-piridinium-jodid (CMPI) és tributil-amin jelenlétében (1.13 egyenlet 1.lépés) (hozam:89%). Majd a karbonsav-észter lúgos hidrolízise (1.13 egyenlet 2.lépés) után az előzővel megegyező módon alakították át a másik karboxilcsoportot 3-amino- propánsavval (1.13 egyenlet 3.lépés) (hozam: 62%) [22].
Fe Fe
NH O
OH O
OMe
O
HN Boc OR
O
Fe
NH O
H N
Boc HN
O
COO Et
R = Me R = H 1.
2.
3.
(1.13)
1.4. N-Ferrocenoil-aminosavak és –peptidek előállítása
A ferrocénkarbonsav amidszármazékai között különösen fontosak a N-ferrocenoil- aminosavak és –peptidek. Szolgálhatnak bioszenzorként (ld. 1.2.2. fejezet), de alkalmasak lehetnek a fehérjék bizonyos szerkezeti elemeinek modellezésére is.
Az első ferrocéntartalmú aminosav- és dipeptid származékokat (Fc-CO-Gly-OMe, Fc-CO- Gly-OH, és Fc-CO-Gly-Leu-OEt) Schlögl állította elő 1957-ben [23]. Ahhoz, hogy a peptidkötést
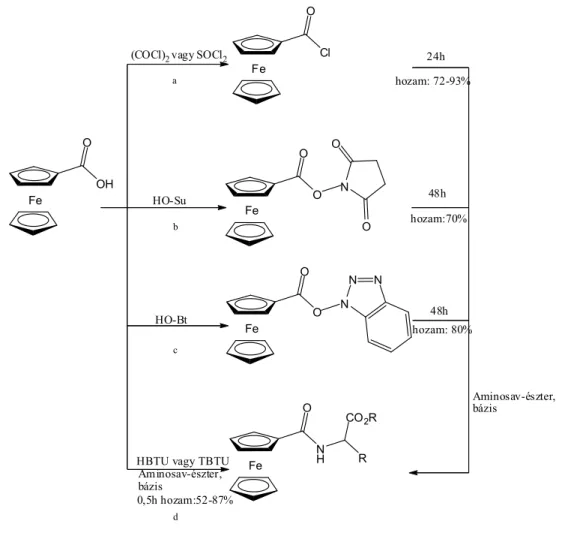
enyhe körülmények között ki lehessen alakítani, a savat aktiválni kell. A ferrocenoil-aminosavak szintézise során a ferrocénkarbonsav aktiválására több megoldást is alkalmaztak (1.5 ábra).
Az aminosavak acilezésére alkalmas vegyület a karbonsav-klorid, melynek kialakítása történhet oxaloil-kloriddal, vagy tionil-kloriddal (1.5 ábra „a” reakcióút) [23, 24].
További lehetőség a karbonsav átalakítása szukcinimiddé (1.5 ábra „b” reakcióút), vagy benzotriazol-észterré (1.5 ábra „c” reakcióút). Reakcióidő 2 nap, a nyerstermék hozama 70-80%
[25].
Fe
OH O
Fe
Cl O
Fe
O O
N O
O
N N N Fe
O O
Fe
NH O
R CO2R (COCl)2vagy SOCl2
HO-Su
HO-Bt
HBTU vagy TBTU Aminosav-észter, bázis
Aminosav-észter, bázis
a
b
c
d
48h
48h hozam:70%
hozam: 80%
0,5h hozam:52-87%
hozam: 72-93%
24h
1.5 ábra Ferrocénkarbonsav aktiválásának lehetőségei
A karbonsav aktiválása történhet HBTU-val, vagy TBTU-val is (1.5 ábra „d” reakcióút). Ez utóbbi megoldásnak több előnye is van: a kívánt kapcsolás rövid idő alatt végbemegy, nem szükséges vízmentes oldószerek használata, megfelelő extraktív feldolgozással a kapcsoló reagensek eltávolíthatóak, ezáltal kiváló hozammal, nagy tisztaságú terméket nyerhetünk (reakcióidő: 30 perc, hozam: 52-87%) [26].
