CHAPTER 6
Fluorocarbon Chemistry
B Y J. H. SIMONS
Fluorine Research Center, The University of Florida, Gainesville, Florida
A N D
T. J. B R I C E
Central Research Department, Minnesota Mining and Manufacturing Company, St. Paul, Minnesota
Page
Introduction 334 Fluorocarbon Chemistry 334
Basis of Properties of Fluorocarbons 335
Fluorocarbon Derivatives 337 Theoretical Considerations 338 Utilitarian Applications 339 Developments in Methods of Producing Fluorochemicals 340
The Electrochemical Process 340 Catalytic Fluorination 342 The Metallic Fluoride Process 344 The Fluorination of Carbon 345 Liquid Phase Fluorination 345 Exchange Reactions 346 The Chemical and Physical Properties of Fluorocarbons 347
The Chemical Properties of Saturated Fluorocarbons 347
The Physical Properties of Fluorocarbons 348 The Purification of Fluorocarbons 349 Surface Properties of Fluorocarbons 351
Viscosity 352 Rotational Isomerism 353
Solubility 353 Energy of Vaporization 357
Conductivities in Acetforic Acid 358 High-Temperature Equilibria 358 Thermodynamic Properties 362
Refractive Index 363 Fluorocarbon Derivatives 364
Fluorocarbon Chlorides and Bromides 364
Fluorocarbon Iodides 366 Fluorocarbon Aldehydes and Ketones 371
Fluorocarbon Olefins 375 Fluorocarbon Carboxylic Acids 383
333
334 J. H. SIMONS AND T. J. Â RICE
Bibliography
Fluorocarbon Nitrides and Other Nitrogen-containing Compounds Alcohols
Fluorocarbon Esters
Fluorocarbon Oxides and Ethers Containing Fluorocarbon Groups Sulfur Compounds
Alkforyl Aromatic Compounds
Styrene Derivatives Polymers and Polymerization
Amines, Phenols, and Nitro Compounds Halogen Derivatives
Page 391 393 394 396 398 399 401 402 403 403 437
Introduction
FLUOROCARBON CHEMISTRY
Since the publication of Volume I, there has been considerable published material relative to fluorocarbon chemistry, and many addi
tional fluorocarbon derivatives have been cited in publications. This has made it desirable to attempt to bring the subject up to date with this chapter. Because this field is advancing so rapidly, much additional information will be available between the writing of this manuscript and the publication of this volume.
The pattern for the development of this branch of chemistry is beginning to take form. The electrochemical process has been shown to be suitable for large-scale industrial use. Two types of products can be produced ; (a) inert substances such as the fluorocarbons themselves, and the fluorocarbon oxides, nitrides, etc. ; and (i>) reactive substances such as the fluorocarbon carboxylic acids or acid halides. From the latter, other series of reactive substances can be produced such as fluorocarbon iodides, aldehydes, nitriles, and olefins, as well as fluorocarbon 1,2-dihy- droalcohols and amines. The reactive substances provide the means of establishing a synthetic fluorocarbon chemistry. The reactions employed for the syntheses will not be those of organic chemistry, but will be either entirely new methods or drastic modifications of known organic or inorganic reactions.
The nomenclature for the fluorocarbons and fluorocarbon derivatives remains in an unsatisfactory state.* In the laboratories of a large indus-
* A division of the Committee on Organic Chemical Nomenclature of the American Chemical Society, namely, the Committee on Nonfunctional Derivatives of Hy
drocarbons, named a subcommittee of itself to consider nomenclature of fluorine- containing organic compounds. It is unfortunate indeed that this subcommittee far exceeded its jurisdiction and considered the naming of fluorocarbons and fluorocarbon derivatives. First, a decision had to be made as to whether fluoro
carbons were organic, and this committee naturally could not render an unbiased decision on that question. Second, it had to be decided that fluorocarbons were
FLUOROCARBON CHEMISTRY 335 trial company where most of the fluorocarbon derivatives are being made, the use of the "perfluoro" system of naming is leading to many errors and much confusion. These names are so cumbersome and am
biguous that they are causing many difficulties and are retarding progress.
Some people are beginning to call the fluorocarbons and their derivatives by the name fluorochemicals. This will be very satisfactory, if the name is used for the fluorocarbon domain and is not extended to include organic compounds. A better name for the field has recently appeared. I t is forbonic chemistry for the subject and forbons for the substances.
Forbonic is a contraction of fluoroc&rbonic. Forbons would include both the fluorocarbons and the fluorocarbon derivatives.
BASIS OF PROPERTIES OF FLUOROCARBONS
There are a number of reasons why fluorocarbons are very different from hydrocarbons (397). When heated, hydrocarbons lose hydrogen to form cracked products, unsaturated substances, or carbon. The fluoro
carbons cannot be expected to do this at any reasonable temperature.
They are, in fact, easily made by the reaction of carbon with fluorine whereas hydrocarbons are not easily made from the elements. From bond energies we can make a rough calculation of the reactions to form an olefin from either a hydrocarbon or fluorocarbon.
R—CH2—CH2—R — R — C H = C H — R + H2 Bonds [(C—C)58.6 + 2(C—H) 174.6 = 233.2]
-> [(C=C)100 + (H—H) 103.4 = 203.4]
Φ C F2— C F2— Φ —> Φ —CF—CF— Φ -r* F2 Bonds [(C—C)58.6 + 2(C—F)214.0 = 272.6]
- * [(C=C)100 + (F—F)63.5 = 163.5]
It is seen that the removal of fluorine from a fluorocarbon to form an olefin is much more difficult energetically than to remove hydrogen from a hydrocarbon. As 109.1 kcal. is much greater than the carbon-carbon bond energy, when the necessary energy is supplied, rupture of the molecule would be expected rather than the formation of the unsaturated sub
stance. The reverse is true with hydrocarbons. It is interesting to make a similar calculation for the removal of hydrogen fluoride from an organic fluoride.
derivatives of hydrocarbons, which they most certainly are not. It is a statement of fact that this committee was formed not to deliberate the question of nomen
clature, but rather was organized to place the stamp of apparent legality on a prejudiced decision made prior to its formation. Any statements or edicts that the committee may issue should not be considered as binding on any scientific writer or editor.
336 J. H. SIMONS A N D T. J. Β RICE
R—CH2—CHF—R —• R — C H = C H — R + H F Bonds [(C—C)58.6 + (C—H)87.3 + (C—F) 107.0 = 252.9] —
[(C=C)100 + (H—F) 147.5 = 247.5]
This illustrates why a hydrocarbon monofluoride is so readily decom
posed. The actual experimental conditions are even more favorable for decomposition of aliphatic fluorides than this indicates. The hydrogen fluoride produced is a powerful catalyst for the polymerization of the olefins with the actual product being tars and polymers.
Removal of hydrogen atoms from the carbon skeleton, either singly or in pairs, is more easily accomplished than the removal of fluorine atoms.
