CHAPTER 2
Physical Chemistry of Fluorocarbons
B Y T . M . R E E D I I I Department of Chemical Engineering The University of Florida, Gainesville, Florida
I. Introduction • • • • 133
II. Vapor Pressures 135 I I I . Interaction of Polyatomic Molecules 141
IV. Virial Coefficients and Equations of State for Gases 147
V. Critical Constants 1 55
V I . Equation of State for Liquids 158
V I I . Liquid Compressibility 1 66
V I I I . Compressibilities of Liquid Mixtures • • • • 171
I X . Acoustic Velocity 173
X . Phase Transitions I75
X I . Liquid Viscosities 1^1
X I I . Gas Viscosities I86
X I I I . Viscosities of Liquid Mixtures I86
X I V . Surface T e n s i o n s I88
X V . Surface T e n s i o n of Mixtures 1 91
X V I . Polarizabilities and Ionization Potentials 192 X V I I . Phase Equilibria of Mixtures
A . V a p o r - L i q u i d Equilibria 197 B. Solubility of Iodine 205
C. Heat of Mixing 8 2 0
D . L i q u i d - L i q u i d Equilibria 210
E . Gas Solubility 211 F. V o l u m e Change or M i x i n g 214
G. Effect of Multicenters o n Interaction 218 X V I I I . Separations of and with Fluorocarbons 219
X I X . T h e r m o c h e m i c a l Data 221
Bibliography 231
I. Introduction
The study of fluorocarbons has not received the attention that the circum
stance of nature and the affairs of the modern economy have allowed the study of hydrocarbons. Other than the superficial one arising from the presences of carbon atoms in chains in the molecules, there is fundamentally no more similarity between hydrocarbons and fluorocarbons than among hydrocarbons and any other compounds of the 100-odd elements. It has
133
134 T. M. REED III
been known for many years that compounds containing hydrogen, although commonly encountered in chemistry, are not representative of compounds that do not contain hydrogen; and were it not for its extreme familiarity, the properties associated with hydrogen would be regarded as spectacular.
Because of its fundamental and familiar role in chemistry the most en
thusiastic description of hydrogen and its compounds amounts to putting it in a class by itself. Quantum mechanics has shown us that hydrogen together with helium are indeed in a class by themselves primarily be
cause of their small mass, small size, and small nuclear charge. These un
usual properties are manifested to a more or less degree in compounds containing hydrogen. Were it not for the fact that fluorine is the most electronegative of all the elements, its compounds would be ordinary.
They are in fact not unusual in their physical properties compared with compounds not containing hydrogen, but they are extremely stable chemically because of the presence of fluorine.
Setting aside their economic importance, hydrocarbons are interesting in the study of intermolecular interaction in that they allow a wide latitude in molecular structure while retaining rather simple and uniform chemical constitution. But because of the peculiar properties of hydrogen, they are not the most general type of molecule to use in such a study. Fluorocarbons are much more to be desired in an initial attack on the effects of molecular structure on physical properties. This idea has been substantiated in recent years by studies of the physical properties of pure fluorocarbons and of fluorocarbons in mixtures with other kinds of molecules. In this chapter these data are presented and discussed briefly.
A few properties of fluorine are included in this chapter and more of them are discussed in Chapter 1. The physical and molecular properties of fluorine are reviewed comprehensively by Wicke and Franck<1 7 6). This article should be consulted for further references to these properties.
The fluorocarbon alkanes, or alkforanes, are particularly interesting nonpolar compounds. These molecules possess ionization potentials that are significantly larger than ionization potentials for hydrocarbon mole
cules. Experimentally, solutions of fluorocarbons with hydrocarbons exhibit unusual properties manifested by very nonideal behavior. Through the theory of dispersion forces, these unusual properties have been success
fully explained as arising specifically from the difference between the ionization potentials of the hydrocarbon and the fluorocarbon species.
In the first approximation, the details of molecular structure appear to be unimportant variables in the fluorocarbon molecule, but very important variables in the hydrocarbon molecule. The degree to which these approxi
mations are actually valid, and the deviations produced by structural effects, offer an important and interesting approach to a study of mixtures
PHYSICAL CHEMISTRY OF FLUOROCARBONS 135 of these molecules. The initial attack should be made on the properties of the pure compounds themselves, for it is the ultimate aim to be able to derive the properties of mixtures from those of the constituent molecules.