Beck és munkatársai egy másik érdekes megoldást választottak az N-ferrocenoil-aminosav- észter előállítására. A karbonsavat EDC-vel aktiválták és ezzel egyidőben hozzáadták az aminosav-észtert, emellett katalitikus mennyiségű 4-(dimetil-amino)-piridint (DMAP), valamint bázisként N-metil-morfolint használtak. A reakció hátránya a hosszú reakcióidő (1-3 nap) (hozam 73-78%) (1.14 egyenlet) [27].
Fe
OH O
CH2Cl2 ClH3NCHR1COOR2 EDC N-Metil-morfolin
[DMAP]
, Fe
O NH
R1 OR2 O
(1.14) Az előző fejezetben tárgyalt lítium-ciklopentadienid és izocianát reakcióján alapuló módszert alkalmazva szintén állítottak elő ferrocenoil-aminosav-észtereket. Aminosav-észter- hidrokloridot feleslegben lévő difoszgénnel reagáltattak, hogy megkapják a kívánt izocianátot, majd a karboxamidok előállításánál leírt módon jutottak a mono-, illetve 1,1’-dikarbamoil- ferrocénhez [17].
Természetesen az itt tárgyalt módszerek mindegyike alkalmas 1,1’-diszubsztituált ferrocenoil-aminosav-észterek előállítására is, a megfelelő dikarbonsavból kiindulva.
Olyan 1,1’-heterodiszubsztituált ferrocenoil-aminosav származékok szintézisére, amikor két különböző aminosav, vagy peptid kapcsolódik a ferrocén két Cp gyűrűjéhez — a hasonló szerkezetű amidokhoz hasonlóan — jóval kevesebb példát találunk az irodalomban. Az általunk közzétett eredményeken kívül (melyeket a kísérleti részben ismertetek) mindössze három olyan publikáció született, amely a nem szimmetrikusan helyettesített 1,1’-ferrocén-dikarbonsav aminosav-, vagy peptid-származékait tárgyalja. Az egyiket Kimura és munkatársai tették közzé, akik sajnos a szintézis részleteit nem közölték [28]. A másik két közlemény Kraatz, illetve Metzler-Nolte nevéhez fűződik.
Kraatz az 1,1’-ferrocén-dikarbonsavat reagáltatta egyidejűleg Pro3-OBzl-rel és hidroxi- benzotriazollal (HOBt). Az így kapott termékelegyből oszlopkromatográfiával elkülönítette a Fe[C5H4-CO-Pro3-OBzl][C5H4-CO-OBt]-t (hozam: 45- 55%). Majd következő lépésként az OBt- csoportot cserélte a másik peptidre (Val-Val-Sta-Ala-Sta-OEt) (hozam: 55- 59%) [29].
Metzler-Nolte az 1,1’-ferrocén-dikarbonsavat HBTU és HOBt 1:1 arányú elegyével aktiválta, bázisként diizopropil-etil-amint használt. Ehhez adta a kétféle aminosavészter hidrokloridjának (L-Ala-OMe.HCl és L-Phe-OMe.HCl, illetve D-Phe-OMe.HCl és L-Ala- OMe.HCl) a ferrocén-dikarbonsavra nézve fél ekvivalens, 1-1 arányú elegyét. Hat óra elteltével megismételte az aktivációs lépést, majd újabb adag aminosavészter hidroklorid elegy hozzáadását követően egy teljes éjszakán át kevertetve, 29% és 23%-os hozammal állította elő a megfelelő heterodiszubsztituált ferrocénszármazékokat. [30]
1.5. Palládium-katalizált reakciók
Az elmúlt évtizedekben az átmenetifém-organikus kémia fejlődése a szerves szintézisutak forradalmi változását idézte elő. Az ismert átmenetifém-organikus vegyületek közül a palládiumkomplexek váltak a legfontosabb katalizátorokká akár az alapvető nyersanyagok, a finomvegyszerek, vagy bonyolultabb, a természetben is előforduló anyagok előállítását vesszük figyelembe. A palládiumkatalizátorok jelenlétében lejátszódó átalakulások sokfélesége, a reakciók kitűnő kemo-, regio- és sztereoszelektivitása lehetővé teszi olyan rendkívül összetett molekulák szintézisének megvalósítását, amely a klasszikus szerves kémia módszereivel nem, vagy csak nagyon nehezen oldható meg.