This is only a partial explanation of the failure of fluorocarbons to take part in organic chemical reactions. The almost perfect covering power of the fluorine atoms for the carbon skeleton protects the internal force fields and necessitates a much higher energy of activation to initiate reaction for fluorocarbons than for hydrocarbons. This physical covering and protection of the carbon skeleton also prevents the fluorocarbons from taking part in another entire class of reactions available for organic compounds. This class is the one in which an important feature of the mechanism is the attachment of the entering atom or group to the carbon atom prior to the detachment of the leaving atom or group (the back side approach mechanism). With more than one fluorine atom bonded to a carbon atom there is insufficient room for an entering group or atom prior to detachment, thus making such mechanisms possible only with much greater activation energies. A great number of organic chemical reactions depend upon a reasonable reaction rate at convenient temperature on the reduction of activation energy by acid catalysis. Organic compounds with their labile hydrogen atoms associate and cooperate with the hydro
gen ions of acid catalysts in manners not at all available for fluorocarbons and their hydrogen-free derivatives. Thus these substances cannot be expected to be amenable to organic chemical techniques of reactions.
Experimentally, hydrocarbons do not have a detectable dipole moment. As all hydrocarbons can be considered derivatives of methane with hydrocarbon radicals replacing hydrogen atoms with some cycliza- tion and olefin or aromatic formations, the lack of a measurable dipole moment means that electrical symmetry is not disturbed by replacement of a hydrogen atom with a hydrocarbon radical. The replacement of the hydrogen atom by a halogen atom or a fluorocarbon radical does, how
ever, introduce a dipole moment. In the fluorocarbon the replacement of a fluorine atom with a fluorocarbon radical does not change the electrical symmetry as indicated by the failure to detect a dipole moment (137).
The replacement of a fluorine atom with a hydrogen atom or hydrocarbon
337 radical does, however, introduce a dipole moment. It is apparent, there
fore, that the hydrocarbons can be considered derivatives of methane; but fluorocarbons are derivatives of methforane. An organic ether can properly be considered a derivative of water, and organic ethers have basic properties equivalent to water (343). Fluorocarbon oxides, how
ever, are not derivatives of water. They are not ethers. They are properly derivatives of OF2. Amines are derivatives of ammonia in which one or more of the hydrogen atoms are replaced with organic radicals. There are no such structures as fluorocarbon amines, because a fluorocarbon radical attached to a nitrogen atom is"a replacement for a fluorine not a hydrogen atom. The fluorocarbon nitrides, as such compounds are called in this book, are derivatives of nitrogen trifluoride, not of ammonia. This explains the lack of basic properties of compounds like ( C3F7) 3 N . It is highly erroneous to call them amines. In the same way it is highly incor
rect to consider fluorocarbons as organic compounds, as hydrocarbon derivatives, or as "fluorinated substances/
1
FLUOROCARBON DERIVATIVES
Derivatives of fluorocarbons have been made with fluorocarbon groups attached to many elements other than carbon or fluorine. If the attach
ment is made to the more electronegative elements such as chlorine, oxygen, or nitrogen, a structure results that has a thermal stability and chemical inertness very similar to that of the fluorocarbons themselves.
The fluorocarbon oxides, nitrides, and even the monochlorides have physical properties very similar to the fluorocarbons. They have low surface tensions, high molecular weight for the boiling point, and similar solubility properties. It is only when we proceed down in electronegativity (and increased atomic size) to iodine that reactive substances are found.
Fluorocarbon iodides are reactive reagents.
Fluorocarbon radicals attached to electropositive elements form sub
stances with considerably different properties. Fluorocarbon hydrides have considerable dipole movements, boiling points much higher than fluorocarbons for the same molecular weight, and solubility properties of their own. Methforane and fluoroform form two liquid phases near the boiling point of the former. Fluorocarbon monohydrides are relatively inert chemically, being somewhere between fluorocarbons and hydro
carbons in this respect. With more than one hydrogen atom in the molecule of fluorocarbon, substances result that hydrolyze relatively easily, probably by the initial removal of hydrogen fluoride. Electroposi
tive elements of larger atomic size, such as mercury and arsenic, form much more reactive fluorocarbon derivatives.
Reactive fluorocarbon derivatives in which the reactive grouping
338 J. H. SIMONS A N D T. J. Β RICE
contains a carbon atom attached to the fluorocarbon radical, such as
— C 0 2H, —CHO, —CN, — CH2OH, and —CH2NH2, have now been made in significant numbers. As would be expected, the reactions of these substances have properties in common with organic substances contain
ing the same reactive group, as this carbon atom is attached to no fluorine atoms. The properties are markedly different, however, due to the difference in effect of the fluorocarbon and hydrocarbon radicals.
Fluorocarbon carboxylic acids are much more acidic than the correspond
ing organic acids. In general, the larger number of —CH 2— groups separating a functional radical from a fluorocarbon portion of a hybrid molecule, the more similar this functional group will be to a like one in an organic molecule. The 1,1-dihydrofluorocarbon alcohols and amines exist although they are somewhat different in properties from organic alcohols and amines. Fluorocarbon alcohols and amines in which there is no —CH2— group to separate the functional radical from the fluoro
carbon chain are as yet unknown. When isolated, they will probably be found to be unstable. The fluorine system analog of the alcohol or amine structure (i. e., — O F i and —NF2) are stable in fluorocarbons but not in organic compounds.
The influence of a fluorocarbon radical on an otherwise organic molecule is, however, experienced throughout the entire molecule. In general, it imparts resistance to reaction, particularly oxidation (413).
THEORETICAL CONSIDERATIONS
The properties of the fluorocarbons and their derivatives are proving of great value in testing and advancing theories of chemical and physical behavior. As the fluorocarbon domain of substances increases, it will become of increasing theoretical interest. Highly significant publications are already appearing.
Modification of chemical theories are necessary to correlate the tre
mendous inertness of the fluorocarbons. As we proceed from acetic acid through chloroacetic acid, dichloroacetic acid, to trichloroacetic acid, the ease of the haloform reaction increases. This has been related to the elec
tronegative characteristic of the chlorine atom. Some modification in this theory appears necessary, because trifluoroacetic acid does not readily take part in the haloform reaction; and fluorine is certainly more electro
negative than chlorine. The properties characterized by the term aromatic have been ascribed to certain carbon-carbon bond characteristics. Com
pounds such as C eF6, with the same carbon skeleton but with fluorine rather than hydrogen atoms attached to the carbon atoms, do not have these aromatic properties. Apparently, the hydrogen atoms are responsi
ble at least in part. The difference in chemical properties of fluorocarbon
339 oxides and nitrides as contrasted with organic ethers and triamines is striking. Any theory based only on the forces and attachments of the atomic skeleton of the molecule must be modified to account for these differences.
The properties of the fluorocarbon oxides show the need for more detailed theoretical considerations. Consider dibutforyl oxide, ( C4F9)20 ; it has a molecular weight of 454 and a boiling point of 101°. For compari
son, octforane, C4F9— C4F9, has a molecular weight of 438 and a boiling point of 104°. It is apparent that dibutforyl oxide molecules attract one another even less than octforane molecules. This should indicate very weak external molecular fields of force. The normal volume bond angles of the oxygen atom are not at 180°, so that a dipole moment for a com
pound in which carbon atoms are attached to this oxygen atom might be expected. This is not found. If the bond angles of the oxygen atom have been strained to 180°, which would ensure symmetry about the oxygen atom, then the strain produced should contribute to the external force field, and the properties of substance would be other than those found.
This compound shows only nominal solubility for acidic substances such as HC1 and B F3, is not itself soluble in acidic solutions, and is not subject to the ether-splitting reagents used in organic chemical reactions.