The premise is that the properties of the pure materials contain all the in
formation which, when combined with the appropriate theoretical model, will yield the properties of any particular mixture. Fluorocarbons are im
portant in this respect, since essentially they are nonpolar molecules. As such, they represent the simplest examples of molecular interaction associ
ated with a variable structure.
From the theoretical view point the physical properties of matter, other than those held in common with the perfect gas, follow from the statement of the interaction energy between molecules as a function of distance between centers of interaction and of relative orientation of the molecules with respect to one another. The simplest statement of this kind applies to the mutual potential energy, e(r), of a pair of centers as a function of the distance r between centers. The most commonly used potential function reads
e(r) = [€*/(» - m)][m(r*lr)n - n(r*jr)ml (la) or alternately,
€(r) = [€*/(» - m^nn/mmy'ln-m^alr)* - (a/r)™]. (lb) The numbers n and m are the exponents on r-1 corresponding to forces of repulsion and of attraction, respectively, and these potential functions are called (w, m) potentials. In the general case n and m are functions of r.
The parameters €*, r*9 and a are defined as e = — €* and r = r* when de/dr = 0, and r = a when e = 0 and r < r*. The relationship ( r * / a )n _m
= njtn holds. These parameters ultimately depend upon the molecular structure, but are usually treated as empirical correlating parameters obtained from indirect measurements of the mutual potential energy. Such measurements are vapor pressure, virial coefficients, viscosity, and com
pressibility. When the (w, m) potential is (12, 6), it is called the Lennard- Jones potential function:
€L_j = €*[(r*/r)i2 - 2(r*/r)«] = 4e*[(a/r)i2 - (f f/r) 6 ] . (2) This function has been used extensively in correlating properties of mole
cules. It is really successful only for monatomic molecules, which are non- polar and are spherically symmetrical and thus have no mutual orientation effects.
II. Vapor Pressures
A theoretical model which has been successfully applied to liquids and solids of nonpolar molecules is the cell model of Lennard-Jones and Devonshire<94>. This model considers the motion of a molecule within a
136 T. M . REED III
spherical cell defined by adjacent molecules. The value of the lattice energy of a mole of molecules at absolute zero, relative to an energy zero at infinite separation, when all the molecules are located at the center of their spherical cells, is given in this theory as
C/0(12, 6) = (^€*/2)[1.0(«*/i?)4 - 2.4(^*/^)2] (3a) for the (12, 6) potential function, Eq. (2), v* is equal to \/2 (r*)3, and v
is the volume assigned to one molecule. The coefficients 1.0 and 2.4 account for contributions to the lattice energy from repulsive and attrac
tive interactions, respectively, between nearest neighbors as well as be
tween more distant neighbors. The number of nearest neighbors is zy and N is Avogadro's number.
For a (28, 7) potential function, which applies to quasi-spherical molecules such as CH4, CF4, and SF6, the analogous lattice energy is given <64) by this model as
C/0(28, 7) = (Nz€*l3)[0.5(v*lv)M* - (3b) A form of the vapor pressure-temperature relationship that may be
derived from this model, as well as from the kinetic theory model and from the classical oscillator model(m>, is
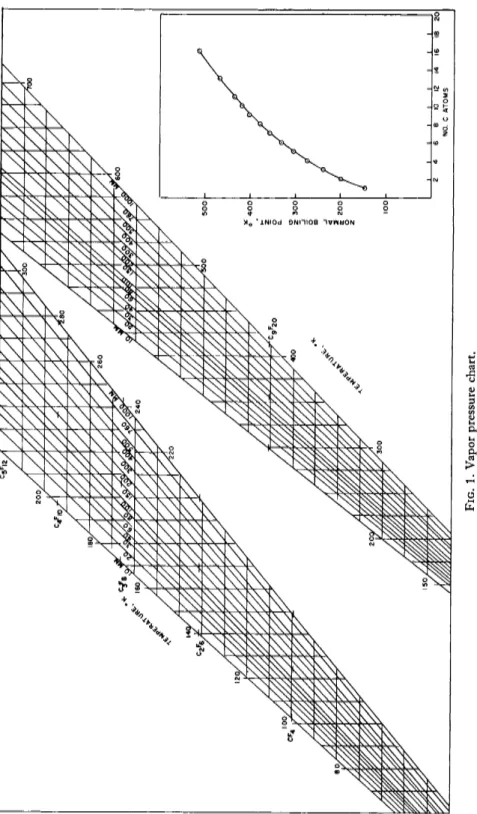
log P = a + b log T + cjT (4) The vapor pressure as a function of temperature has been reported for
several fluorocarbons below ten carbon atoms per molecule. The empirical relationships are given in Table I. Figure 1 may be used to estimate vapor pressures of fluorocarbons. The coefficient c in Eq. (4) is particularly interesting in that it gives a value for the potential energy (or lattice energy) of a molecule in the condensed phase. From either the kinetic theory, the oscillator theory, or the cell theory
2.303/fc = - (Ug- U0), (5) where Ug and Uo are the molar potential energies in the gas phase and
in the liquid phase, respectively, at absolute zero of temperature, and R is the perfect gas law constant. If we take Ug as zero,
2.303 Rc = U0y (6)
which may be regarded as an experimental value for Uo. Rough values for a, b, and c, have been estimated for some fluorocarbons and for some hydrocarbons as shown in Table II.