A palládiumkatalizátorok többek között lehetővé teszik, hogy egy molekula telítetlen szénatomján új szén-szén kötést hozzunk létre (Heck reakció-, vagy különböző fémorganikus reakciópartnerekkel lejátszódó kapcsolási reakciók segítségével), vagy C-heteroatom (C-N, C-P, C-S, C-O stb) kötést alakítsunk ki a palládiumkomplex jelenlétében lejátszódó nukleofil szubsztitúció révén [31a,b,c]. További előnyük, hogy képesek különböző kis molekulák, köztük a szén-monoxid aktiválására, mely így közvetlenül beépíthető valamely szerves molekulába. Az utóbbi folyamat karbonilvegyületek- vagy karbonsavszármazékok képződéséhez vezet.
A fenti reakciók kiindulási vegyületei a legtöbb esetben aril- vagy alkenil-halogenidek, illetve aril- vagy enol-triflátok, melyek a katalitikusan aktív palládiumkomplexszel reakcióba lépve szén—palládium kötést tartalmazó aril- vagy alkenil-palládium komplexek képződéséhez vezetnek. A központi palládiumatomhoz kapcsolódó telítetlen szénatomok a palládium
„közvetítésével” reakcióba léphetnek különböző nukleofil reakciópartnerekkel (alkénekkel, fémorganikus reagensekkel, aminokkal, stb.). A halogénszármazékok közül — bár ezek a
legdrágábbak — kitűnő reakciókészségük miatt a jódvegyületeket alkalmazzák elsősorban.
Számos példa található azonban a brómszármazékok átalakítására is, az utóbbi időben pedig egyre többen foglalkoznak a legkönnyebben hozzáférhető aril-kloridok reakcióival [32, 33].
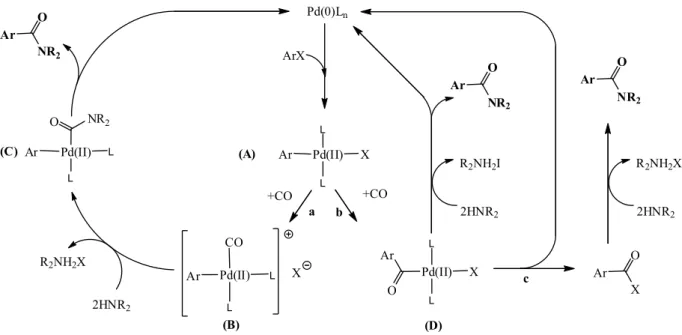
A bevezetésben említett reakciók esetében — legyenek azok egy elektrofil és nukleofil reakciópartner között végbemenő kapcsolási reakciók, vagy szén-monoxid jelenlétében lejátszódó karbonilezési reakciók — az aktív katalizátor valamilyen Pd(0)-komplex. Az aktív részecske a reakcióelegyben alakul ki, a fő kérdés általában az, milyen palládiumvegyületek hozzáadásával érhetjük el a legjobb eredményt. A leggyakrabban használt katalizátorrendszerek vagy már eleve 0-oxidációfokú palládium komplexet tartalmaznak, ilyen például a Pd(0)(PPh3)4
vagy a Pd(0)(dba)2 + nL rendszer, vagy olyan Pd(II)komplexek vagy Pd(II) sók, amelyekből a reakcióelegyben „in situ” alakul ki az aktív részecske valamilyen redukálószer (pl. egy amin (1.15 egyenlet), vagy a ligandumként használt foszfán (1.16 egyenlet)) hatására [34, 35].
Redukció Et3N-al:
PdL2X2 PdL2X
N H
Et2
-hidrid elimináció
NEt2X -
HPdL2X
reduktív elimináció
PdL2 + HX ligandum
csere
X
(1.15) Redukció PPh3-al:
Pd(OAc)2 PdOAc
Ph3P Ph3P
OAc
O O
P Pd
PPh3 OAc
Ph3
'Pd-PPh3' Ac2O
OPPh3 +
+ reduktív elimináció ligandum
csere
(1.16)
A fejezet további részében a karbonilezési reakciókon kívül kizárólag azoknak a palládium- katalizált homogénkatalitikus reakcióknak a bemutatására szorítkozom, melyeket már alkalmaztak az általam is kiindulási anyagnak választott jód-, illetve 1,1’-dijód-ferrocén átalakítására. Jód-ferrocén és 1,1’-dijód-ferrocén karbonilezését kutatócsoportunk valósította meg első ízben. A munkám jelentős részét képező karbonilezéssel kapcsolatos általános tudnivalókat az 1.5.2. fejezetben tárgyalom.