A liquid fluorocarbon such as pentforane or butforane has physical properties such as boiling point, freezing point, heat of vaporization, viscosity, dipole moment, and polarizability that are not greatly different from the hydrocarbon of the same carbon skeleton. Both casually and by the application of published theories, it might be expected that ideal or near ideal solution would be found for the fluorocarbon-hydrocarbon binary mixture. The actual deviations from Raoult's law are so great that a new concept, that of interpénétration, was advanced to provide an explanation. This is adding a new approach to the study of the liquid state. The properties of the fluorocarbons and their derivatives can be expected to contribute significantly to further theoretical developments not only for the liquid state, but throughout natural philosophy.
UTILITARIAN APPLICATIONS
Recently, there has been a very considerable amount of popular (8, 213) and semipopular (9, 10, 193) articles concerning fluorocarbons and their utilization for commercial purposes or consumer goods. The uses for these substances will, of course, come about because of the desir
able properties of the product. A discussion of what can be expected is given in a Chemical Engineering report (397). Although the present cost of fluorocarbons or fluorocarbon derivatives is $5.00 per pound and
340 J. H. SIMONS AND T. J. Â RICE
upward, this price can be expected to decrease as demand and production increase.
Developments in Methods of Producing Fluorochemicals
T H E ELECTROCHEMICAL PROCESS
The first commercial plant for the production of fluorocarbons and their derivatives by the electrochemical process was put into operation near Hastings, Minnesota, in October, 1951. This is a small plant produc
ing about 250 pounds of fluorocarbon product per day and using a 10,000- ampere cell operating at about 5 volts. This cell is about 6 ft. high and 4 ft. in diameter. The electrode pack consists of a series of alternate nickel anode and iron cathode plates spaced about % inch apart, and there is no separation of the anode and cathode regions of the cell.
Prior to the construction of the large commercial cell, a 2000-ampere cell had been operated continuously for several thousand hours without shutdown, with no indication of the accumulation of undesirable by
products, and with substantially no corrosion of the cell or electrodes.
Normally, the cell operates at about room temperature and atmospheric pressure, with some cooling provided by coils of tubing in the cell through which a liquid coolant is passed. The vapors from the cell consist of hydrogen, hydrogen fluoride, and gaseous fluorocarbons. A condenser strips the hydrogen fluoride from the gas stream and returns it to the cell.
The gaseous fluorocarbon products are subsequently removed from the hydrogen. The liquid products are more dense than the electrolyte and insoluble in it. They are removed from the bottom of the cell. The ease of separation of the products, either from the gas stream or as a liquid from the bottom of the cell, follows from the very convenient fact that fluoro
carbon derivatives are insoluble in liquid hydrogen fluoride whereas many organic compounds are very soluble. The fluorocarbons themselves are less soluble in the electrolyte than hydrocarbons.
This process is eminently suitable for large-scale industrial use. It is potentially a low-cost process in large installations, so that when the demand is sufficient, competitive prices will prevail. I t produces the product in one step continuously in a piece of equipment of very high volume efficiency. Either continuous or batch charging of raw materials is employed. It is highly flexible as many different products can be ob
tained from the same equipment by varying raw materials and process variables, although high yields of specific products can be secured.
The process is not one of fluorination, as elementary fluorine is not made in the cell even in minute quantities under normal operating condi
tions; it is certainly not "electrochemical fluorination." The products are fluorochemicals, i.e., members of the fluorocarbon domain, but they
are not "fluorinated," as they do not result from a process using fluorine in a fluorination reaction.
The cell produces a large variety of saturated fluorochemicals, cyclic and noncyclic (119, 398, 401), such as C F4, C2F6, C3F8, C4F i0, C5F i2, C6F i2, CeF i4, C7Fi6, CgFis, C9F20, and C i0F2 2; hydrogen-containing prod
ucts such as C H3F , C H2F2, C F3H , C2F4H2, C2F5H , C3F6H2, and C3F7H ; oxides such as C F3O C F3, C2F5O C2F5, C4F9O C4F9, C 5 F 1 1 O C 5 F 1 1 , C4F9- OCF3, C F3O C F2C F2O C F3, C2F5O C2F4O C2F4O C2F5, C e F n O C F , , C4F80 , and C5F1 0O ; acid fluorides such as CF3COF, C2F6C O F , C3F7C O F , C s F n C O F , C7FnC O F , C9F1 9C O F , C6FuC O F , and C6F n C F2C O F ; nitrides such as ( C F3)3N , ( C2F5)3N , ( C3F7)3N , ( C4F9)3N , ( C5Fn)3N , ( C F3)2N C6FU, ( C ^ N C F ^ F n , and ( C2F5)2N C3F7; and other sub
stances such as F C5H4N , C6F n N F2, C5F1 0N F , and C F3( C F2)7S F . I t has been shown that, from the acid fluorides, the fluorocarbon carboxylic acids can be easily prepared. By simple reactions the acids can be con
verted to aldehydes, 1,1-dihydroalcohols, 1,1-dihydroamines, bromides, iodides, and olefinic compounds. The iodides can be used to prepare Grignard compounds or mercury, phosphorous, sulfur, and other deriv
atives. The olefins, aldehydes, alcohols, etc. are also sources of other prod
ucts. Thus the electrochemical process will enable an entire syslem of synthetic fluorocarbon chemistry to be developed, as well as provide the source materials for hybrids between fluorochemicals and organic chemical substances.
A large variety of starting substances can be used to produce any given product. For example, C8F18 can be produced from C8H i8, C9H i0C O2H , C8Hl e( C 02H )2, C8H1 7N H2, C7H1 5C N , C8H1 7S H , C8H1 7O H , etc. A mix
ture of starting materials can be used, sometimes highly advantageously.
Several products can be made at the same time or several raw materials used to give the same product. The process variables can be altered over wide limits. In general, the process is highly flexible and highly adaptable.
I t should be emphasized, however, that the structure of the product or products may, and frequently are, quite different from the structure of the raw material.
The versatility of the process is such that compounds of widely differ
ent molecular weights, boiling points, and structure can be prepared in a single cell without changes of its physical construction and with only minor changes in processing equipment. The wide range possible is illus
trated in the fact that fluorocarbon carboxylic acids extending from C F3C 02H to C i3F2 7C 02H have already been produced, with cyclic, branched, or straight-chain structures and containing dibasic acids in the series. These have been produced from both aromatic and aliphatic starting materials (236).
342 J. H. SIMONS AND T. J. Â RICE CATALYTIC FLUORINATION
The catalytic fluorination process has been used extensively in the preparation of compounds containing reactive groups and large percent
ages of fluorine.
In an extension of work on acetone reported by Fukuhara and Bigelow in 1941 (131), Holub and Bigelow (222) have fluorinated several ketones over copper gauze at about 100°. The fluorocarbon ketones C F3C O C2F5 and (CF2)4CO and the mono and dihydro ketones
C4F7HO
and C F6H20 (from methyl ethyl ketone) were isolated in small yields, 15% in the case of C F3C O C2F5. Catalytic fluorination is superior to fluorination byC0F3
for the preparation of fluorocarbon ketones;C0F3
completely destroys the organic ketone.The preparation of C F3S F5 (10%) and C F7H S (15%) from C H3S H using silver-plated copper packing at 200° has been reported by Silvey and Cady (396). Bigelow and Tyczkowski (45) obtained C F2( S F5)2 from CS2 and F2.