The cell model has been modified by Prigogine and collaborators*131) to apply to so-called r-mer molecules. An r-mer molecule is one com
posed of repeating identical units in a chain each unit being of volume v.
The lattice energy Uo is then written as
Uo = (Nqzc^l2)[\.^lvf - 2A{v*lvf]y (7)
FIG. 1. Vapor pressure chart.
TABLE I VAPOR PRESSURE EQUATIONS (P in mm Hg; logarithm base = 10; Tin °K; (1) = liquid; (s) = solid) Compound logP T range P range Reference CF 4 (1) 5.04420 - 701.73/T + 1.75 log T - 0.0076715T 104-146 10-800 149 C 2F 6 (1) (a) 7.376 - 875.7/T 174-196 210-790 150 (b) 14.22568 - 1125.329/T - 2.33917 log T - 0.00109858T 195 760 122 C 2F 6 (s) - 2227.0/'T - 0.0915 log T - 0.05161 T + 24.307 — — 122 CaF 8(l) 247.06220 + 0.099584T - 102.0199 log T - 6,044.6163/T 181-238 24-800 32 «-C 4Fio (1) (a) 54.4313 - 2589.94/T - 18.5852 log T + 0.0118856T 233-383 125-16,400 18 (6) 26.1077 - 2039.6/T - 6.4539 log T — — 157 W-C5F12 (1) (a) 6.88375 - 961.192/T - 75512.4/T2 286-338 400-2400 5 (b) 22.2092 - 2108.0/ T - 4.9814 log T — — 155 (c) 57.57966 + 0.012276T - 19.61098 log T - 2948.5076/T 221-303 9.3-784 32 iso-C 5Fi2 (1) (a) 6.77319 - 904.909/T - 83581.8/T2 289-338 450-2300 5 (b) 23.69191 - 5.47602 log T - 218.95414/T 228-305 15.8-820 32 «-C 6Fi4 (1) (a) 6.8637 - 1075.6/(T - 60.34) 284-342 116-1121 164 (b) 6.89190 - 1090.52/(T - 58.34) — 273-766 39 (c) - 182.17626 - 0.062970T + 78.32742 log T + 2825.8877/T 256-333 24-844 32 2-(CF 3)C 5Fii(l) (a) 6.8825 - 1093.77/(T - 57.59) 277-342 81-1071 164 (b) - 45.38399 - 0.022957T + 23.07089 log T - 751.08410/T 253-332 20.3-792 32 3-(CF 3)C 5Fii(l) - 5.29114 - 0.010831T + 6.77420 log T - 1759.2707/T 244-328 10.8-691 32 2, 3-(CF3)2C 4F 8(l) 800.65548 + 0.214365T - 316.48588 log T - 23,585.531/T 275-333 67-780 32 W-C7F16 (1) (a) 6.96493 - 1196.067/(T - 62.80) 271-379 17-1529 120 (b) 8.0166 - 1824/T 293-355 61-760 62 C9F20 (1) 8.0534 - 2072/T 355-401 160-760 135 W-C16F34 (s) 14.61 - 5464/T 288-303 4.8 x 10"5 -15 38 x 10-5 138 T. M. REED III
C2F4 (1) C 3F 6 (1) cyclo-C4F8 (1) cyclo-CsFio (1) cyclo-CsFio (s) cyclo-CeFi2 (1) cyclo-CeFi2 (s) cyclo-(CF3)C 6Fn(l) cyclo-(C2F5)C6Fn (1) F 2 (1) NF 3 (1) SF 6 (1) SeF 6 (s) TeF 6 (1) TeF 6 (s) WF 6 (1) WF 6 (s) 7.618 - 931.9/T 7.44806 - 1060.757/(T - 10.66) (a) 14.75709 - 2025.845/JT - 21.85656 x 10"3 T - 2.16552 x 10-5 T2 (b) 6.70267 - 1315.906/T + 8.778482 x 10"3 T - 1.739691 x 10"5 T2 (a) 6.98522 - 1013.820/T - 59041.9/T2 (b) 38.55794 - 10.66588 log T - 2756.7223/T - 1.57895 + 3.55788 log T - 1270.2262/T (a) 7.59819 - 1527.30/T (a) 8.67643 - 1889.12/T (b) - 195.30623 - 0.063257T + 83.118305 log T + 3232.6375/T (a) 7.8645 - 1740.7/T (b) 7.75062 - 1702.26/T (c) 2.88081 + (1 - 349.456/T)(0.907847 - 10.32816 x 10~4 