1.5.1. A jód-ferrocén és a dijód-ferrocén palládium-katalizált reakciói
Bár a ferrocén reakcióiban az aromás vegyületekhez hasonló módon viselkedik, és az aril- halogenidek palládium-komplexek jelenlétében lejátszódó homogénkatalitikus reakcióit szívesen alkalmazzák aromás származékok szintézisére, a jód-ferrocén és dijód-ferrocén hasonló átalakítására viszonylag kevés példát találunk az irodalomban. A ferrocénszármazékok szintézisének legelterjedtebb kiindulási anyaga még mindig a ferrocénkarbonsav, illetve annak aktivált származékai.
1.5.1.1. Kapcsolás fémorganikus reakciópartnerekkel
Új szén-szén kötés kialakítása minden időben a szintetikus szerves kémia meghatározó átalakításai közé tartozott. A klasszikus C-C kötést létrehozó módszerek (pl. Grignard reakció, Wittig reakció, cikloaddíció stb.) mellett az elmúlt 30 évben az átmenetifémek (Pd, Ni, Cu) által katalizált keresztkapcsolási (cross-coupling) reakciók jelentősen kiterjesztették a szintetikus lehetőségeket.
A fémorganikus reagensek jelenlétében lejátszódó kapcsolási reakciók általában palládium(0) vagy nikkel(0) katalizálta reakciók. A kapcsolási reakció mechanizmusát leegyszerűsítve az 1.6 ábra mutatja be.
Pd(0)L2
R1
Pd(II) X L
R1 L
Pd(II) R2 L
R1 L Pd(II)
L R2
R1 L
X
R2M R1 R2
MX (1)
(2) (3)
(4)
1.6 ábra A fémorganikus reakciópartnerek jelenlétében lejátszódó kapcsolás általános mechanizmusa
A folyamat nyitó lépése egy szerves (általában halogén-) vegyület oxidatív addíciója az alacsony oxidációs állapotú átmenetifémre (1.16 ábra 1. lépés). A kapcsolni kívánt másik szerves
molekularészlet a keletkező intermedierről ún. transzmetallálási lépéssel kerül az átmenetifémre (1.16 ábra 2. lépés). Általában ez a reakció a folyamat sebesség-meghatározó lépése. A transz elhelyezkedésű csoportok egy gyors izomerizációs lépés eredményeként cisz-helyzetbe kerülnek (1.16 ábra 3. lépés), majd a záró reduktív eliminációs lépés (1.16 ábra 4. lépés) eredményeképpen kialakul a kapcsolt termék és a katalitikus ciklus bezárul az alacsony oxidációs állapotú átmenetifém újraképződésével.
Az elektrofil reakciópartner, amelynek oxidatív addíciójával a folyamat elindul, elsősorban aril/alkenil-halogenid, vagy -triflát. A fémorganikus reakciópartnerek között találunk ón- (Stille kapcsolás) [36], bór- (Suzuki kapcsolás) [37-41], cink-, alumínium- és cirkónium- (Negishi kapcsolás) [42, 43], szilícium- és germánium- [44, 45] lítium- és magnézium- [46], valamint indiumvegyületeket [47].
A keresztkapcsolási eljárások legelterjedtebb és részleteiben is ismert képviselője a Stille nevével fémjelzett reakció. A folyamatban a ligandumcsere az ónorganikus vegyület és a palládiumkomplex között játszódik le (1.17 egyenlet).
X + R2SnR33
R1 R1 R2 + R33SnX
R1 = alkenil, aril
R2 = alkenil, alkinil, allil, aril, benzil R3 = alkil
X = I, Br, OTf [Pd]
(1.17) A kapcsolási reakcióban az ónatomon lévő csoportok közül csak az egyik vesz részt.