A process for preparing trifluoroacetyl fluoride by the fluorination of acetone has been described by Haworth, Stacey, and Appleton (191). The reaction was carried out by mixing a 1:1 mole ratio of nitrogen and fluorine with slightly less than the calculated amount of acetone at 200°
in a reactor packed with gold-plated copper turnings. The yield of acid was about 22%.
The direct fluorination of acetyl fluoride by Miller and Prober (311) gave the mono and difluoro acid fluorides in a 6:1 ratio ; only traces of trifluoroacetyl fluoride were found. Cucolo and Bigelow (100) obtained C2F6N F2 and a corrosive gas C F2 = N F from acetonitrile and fluorine.
A series of aromatic compounds containing increasingly larger per
centages of fluorine have been fluorinated (138). Low temperatures, 66 to 92°, were used so that differences in the reactivities of the members of the series could be observed. The fluorination of 1,4-difluorobenzene was largely a combustion reaction; CeFu was the only significant product.
Benzotrifluoride, in contrast, suffered little combustion but formed a large amount of a viscous polymer.
1,4-Dimethforylbenzene and 1,3,5-trimethforylbenzene gave the most interesting results. Little fragmentation was noticed in either instance;
some polymerization (28%) occurred with the first compound, but virtually none with the latter. The products from 1,4-dimethforylberizene were chiefly C8 compounds containing five to six more fluorine atoms per molecule than the starting material. Because of the complexity of the mixture and the many structural and stereoisomers possible, it was found
difficult to assign definite structures to the compounds isolated. They can be accounted for by the process of addition of fluorine and hydrogen fluoride to the ring, followed by some replacement of hydrogen atoms by fluorine atoms. The products from 1,3,5-trimethforylbenzene were similar and equally hard to untangle. They were largely C9 compounds both saturated and unsaturated; they contained from four to seven more fluorine atoms per molecule than the starting material. The compounds isolated and the structures tentatively assigned are included in the table of compounds at the end of the chapter.
The vapor phase fluorination of trichloroethylene has been studied (181). The addition of fluorine and of chlorine fluoride were believed to be the important initial steps with the replacement of chlorine a minor initial step which also served to provide a source of chlorine fluoride.
Following the initial addition reaction, a variety of substitution reactions take place. The products were largely a mixture of seven two-carbon chlorofluorocarbons with smaller amounts of fragmentation and poly
merization products. The fluorination of trichloroethylene in the liquid phase has previously been reported; extensive polymerization occurred.
In a similar manner the products of the vapor phase fluorination of CHC1=CC1—CHF2 have been considered to arise from the initial addition of F2 or C1F followed by substitutive fluorination or chlorination (182). Under mild conditions CHFC1—CC12—CHF2 and CHC1F—
CFC1—CHF2 were the major products; more vigorous conditions led to CFC12—CFC1—CF3 (or its isomer CF2C1—CC12—CF3) and CF2C1—CFC1—CF3.
A series of papers by Smith and coworkers on the investigation of catalytic fluorination for the production of fluorocarbons carried on in England during World War I I have recently appeared (176, 177, 321, 322). They found, as had Cady and von Grosse (Vol. 1, p. 424), t h a t plated copper turnings made the best reactor packing for catalytic fluorination (321, 322). Gold plating gave the best results followed by cobalt, silver, nickel, and brass, all of which were good, and mercury, chromium, rhodium, and iron, which were poor (322). Musgrave and Smith (321) also found that fluorocarbons could be produced by the fluorination of carbides such as CaC2, T h C2, and UC2.
A further investigation of gold-plated packing led to the conclusion that better yields of fluorocarbons having the original carbon structure could be obtained using hydroaromatic hydrocarbons than by using the parent aromatic hydrocarbon (177). A 2 1 % yield of C8F i6 could be obtained from ethylcyclohexane while ethylbenzene gave only 9.6%.
These yields are lower than the 40 to 90% yields obtained by Cady using
344 J. H. SIMONS AND T. J. BRICE
silver-plated copper packing. Methforylbenzene gave polymeric products that could be stabilized with AgF2 to yield fluorocarbon oils and resins (415).
The gold plating serves an important function, perhaps that of a halogen carrier. The plating was slowly attached during the process and auric and cupric fluorides were carried to the base of the reactor, a process accompanied by diminishing yields of fluorocarbons.
The physical properties of a number of fluorocarbons were also reported (176, 179, 321).
T H E METALLIC FLUORIDE PROCESS
This process was used extensively during World War II for the produc
tion of fluorocarbons. (See Vol. I, p. 426.) The development of the cobalt trifluoride process in England has been reported by Haszeldine and Smith (178) and others (21, 278). They found it better suited to the production of fluorocarbons than catalytic fluorination. Higher yields of fluorocar
bons were obtained from unsaturated than from saturated hydrocarbons.
As expected, the process has not proved suitable for normal com
mercial production because of its great expense. It has been widely used on a laboratory scale, however, and a number of different classes of carbon-fluorine compounds have been prepared, chiefly fluorocarbon derivatives of N F3. Cobalt trifluoride is the fluorinating agent generally used. The yields of the fluorocarbon analog of the organic starting mate
rial are usually low.
The process has been used to prepare ( C F2)6N F from pyridine (0.2%
yield), c - C6F n N F2 from aniline (0.2%), ( C F3)2N F from dimethyl- aniline, and C F3N F2 from methylaniline and from methylamine (157).
A number of aliphatic tertiary amines gave the corresponding Φ3Í com
pounds (160). ( C F3)2N F was obtained in 40 to 70% yield by Thompson and Emeleus from trimethylamine (432).
A derivative of sulfur hexafluoride, C F3S F5, has been prepared from methyl mercaptan and from carbon disulfide; CS2 and C o F3 at 200 to 250° gave a 40% yield of C F3S F5 (396). By this process C6 and C6 fluoro
carbons were prepared for physical property studies by Cady and coworkers (19, 420).
Attempts to use cobalt trifluoride to prepare fluorocarbon ketones have been completely unsuccessful, only cleavage products being isolated.
At first glance, the metallic fluoride process would seem better suited to the fluorination of fragile molecules such as ketones, since it is a less exothermic process than direct fluorination. It has been postulated that the complete destruction observed was a consequence of the formation
of a complex of Co(III) and the carbonyl group (222). The a cleavage process then occurred and the organic fragments were fluorinated.
Attempts to prepare fluorine-containing sulfur and nitrogen compounds by the action of C o F 3 with thionaphthene and 2-methylindole were unsuccessful (316).
The heat of fluorination of C o F 2 to CoF3 and the heats of reaction of C o F 3 with hydrogen and with bis(methforyl)benzene have been measured (232). The results generally confirm previous estimates (see Vol. I, p. 428), although the reaction with ( C F 3)2C6H4 was complicated by side reactions.
T H E FLUORINATION OF CARBON
The reaction of fluorine with graphite was carried out under a variety of conditions by Rudorff and coworkers (373, 374, 375, 376, 377), and evidence for the formation of solids other than the well-known (CF)X ob
tained. Fluorine mixed with hydrogen fluoride reacted at room tempera
ture to give a solid whose analysis corresponded to (C4F)X. Its chemical properties were similar to those of (CF)X. The x-ray pattern differed from that of ( C F ) x , but the structure was similar in that the fluorine atoms were arranged in layers between two planes of carbon atoms.