Szerencsére az ónról a legkülönbözőbb csoportok vihetők át szelektíven, hiszen az alkil-transzfer sebessége jóval kisebb, mint a telítetlen csoportoké. Így az általánosan alkalmazott ónreagensek nem szimmetrikusak, három alkilcsoportot (metil-, vagy butil-) negyedikként pedig alkinil-, alkenil-, aril-, benzil- vagy allilcsoportot tartalmaznak. A módszernek számos előnye van: a kereskedelmi forgalomban sokféle trialkil-ón származék megtalálható, továbbá ezek a vegyületek általában kevéssé érzékenyek oxigénnel, vagy nedvességgel szemben, a reakció szelektivitása kitűnő, emellett enyhe reakciókörülmények között, számos funkciós csoport (OH, NH, COOH, CHO) jelenlétében végbemegy anélkül, hogy szükség lenne védőcsoportok alkalmazására [36].
Mindezek a tulajdonságok elsősorban az ón—szén kötés kis polaritásának köszönhetők. A kapcsolást különösen eredményesen használják több funkciós-csoportot tartalmazó, bonyolult
szerkezetű molekulák átalakítására. A Stille-kapcsolás hátránya viszont az ónvegyületek közismert toxikus hatása és a reakcióban keletkező trialkilón-halogenidek eltávolításának problémája.
Ferrocénvázas vegyületek előállításánál is találunk néhány olyan példát, ahol Stille- kapcsolást alkalmaztak, azonban ezekben az esetekben a ferrocénszármazék az ónorganikus reakciópartner (ferrocenil-tributil-sztannán) nem pedig a halogenid. A Stille-reakció tárgyalását ebben a fejezetben az teszi mégis indokolttá, hogy munkám során általam előállított jód-ferrocén- származékok átalakítására alkalmaztam ezt a kapcsolási reakciót. (Lásd kísérleti rész 2.3.1 fejezet 2.6 egyenlet)
A Suzuki-reakció talán a keresztkapcsolási reakciók legsokoldalúbb fajtája. A felhasznált bórorganikus vegyületek szintén csökkent nukleofil karakterrel rendelkeznek (ezért számos funkcióscsoport jelenlétében kivitelezhető a kapcsolás védőcsoportok kialakítása nélkül is), valamint oxigénnel és nedvességgel szemben stabilak. A módszer további értékes tulajdonsága, hogy a kiindulási boronsavak és származékaik nem toxikusak, a reakció vizes közegben is elvégezhető és a képződő bórsav nem zavarja a tisztítási eljárásokat. Fontos megemlíteni, hogy a ligandumcsere során — más keresztkapcsolási reakcióktól eltérően — egy ekvivalens bázis jelenlétére van szükség. Mivel a szerves bórszármazék nukleofil ereje nem elég nagy, így az organopalládium-halogenidet kell elektrofilebbé tenni, a halogenidet alkoxi- vagy karbonátionra cserélve. Feltételezések szerint a reakcióközegben jelenlevő alkoxid/karbonát/hidroxidionok nem csak a palládium elektrofil jellegét fokozzák, de a bórorganikus vegyület nukleofil jellegét is oly módon, hogy egyensúlyi addícióban anionos boronátkomplexet képeznek vele (1.18 egyenlet).
Alkáli-fluoridokat szintén eredményesen használtak „Suzuki-bázisként” [37-41].
R Pd X
NaOR'
NaX
R Pd OR'
R"B(OH)2
R'OB(OH)2
R Pd R"
Pd(0) RR"
NaOR' : NaOH, Na2CO3, NaHCO3, NaOEt; EtOH, TlOH, Ba(OH)2, Cs2CO3,CsF, KF (1.18) Aril-ferrocének előállítására a Suzuki-kapcsoláson belül is több lehetőségünk van. Először is a ferrocenilcsoportot képviselheti egyrészt az aril-boronsav, másrészt a haloferrocén is. Mivel a
ferrocén-boronsav és a jód-ferrocén (vagy bróm-ferrocén) elérhetőségét, stabilitását tekintve nem nagyon különböznek egymástól, a választást inkább a másik reakciópartner kezelhetősége határozza meg. Döntő jelentőségű a katalizátorrendszer, a bázis, illetve az oldószer megfelelő megválasztása. Knapp és Rehahn összehasonlította a ferrocén-diboronsav / fenil-halogenid és dihaloferrocén / fenil-boronsav reakciópartner párok reakcióit két különböző rendszerben (A:
toluol / 2M Na2CO3-oldat / Pd(PPh3)4; B: DME / 3M NaOH-oldat / PdCl2(dppf)). Az A módszer alkalmazásakor a dijód-ferrocén : fenil-boronsav pár esetén volt jóval nagyobb a konverzió (55%
/ 25%), míg a B esetén mindkét reakciópartner pár alkalmazása azonosan jó eredményt mutatott (konverzió: 90%) [48].
Imrie jód-ferrocént reagáltatott különféle szubsztituált fenil-boronsav származékokkal Suzuki-reakcióban. A paraméterek fokozatos változtatásával meghatározta az optimális reakciókörülményeket: 90%-os etanol mint oldószer, palládium-acetát katalizátor, és nátrium- karbonát, vagy bárium-hidroxid bázis bizonyult legelőnyösebbnek (1.19 egyenlet) [49].
Fe I
(HO)2B Fe
+
X
X Pd(OAc)2,Na2CO3
vagy Ba(OH)2 EtOH-H2O
X= OCH3, CH3, H, C6H5, F, Br, COCH3, CHO, CF3
(1.19) A benzol gyűrűhöz kapcsolódó elektronküldő szubsztituensek növelik a reakció sebességét azáltal, hogy ezek a csoportok nukleofilabbá teszik a fenil-boronsav származékot. Elektronszívó szubsztituens esetén az ellenkezője tapasztalható.
A keresztkapcsolási reakciók következő változata, amelyet jód-, illetve dijód-ferrocén átalakítására is használtak, a cinkorganikus reagensek jelenlétében lejátszódó Negishi-reakció. A cinkorganikus vegyületek alkalmazása mellett szól, hogy viszonylag könnyen hozzáférhetőek, a funkciós csoportok egy jelentős részénél nincs szükség védőcsoportok alkalmazására, és a cink és palládium közti ligandumcsere már szobahőmérsékleten is gyorsan lejátszódik. Hátrányuk, hogy nedvességre és oxigénre érzékenyek.
Jód-, illetve 1,1’dijód-ferrocén és egy cinkorganikus szerin származék palládium katalizált kapcsolási reakciójában az 1.20 egyenletben látható termékek keletkeztek. 1,1’-Dijód-ferrocén
kiindulási anyag esetén a cinkorganikus reagens feleslegének növelése a diszubsztituált ferrocén származék keletkezését jelentős mértékben elősegítette [50].
Fe I
X
Zn NHBoc
CO2Me Pd2(dba)3, (o-Tol)3P
Fe
Fe MeO2C
NHBoc
NHBoc MeO2C
NHBoc
CO2Me +
X= H, I
(1.20) 1.5.1.2. Kapcsolások terminális alkin reakciópartnerrel
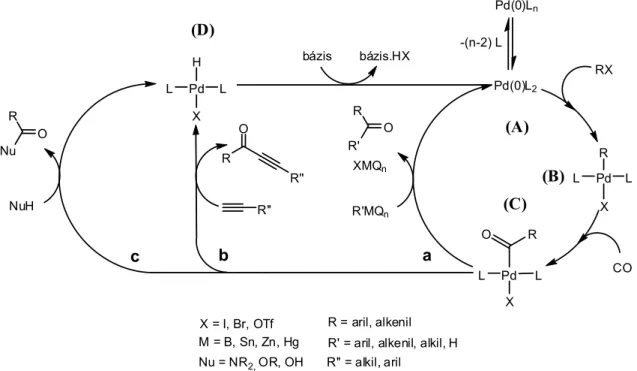
Aril-alkinek és konjugált eninek legkényelmesebb előállítási módja az aril- vagy alkenil- halogenidek palládium katalizált kapcsolása terminális alkinekkel. Sonogashira nevéhez fűződik az a felismerés, hogy in situ előállított alkinil-réz reagensek az előbb említett kapcsolási reakcióba vihetők (1.21 egyenlet).