The reaction of graphite with anhydrous H F in the presence of an oxidizing medium such as K 2C r 20 7 or fluorine yielded a blue material corresponding to [C24]
+
HF2~-4HF. The x-ray pattern indicated that the bifluoride structure was present (373).
The reaction of carbon with C1F3 at 350° and higher gave a mixture of fluorocarbons and chlorofluorocarbons such as CF2C12, C2F5C1, and C3F7C1 (95).
LIQUID PHASE FLUORINATION
Liquid phase fluorination has never been widely used because of the difficulties involved (see Vol. I, p. 420). A process utilizing the known tendency for polymerization during liquid phase fluorination has been developed for the preparation of fluorocarbons (418). Dilute fluorine was allowed to react at room temperature with a fluorocarbon solution of a hydrocarbon. Under these conditions aromatic and aliphatic hydro
carbons polymerized without much fluorination while compounds con
taining about 70% fluorine did not polymerize appreciably. Fluorine- containing substances such as benzotrifluoride were simultaneously polymerized and fluorinated. In this manner small quantities of fluoro
carbons boiling in the lubricating oil range have been made from benzo
trifluoride. The process is not suited to commercial production or to the preparation of pure compounds.
346 J. H. SIMONS AND T. J. BRICE EXCHANGE REACTIONS
There are three types of exchange reactions to be considered : S warts- type reactions, intermolecular rearrangements (disproportionation), and intramolecular rearrangements. The first two are well known, the third process has probably occurred in previous experiments but a critical examination of it has only recently appeared.
The Swarts reaction—the replacement of other halogens by fluorine using metal fluorides—has been used under increasingly drastic conditions and more extensive replacement is possible than has previously been reported. This is illustrated by the preparation of CF3Br from CBr4, SbF3, and bromine at 180 to 220° and 60 p.s.i. (438) and the replacement of the last chlorine atoms in chlorofluorocarbons by the use of S b F5 at 175° (35).
C r F3 has been found to be a superior catalyst in exchange reactions; com
plete conversion of chlorofluoromethanes to C F4 can be achieved with H F at 880° using it as a catalyst (36).
The use of aluminum halides in disproportionation reactions has been summarized by Park (Vol. I, p. 535). These processes are commercially feasible methods for making CF3C1, C F3H , and other fluorocarbon
derivatives.
Evidence for a companion reaction, the intramolecular rearrangement of chlorofluorocarbons in the presence of A1C13, has been disclosed by Miller et al. (308). In an experiment on the preparation of CF2C1CC13 from CFC12CF2C1 and A1C13, they discovered that in addition to the desired product (40%) they had made CF3CC13, an isomer of the starting material, in 50% yield. That the rearrangement was intramolecular was indicated by experiments using radioactive chlorine. When CF2C1CFC12 was treated with A1C13 containing radioactive chlorine, CF3CC13 was obtained in 20 to 30% yield and it did not contain any appreciable amount of radioactive chlorine. Evidently the rearrangement had pro
ceeded without chlorine exchange with the A1C13. AlBr3 also effects the rearrangement of CC12FCF2C1 to CC13CF3 (24). Attempts to fluorinate allylic chlorine compounds using antimony salts have led to rearrange
ment of the starting materials. Whether or not these rearrangements are intramolecular is unknown. All of the rearrangement reactions lead to compounds having the fluorine atoms bunched into the very stable C F3 and C F2 groups wherever possible.
I t has been known for a long time that A1C13 can replace chlorine for fluorine under drastic conditions. AlBr3 and All3 have been found to be more reactive than A1C13 (24). CF2C1CFC12 and AlBr3 yielded C2F3Cl2Br, C2F2Cl3Br, and CCl3CBr3; CF2—CF2—CFC1—CFC1 and C F2= C F C 1 were reported to be unaffected.
The Chemical and Physical Properties of Fluorocarbons
T H E CHEMICAL PROPERTIES OF SATURATED FLUOROCARBONS The remarkable stability of saturated fluorocarbons is amply demon
strated by the relatively few known chemical reactions. All of the chemi
cal reactions here reported are high-energy reactions.
Walker and Gibson (437) pyrolyzed C7F ie at 700 to 900° and found the products to be largely C i to C4 fluorocarbons both saturated and olefinic.
Rogers and Cady (367) found stainless steel and tungsten reacted rapidly with C F4 at 1000°, but that platinum was only slightly affected even up to 1500°; they then pyrolyzed n - C6F i2 by bringing it in contact with a platinum filament maintained at temperatures from 840 to 1325°, and examined the products. Noticeable reaction started at about 900°;
the products here were largely C3 and C4 compounds. Increasing tem
perature favored the formation of C2Fe, which was the dominant product above about 1150°. Very little C F4 was found at any temperature. The same products with some solid substance in addition was obtained from propforane, normal butforane, and cyclopentforane (419). The decom
position of propforane was found to be first order with an activation energy of 84 kcal.
Wilson (445) studied the hydrolysis of methforane in a nickel tube 0.5 inch O.D. and 24 inches long, coated on the inside with a 0.005-inch layer of gold. With about one-half per cent of water and 760 to 1000 mm.
of C F4 pressure, a slow and measurable reaction was found at 850°. The rate of reaction was found to follow the equation
where PHF , ŃΗ,Ο, and PCF4 are the pressures in millimeters of H F formed and of H20 and C F4 entering the reaction zone. Κ = 7.88 X 1 0 ~
n at 1123°K and 31.2 × 1 0 "
11
at 1148°K, giving an energy of activation for the reaction of 60 kcal. per mole C F4. The results of these studies indi
cated that the reaction was heterogeneous with some catalytic effect of the gold surface. As nickel, platinum, and copper were found to give greater catalytic effects for this reaction, a study of the homogeneous hydrolysis reaction does not appear possible in metal tubes. In the same apparatus Wilson found that propforane alone at one atmosphere pres
sure and 550° furnished slowly a product which gave fluoride ion in aqueous solution. Pentforane in the same equipment gave evidence of a similar reaction but at one-third the rate, and cyclopentforane at one twenty-fourth the rate. The nature of this reaction is obscure.
dt = K ( 2 P H
3O ) 2
( P C F
4) 2
'
;
2 . 3 3
348 J. H. SIMONS AND T. J. BRICE
The irradiation of a mixture of C 2F4 and mercury vapor with 2536 A light has been reported to produce cyclopropforane; insufficient evidence has been presented to properly judge the claim (11, 12).
An interesting experiment on the attempted reaction of C F 4 with excited hydrogen and xenon atoms has been described by Dacey and Hodgkins (101). H ( 6
3
P i ) and H ( 6
!
P i ) were without effect but X e ( 3
P i ) decomposed C F 4. The quantum efficiency with Xe(
3
P i ) was about 1; the products are described only as "fluorine" and an unidentified solid. They concluded that the energy necessary to break the first C—F bond is more than 154 and less than 194 kcal. per mole.
A study of the formation of methforyl radicals, C F 3, by the reaction of C F 3I, CF3Br, or CF3C1 with atomic sodium has led to the conclusion that the activation energies of the respective reactions are 1.7, 2.3, and 7.4 kcal., respectively (219). The molecular species C F 2 has been shown to have a half-life of one second or more under a designated set of experi
mental conditions (258).