X + R1
R1 = alkenil, aril, acil, R2 = alkil, aril, trialkil-szilil X = I, Br, OTf
[Pd]/Cu HC CR2
bázis R1C CR2 + bázis.HX
(1.21) Ez a folyamat végső soron két katalitikus reakció: a palládiumkomplexek réz-katalizált alkinilezése (1.7 ábra „B” ciklus), valamint az sp2-szénatomokat tartalmazó halogenidek és terminális acetilének palládium jelenlétében lejátszódó kapcsolása kombinációjaként fogható fel (1.7 ábra „A” ciklus). Bár a reakció valószínűleg az 1.5.1.1. fejezetben bemutatott fő lépéseket (oxidatív addíció, fémek közti ligandumcsere, reduktív elimináció) követve játszódik le, pontos mechanizmusa nem ismert. A katalitikusan aktív részecskék szerkezetével és a CuI kokatalizátor valódi szerepével kapcsolatban nem állnak rendelkezésünkre biztos adatok. Az oxidatív addíciót követően a palládium(II)komplex reakcióba lép a terminális acetilénnel, valószínűleg egy átmenetileg képződő réz-acetilid intermedier közreműködésével, majd a keletkező alkinil-
palládium(II) vegyületből reduktív eliminációval képződik a termék. Katalizátorként leggyakrabban a PdCl2(PPh3)2 komplexet használják. Feltételezik, hogy a katalitikusan aktív részecske ekkor a Pd(II)komplex és az acetilén reakciójában képződő Pd-acetilid komplexből jön létre reduktív elimináció útján (1.7 ábra „C” ciklus). Az előbb említett palládiumvegyületen kívül általánosan alkalmazzák még a Pd(0)(PPh3)4, illetve Pd(0)2(dba)3 + foszfán típusú katalizátorokat is. A kis reakciókészségű aril-bromidok átalakításánál gyakran a 2/1-nél nagyobb foszfán/palládium aránnyal dolgoznak, hogy a szükséges magasabb hőmérsékleten elkerüljék a katalizátor bomlását, fémpalládium kiválását [51].
R2 C CH
C C R2 R1 Pd(0)L2
R1 X
amin.HX
CuX R2 C CCu Pd(II)
R1
X
L L
C R2 Pd(II)
R1
C
L L
Pd(II) L
L
Cl Cl
Pd(II) L
L
C C
C R2
C R2
C CC
R2 C R2
CuX R2 C CCu
R2 C CH amin.HX
"A" ciklus
"B" ciklus
"C" ciklus
1.7 ábra A Sonogashira kapcsolás mechanizmusa
Mivel a kokatalizátorként használt réz-sók oxidatív körülmények között elősegítik az alkinek úgynevezett Glaser-típusú homokapcsolását, mely a megfelelő szimmetrikus diint eredményezi melléktermékként, az utóbbi néhány évben számos kutatócsoport koncentrált a réz- mentes Sonogashira kapcsolás megvalósítására [52-55].
Ferrocénvázas vegyületek esetében a jód-ferrocén, illetve a dijód-ferrocén és a legkülönbözőbb terminális alkinek (fenil-acetilén, acetilén [56], 1,4-dietinil-benzol [57, 58], 4- etinil-piridin, 5-etinil-bipiridin [59]) kapcsolása, változatos ferrocénvázas acetilén származékok képződését eredményezte 45-96%-os hozamok mellett.