T H E PHYSICAL PROPERTIES OF FLUOROCARBONS
The general aspects of the usual physical properties of fluorocarbons have been discussed in Vol. I, Chapter 13. Most of the information included here deals with the use of fluorocarbons in theoretical studies or with a type of information not previously reported. Most of the factual information is included in the table of compounds at the end of the
TABLE I
Properties of n-Butforane (4-12) and n-Pentforane (407)
Property n - C 4 F i0 n - C6F i2
Boiling point 270.96°K 302.4°K Freezing point 144.96 ± 0.05°K 147.51°K
Crystal transition 144.51°K Vapor pressure log Ρ = - 2 0 3 9 . 6 / Γ log Ρ = - 2 1 0 8 . 0 / Ύ
equation - 2.8024 log Τ - 4.9814 log Τ Ρ = mm. Hg. at + 26.1077 + 22.2092
0°C g - 980.665 Τ = °K Low pressure gas
equation PV(1 + 0.041P) - RT Liquid density D - 1.6484 D - 1.6195
Τ - °K + 3.18 X 10~
3
(259.88 — T) + 3.375 X 10"
3
(293.16 - T) - 0.0003 Χ 10"
β
(259.88 - T 7
) 8
- 6.374 X 10"
e
(293.16 - T)»
Molal volume 144.4 cc. at 259.95°K 177.8 cc. at 20°C Ev uncorrected 5480 cal. at 259.95°K 6081 cal. at 300°K Electron polarizability 9.47 A
8
10.94 A 3
Dielectric constant 1.68
349 chapter. The use of infrared spectroscopy in fluorocarbon chemistry is described in a separate section.
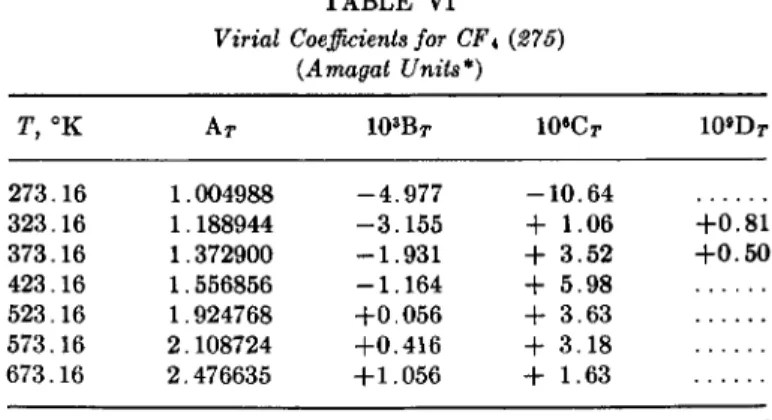
Two fluorocarbons have been made in very high purity and the properties determined. These are given in Table I.
T H E PURIFICATION OF FLUOROCARBONS
The problems of purification and of establishing criteria of purity are important in fluorocarbon chemistry, probably more so than in most other fields of chemistry. There are two kinds of purity with which we are concerned. First, as to whether the sample consists essentially of a single molecular species, and second, as to whether the sample contains impurities which will be harmful in the application for which the fluoro
carbon is intended. I t is quite possible to prepare a sample which, by the usual standards of purity, is essentially a single molecular species and yet contains enough harmful impurities to seriously impair its usefulness.
Conversely, a sample may obviously not be a single molecular species and yet contain such small amounts of harmful impurities as to be superior to the first sample. Both aspects of the problem of purity are important.
The definition of what constitutes a harmful impurity depends on the proposed use for the material. Saturated fluorocarbons and inert deriva
tives such as Φ3Í and Φ 20 , where Φ is a fluorocarbon radical, find uses chiefly because of their remarkable chemical resistance, low intermolecu- lar forces, and electrical inertness. Even minute traces, a few parts per million, of chemically unstable or polar impurities can nullify the ad
vantages gained by the use of the fluorocarbon.
Methods of purification have been refined and standards of purity, particularly in regard to polar impurities, have risen since fluorocarbons were first produced. The early methods of purification were a combina
tion of chemical and physical methods. The usual procedure was to refluorinate crude materials under increasingly drastic conditions, scrub out the acid gases, and refractionate. This method is not well suited to the purification of high molecular weight fluorocarbons because the condi
tions are so drastic that substantial amounts of material are destroyed.
Refluorination is not needed by the products from the fluorination of carbon or from the electrochemical process, since they are free of major amounts of polar impurities.
Chemical methods of establishing purity include the determination of the fluorine, carbon, and hydrogen content, and of the amount of hydrolyzable fluorine. Physical methods have also been used: refractive index, molar refraction, dielectric constant, dielectric loss, molecular weight, and constancy of boiling point are the chief properties measured.
350 J. H. SIMONS A N D T. J. Β RICE
In this manner a fair idea of the purity can be obtained ; questions as to the presence or absence of close-boiling isomers or of trace amounts of polar compounds still remain.
A physical procedure much used in the determination of purity is the analysis of cooling or melting curves. The amount of impurity can be determined to within about ± 0 . 0 1 % . Trace amounts of impurities remained undetected. Other disadvantages are (a) the method does not distinguish between harmful and harmless impurities and (6) the time and effort needed to obtain such data is usually prohibitively large.
Infrared absorption spectra provide a rapid method of establishing the purity insofar as a single molecular species is concerned and are also helpful in detecting fairly small amounts of some polar impurities.
The purification of n-heptforane has been carried out by Blumkin et al. (47). They found that careful fractionation of crude n - C7F i6 pre
pared by the cobalt trifluoride process gave a material of 98.5 mole % purity. Equilibrium melting in a low-temperature centrifuge converted C7Fi6 of 8 4 % purity to 98.4% purity. Selective adsorption of the impuri
ties, probably hydrogen-containing compounds, on silica gel increased the purity to 99.97 mole %. They considered that fractional crystallization of distilled material followed by silica gel treatment was the most prac
tical and efficient method of obtaining pure n - C7F i6. The purities were determined by melting point studies (327).
Similar conclusions as to the value of silica gel treatment were reached by Hildebrand, Fisher, and Benesi (215). However, even after silica gel treatment, mass spectrometric analysis of their C7F i6 showed the presence of impurities believed to be C7F15H and C7F i4H2. No determination of the percentage of impurity was made, but it must have been extremely low in view of the small amounts of such impurities detected by Oliver and Grigard after the silica gel purification of n - C7F i6 by Blumkin. Mass spectrometric analysis then offers a sensitive and rapid method of detect
ing such polar impurities in cases where the necessary equipment is available.
In summary, the purification of saturated fluorocarbons and the inert derivatives, like fluorocarbon oxides and nitrides, is best accomplished by careful fractionation of base-stable crude followed by a silica gel treatment. Fractional crystallization will be needed to separate out close- boiling saturated isomers, if it is necessary that they be removed. The purity can be established by analysis of cooling curves or by infrared spectra when a reference curve is available. Mass spectrometric analysis is also valuable in determining the purity of the sample. Electrical measurements—dielectric constant and dielectric loss—can be used to detect trace amounts of polar molecules.
351
SURFACE PROPERTIES OF FLUOROCARBONS
The surface tensions of a number of fluorocarbons have been reported in Vol. I, p. 438. Precise values for a number of fluorocarbons at various temperatures have since been reported by Rohrbach and Cady (368).
Their data are contained in Table I I .