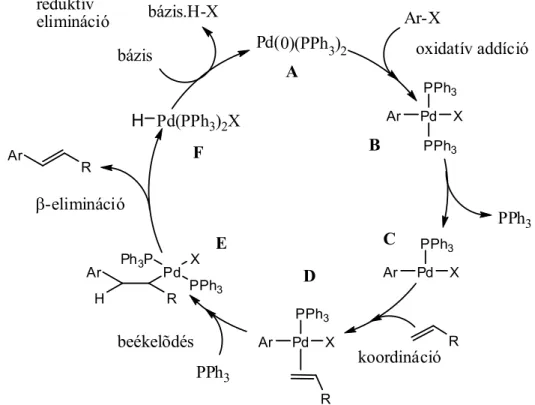
1.5.1.3. Heck reakció
A -kötésű átmenetifém-organikus vegyületekből kiinduló szén-szén kötés kialakulásához vezető folyamatok közül a keresztkapcsolási reakciók mellett szintén nagy jelentőségű az alkén karbopalladálásából, majd az azt követő -eliminációból álló folyamat, amelyet felfedezőjéről Heck-reakciónak neveztek el. A 1.8 ábrán látható katalitikus ciklus első lépése ebben az esetben is az aril-halogenid és a palládium (A) között lejátszódó oxidatív addíció. A kialakult σ-kötésű palládiumorganikus vegyülethez (C) koordinálódik az alkén, majd a beékelődést követően olyan alkilpalládium-komplexhez jutunk, melyből egy rotáció után kialakul az eliminációhoz szükséges szin-periplanáris Pd-C-C-H elrendeződés (E). Így lehetőség nyílik a β-elimináción keresztül megvalósuló stabilizációra, mely az alkén termék képződéséhez vezet.
Pd(0)(PPh3)2
Pd
R
Pd(PPh3)2X
R Ar R
bázis.H-X Ar-X
PPh3 PPh3 X Ar
Pd PPh3
X Ar
Pd PPh3
X Ar
Pd Ph3P X
PPh3 R H
Ar
H
PPh3
PPh3
bázis oxidatív addíció
koordináció beékelõdés
-elimináció reduktív elimináció
A
B
C D
E F
1.8. ábra A Heck-reakció mechanizmusa
A hidrogén-halogenid eliminációja, mely a hozzáadott bázis hatására megy végbe, a katalitikus ciklus záródását, az aktív katalizátor (A) regenerálását eredményezi [60, 61].
Olyan Heck-reakciókban, amikor a β-elimináció során nem az eredeti C=C kötést regeneráljuk, a reakcióban általában új kiralitáscentrum alakul ki. Ilyen esetekben királis ligandumok alkalmazásával elvileg lehetőségünk van enantioszelektív átalakításokra.
A Heck reakció során elérhető konverzió és szelektivitás természetesen jelentős mértékben függ a reakciópartnerektől, de emellett befolyásolható a körülmények - oldószer, katalizátor, bázis, esetleg valamilyen egyéb adalék - megfelelő megválasztásával. Az eredetileg Heck által közölt Pd(OAc)2/PPh3/Et3N katalizátor—bázis párosításon kívül általánosan alkalmazzák a Jeffery [62] által kidolgozott Pd(OAc)2/K2CO3/nBu4NCl rendszert DMF vagy DMSO oldószerben. Ez utóbbi esetben a kvaterner ammóniumsó szerepe a feltételezések szerint az, hogy a kialakuló palládiumkolloidot stabilizálja, megakadályozza az aggregációt [63]. E két, már-már klasszikussá váló módszer mellett számos katalizátor—bázis—oldószer összeállítást kipróbáltak már [64].
Kasahara és munkatársai írták le először olefinek palládium katalizált reakcióit jód-, illetve 1,1’-dijód-ferrocénnel. A szokásos Heck körülmények között reagáltatták a halogeno- ferrocéneket az alkének legkülönbözőbb képviselőivel: egyszerű olefinekkel (sztirol), akrilsav származékokkal (etil-akrilát, metil-metakrilát, akrilnitril), telítetlen ketonokkal (metil-vinil-keton, fenil-vinil-keton), allil-alkoholokkal (allil-alkohol, 3-butén-2-ol) és telítetlen acetátokkal (vinil- acetát, izopropenil-acetát, acetofenon-enol-acetát) (1.22 egyenlet).
FcI +
[Pd]
bázis C C
H
F c H
R H2C C
H R
(1.22) Allil-alkoholok és jód-ferrocén reakciója minden esetben ferrocén-karboxaldehidet vagy – ketont eredményezett (1.23 egyenlet).
FcI + H2C C H
HC OH
R [Pd]
bázis
H2
C C
H C OH
R Fc
H2 C
H2 C C
O R Fc
(1.23) Vinil- és izopropil-acetát jód-ferrocénnel való Heck-kapcsolása diferrocenil-etilén, illetve