TABLE II
The Variation of the Surface Tensions of Fluorocarbons with Temperature (368) Temperature in °C; surface tension in dynes per centimeter
n-C5Fi2 Iso-CsFn Cyclo-C
bFio n - C7F ie
0 . 4 0 ° — 1 1 . 7 3 6.23°—11.16 10.67°—10.72 15.95°—10.25 20.00°—9.87 2 4 . 9 5 ° — 9 . 4 2
2.43°—12.11 8 . 2 ° — 1 1 . 5 9 12.44°—11.21 17.43°—10.72 20.00°—10.48
10.5°—11.99 12.5°—11.82 15.1°—11.59 17.8°—11.32 2 0 . 0 ° — 1 1 . 0 9
0 . 0 ° — 1 4 . 6 9 2 0 . 0 ° — 1 2 . 6 9 31.05°—11.86 4 1 . 7 ° — 1 0 . 6 0 4 2 . 2 ° — 1 0 . 5 7
A discussion of the surface chemistry of fluorocarbons and their derivatives has been given by Scholberg (383). He points out that although the free surface energies of fluorocarbons are the lowest known, the Eotvos equation can be used to predict values which are approxi
mately correct; they are about 10% lower than the actual values. Schol
berg then goes on to report the results of experiments on the effects of surface-active fluorocarbon derivatives. Dilute solutions of long-chain fluorocarbon carboxylic acids were found to lower the surface tension of water to a greater extent than any other type of compound; values as low as 16 dynes per centimeter were obtained even at low concentrations of the fluorocarbon acids. I t is believed that the surface films are effec
tively gaseous. If this is so, it means that fluorocarbon surface-active compounds may not be very suitable for emulsion formation since the gaseous film will be easily deformed or broken. Experimentally this appears to be the case. Emulsions of fluorocarbons in water stabilized by fluorocarbon surface-active compounds break quickly.
Measurements of the interfacial tensions of fluorocarbons and water and of fluorocarbons and hydrocarbons were made. The interfacial ten
sion to water is high—about 55 dynes per centimeter; to aromatic hydro
carbons it is about 5 dynes per centimeter; and to aliphatic hydrocarbons it is somewhat lower. Since low interfacial tensions are required for the production of emulsions, it should be difficult to form emulsions of fluoro
carbons in water with the usual emulsifying agents. Agents having aliphatic chains should be a little better than those having aromatic
352 J. H. SIMONS A N D T. J. Β RICE
groups. In actual practice it was found that fluorocarbons were difficult to emulsify and that the emulsions were usually not very stable.
Toluene normally spreads on glass with essentially a zero contact angle. Very dilute solutions of the fluorocarbon carboxylic acids in toluene, about 0 . 1 % , cause the angle of contact with glass to be finite.
This is true of acids having more than four carbon atoms. The fluoro
carbon film adsorbed on the glass tends to cause the toluene droplet to contract rather than spread out and wet the glass. This could mean that when a well-oriented, close-packed fluorocarbon interface can be made, it will be repellent to most substances. One way of looking at this is to consider that the low molecular attraction between the fluorocarbon films and the impinging liquid results in little free energy drop on con
tact; this drop is less than the energy that must be supplied to spread the impinging liquid, so it does not spread. There is ample qualitative evi
dence to support this viewpoint. Water, carbon tetrachloride, and aromatic compounds show a large contact angle on a Teflon surface and are thus nonwetting. This nonadhesiveness of Teflon, and of other fluoro
carbon polymers, is of considerable commercial importance and will be increasingly so as these polymers become more widely used. Fox and Zisman (127) have also found Teflon to be an excellent low-energy surface for the study of contact angles and wetting relations of a solid with a wide variety of liquids.
A surface coated with an oriented monolayer of C 9 F 1 9 C O 2 H was found to be even more resistant to wetting than Teflon (385). This is believed to be due to the presence of a surface of essentially methforyl groups. The values of the contact angle of a number of organic substances were meas
ured. It was estimated that the free surface energy of such a film does not exceed 25 ergs per square centimeter.
VISCOSITY
The viscosities of a number of fluorocarbons and fluorocarbon deriva
tives have been measured over ranges of temperature and the data treated by Eyring's rate theory of liquid flow (59, 69). The viscosities of the fluorocarbons having five carbon atoms per molecule are in the order, cyclopentforane > isopentforane > normal pentforane. Fluorocarbons have higher activation energies for viscous flow than hydrocarbons: Evin for normal octforane is 3.24 kcal. per mole while that for normal octane is 2.31. This is related to the higher temperature coefficient of viscosity found for a pure fluorocarbon over the hydrocarbon of the same carbon structure.
The fluorocarbon oxides have lower viscosities and also lower activa
tion energies for viscous flow than fluorocarbons of the same number of
353 carbon atoms. This similarly with other properties of the fluorocarbon oxides is difficult to correlate by present theories.
ROTATIONAL ISOMERISM
The existence of rotational isomers of saturated, straight-chain fluoro
carbons has been demonstrated by Szasz (424) and the energy differences between them calculated. To do this the infrared spectra of n - C e F ^ , n-C«Fi4, and n - C7F ie in the region 9 to 13/x were obtained for the vapor, liquid, and solid phases. A striking simplification of the spectrum occurs abruptly at the freezing point; this permits the separation of the bands belonging to the most stable isomer, which is considered to be the only one present in the solid, from the rest of the spectrum. In addition, quantitative intensity measurements were made of selected pairs of lines belonging to different isomers in the liquid phase. The results of these measurements are summarized in the accompanying Table I I I .
TABLE H I
The Energy Differences between Rotational Isomers in Liquid Fluorocarbons ~(4®4)
Compound
Line pairs, c m .
-1 Temperature range, °K
Energy difference, cal./mole, — ΔΗ 834-881 150-220 460 ± 100 990-1022 150-220 350 ± 150
n-CeFu 795-818 195-280 600 ± 150
818-833 195-280 580 ± 150 n-C7Fie 1030-1058 220-330 600 ± 100
I t was assumed that the extended, zigzag form represents the most stable configuration and is the only one present in the solid. The straight forms of the fluorocarbons are nonpolar, while the " b e n t " forms will possess permanent electric dipoles. This would tend to stabilize the bent forms in the liquid as compared to the vapor. However, the similarity of the vapor and liquid spectra shows that the rotational isomers are present in nearly the same relative concentrations in the two phases.
This observation is in agreement with the fact that the energy differences between the isomers, as shown in the table, are not large.
SOLUBILITY
Experimental data on the solubility properties of fluorocarbons have appeared and theoretical treatments of the data have been made. Fluoro
carbons can be expected to contribute significantly to solubility theories and knowledge of the liquid state. Early studies of liquids of low dielectric constant soon showed that ideas of the liquid state based upon the prop-
354 J. H. SIMONS A N D T. J. B R I C E
erties of water were biased. Water was far from a usual or normal type of liquid. Hydrocarbons available in great variety soon came to be con
sidered the normal for comparison for other liquids or for solubilities.
The fluorocarbons have now shown hydrocarbon liquids to possess properties making them undesirable as the ideal for comparison. Actually the liquified inert gases would be the superior comparison substances, but the fluorocarbons approach them as closely as can be expected of any polyatomic substances. The fluorocarbons being more readily available than the inert gases and having greater variety and higher boiling points will certainly be the best secondary standard for liquid state and solu
bility studies. The variety of molecular structures available will enable the structure contributions to liquid properties to be evaluated unhin
dered by the "interpénétration" recently shown (407) to be prevalent in hydrocarbon liquids.
The solubilities of nitrogen and chlorine in a number of liquids, includ
ing fluorocarbons, have been measured by Gjaldbaek and Hildebrand (139, 140). The solubilities of nitrogen expressed as mole fraction × 10
4 are: in n-heptforane, 0°, 40.1; 25°, 39.1; 50°, 38.0. In methforylcyclo- hexforane, 25°, 31.8; in dimethforylcyclohexforane, 25°, 33.0; 50°, 31.9.
By comparison the solubility of nitrogen in n - C6H i4 is 14.0 at 25°; in benzene, 4.48; and in carbon disulfide, 2.23. The solubility of nitrogen in the fluorocarbons is thus much higher than in organic liquids. This is reasonable only when the interpénétration of the organic liquids is taken into consideration. Nitrogen and fluorocarbons both have low inter- molecular forces, approaching those of the rare gases, and will be com
patible with one another. The organic substances will tend to "squeeze o u t " the nitrogen molecules. Hildebrand has adjusted, on a semiempirical basis, the solubility equation for regular solutions so that calculated solu
bilities are in fair agreement with the experimental values except for fluorocarbons. Nitrogen has been assigned a F of 53 cc. and δ2 of 5.2. I t is possible to calculate fairly well the solubility of nitrogen in nonpolar solvents from the δ value of the solvent by interpolation of the values for known solvents. The failure of fluorocarbons to fit the adjusted solubility equation is partially attributed to their larger molar volume than hydro
carbons; a correction term based on this idea slightly lessens the dis
crepancy but does not remove it.
The solubility of chlorine in n-heptforane is 11.0 mole % at 20° and 9.77 mole % at 25°. The partial molar volume at 0° in n - C7F ie is 49.4 cc.
and 52.7 cc. at 25°; these values are close to the values for liquid chlorine at the same temperatures (48.4 and 50.8). They show that the chlorine molecules do not pack together in the pure state ("interpenetrate") or
" e x p a n d " when in dilute solution in the fluorocarbons as do hydrocar-
bons; the latter point will be considered in detail later. The solubility is less than ideal, which is attributed to the great difference in internal pressures (ä2 = 9.20 for chlorine, 6.10 for n - C7F ie) . Chlorine forms virtually ideal solutions with substances having about the same solubility parameter.
The solubilities of n-heptforane with benzene, carbon tetrachloride, chloroform, ç-heptane, and 2,2,4-trimethylpentane were measured by Hildebrand, Fisher, and Benesi (215). The critical solution temperatures obtained from the liquid-liquid solubility curves were 113.5°, 58.7°, 78.5°, 50.0°, and 23.7°, respectively. The solubility parameter differences calculated from these experimental results were in agreement with the values predicted by solubility theory except for the two saturated hydrocarbons.
Simons and Linevsky (409) determined the solubility of naphthalene, paranitrotoluene, and hexachloroethane in dibutforyl oxide and triprop- foryl nitride. Naphthalene was soluble to a mole fraction of 0.00257 in the oxide at 25° and 0.00402 at 35°; 0.00300 in the nitride at 25° and 0.00461 at 35°. This compares with a solubility of 0.190 mole fraction in dibutyl ether at 25° and 0.304 at 35°, and also the ideal solubility of 0.299 at 25° and 0.384 at 35°. Thus, despite the dipole moment of the ether, naphthalene is more nearly ideally soluble in it than in the fluorocarbon oxide or nitride. The authors hold "interpénétration" of the hydrocarbon molecules responsible. They find nitrobenzene soluble in (C4Ffl)20 to a mole fraction of 0.00268 at 25° and 0.00399 at 35°; in ( C3F7)3N , 0.00323 at 25° and 0.00515 at 35°, compared with ideal solubilities of 0.571 at 25°
and 0.712 at 35°. Again the deviation is large and is assumed to be caused by interpénétration of the molecules of the organic substance. Hexachloro
ethane was found to be 0.00901 mole fraction soluble in (C4F9)20 at 25°, 0.0148 at 35°; in ( C3F7)3N , 0.0111 at 25° and 0.0160 at 35°. These values are nearer the ideal, which are 0.0526 at 25° and 0.0709 at 35°. From the difference in solubility at two temperatures, the heat of solution was calculated. For naphthalene in (C4F9)20, the heat of solution was 8150 cal. per mole, in ( C3F7)3N , it was 7850. This is compared with the ideal value of 4580. For paranitrotoluene in (C4F9)20, the heat of solution was 7320; in ( C3F7)3N it was 8620. This is compared with the ideal value of 4010. The excess of these values above the ideal gives a rough measure of the heat of interpénétration.
The problem of the treatment of the data on solutions of fluorocarbons with hydrocarbons is of considerable theoretical interest. Hydrocarbons are not as soluble in fluorocarbons as calculations based on the solubility equation of Hildebrand would predict. I t has also been noted that satu
rated hydrocarbons are better solvents for iodine and phosphorus than
356 J. H. SIMONS A N D T. J. B R I C E
would be expected from their solubility parameters. The difficulty thus lies in the irregular nature of hydrocarbons. Hildebrand (214) has sug
gested that for practical purposes the solubility parameter for n-heptane be arbitrarily taken as 8.1 instead of the calculated value of 7.45 (the square root of the heat of vaporization per cubic centimeter). If this is done, the solubility equation may be used to calculate hydrocarbon solu
bilities with fair success. Simons and Dunlap (407) also place the blame for solubility difficulties on the hydrocarbons and suggest the concept of
"interpénétration" in order to arrive at satisfactory correlations with experimental fact.
These authors made a complete study of the binary system n-pent- forane and n-pentane with pressure-composition diagrams determined at five temperatures, the liquid phase temperature-composition diagram, volume changes on mixing, and related information. Large positive deviations from Raoult's law were found that could not be accounted for by the difference in internal pressures of the pure components or by use of existing theories of nonpolar substances. The critical unmixing tem
perature calculated by use of these theories from the physical properties of the pure components is 62°K, whereas the experimentally determined value is 265.5°K. As the two components have physical properties and apparent structure so nearly alike, an explanation for the nonagreement of theory and experiment is necessary. Because the great difference in molecular weight of the two components made very precise determina
tions possible, experimental error cannot be advanced as the reason. In order to arrive at a correlation, the authors advanced the concept of interpénétration. This envisioned a meshing or interlocking of the hydro
carbon molecules because of the small apparent size of the hydrogen atoms. This interpénétration was assumed to be small or nonexistent between fluorocarbon molecules or between the molecules of the unlike components. This concept was successful in treating the data both qualitatively and quantitatively, even to the extent of providing an explanation for the great difference in the velocity of sound in fluoro
carbon and hydrocarbon. Simons and Mausteller (412) repeated these experiments for the binary system n-butforane and ç-butane. These pure components are even more alike in phj'sical properties than the five carbon atom substances. The results obtained, however, are quite parallel to those of the previous study and the correlation on the basis of inter- penetration is equally successful.
The system WFe-c-C6Fio is an interesting one to study. The two com
pounds have similar boiling points and molecular weights, which indi
cates that there is a close similarity in the intermolecular forces. They might then be expected to form close to an ideal system despite the great