T + 10.92653 x 10-7 T2 ) 2.88081 + (1 - 374.135/T)(0.921458 - 10.09293 x 10"4 T + 10.0873 x 10-7 T2 ) 7.08718 - 357.258/T - 1.3155 x 1013 /T8 (a) 6.77966 - 501.913/T - 15.37) (b) 7.15285 - 613.33/T (c) 4.64615 - 1.75 log T - 662.05/T + 0.0066007T 7.26927 - 899.46/ T 8.295 - 1231/T 7.091 - 988/T 8.594 - 1342/T 6.88699 - 928.580/T - 67,942.9/T2 (a) 8.1847 - 1533.1/T (b) 10.0682 - 2032.2/T 141-192 10-567 150 232-293 460^900 175 267-388 760-20,800 36 177-274 4.9-985 52 290-329 600-2300 5 285-296 516-793 32 229-281 19.5-425 32 336-394 1140-5320 146 293-333 169-1014 146 252-325 15.9-758 32 272-350 30-760 62 313-353 205-850 146 309-384 149-2026 59 311-411 71-2026 59 53.5-90 1.6-1220 76 89-144 1-764 79 148-233 1060-33,000 79 80-145 0-2-773 109 273-Tc 9500-P c 27 194-226 93-698 85 194-241 48-963 85 194-241 48-963 85 285-324 620-2400 4 265-276 — 4 -265 — 4
PHYSICAL CHEMISTRY OF FLUOROCARBONS 139
140 T . M . R E E D I I I
which is the same as Eq. (3a) except for the factor q. ei* is a constant for the homologous series. The number of first nearest neighbor pairs of the r-mer molecule is qz, given by
qz = rz - 2r + 2 (8) where r is the number of identical repeating units in the molecule, and rz is
the number of nearest neighbor pairs to one unit when it is not in the chain. The term 2r assumes that connecting a unit in a chain eliminates 2 nearest neighbor pairs, and the term 2 is added to account for these two neighbor pairs that remain at the ends of the chain. In the fluorocarbon alkanes the unit is taken as C F 2 — C F 2 , and CF4 is assumed to be equivalent to a C F 2 — C F 2 unit. A molecule of formula C7 ZF2n+2 possesses 1 + (n—1)/2 units. That is, (n— l)/2 units as ( C F 2 ) 2 plus CF3 and F which combined are equivalent to CF4, or one additional unit. The parameter ei* is sup
posedly the same for all r-mers in the series. It is the minimum mutual pair potential energy for a unit, and is approximately equal to €* for the first member in the series. In the hydrocarbon series (ref. 131, p. 343) e* increases slightly with increase in molecular size or number of C H 2 units.
For the fluorocarbon alkanes the first member is CF4 for which e*/& = 152.5°K<75) where k is Boltzmann's constant. The value of ei*/& for other members in the series may be calculated in the following way.
T A B L E I I
APPROXIMATE CONSTANTS I N logio Pmm = a + b l o g i o T + c/T
Molecule Boiling point a -b —c
(°K) ( ° K )
F2 85.0 10.37 1.4 407
C F4 145.2 10.5 1.2 730
C2F6 194.9 13.3 2.2 1060
C3F8 236.5 15.8 3.0 1390
«-C4Fio 271.2 18.4 3.8 1720
w-C5Fi2 302.5 21.1 4.6 2060
«-C6Fl4 330.3 23.7 5.4 2390
W-C7F16 355.7 26.3 6.2 2720
CH4 111.67 10.73 1.53 519
C2H6 184.53 13.64 2.38 988
C3H8 231.09 15.44 2.91 1315
« - C 4 H i o 272.66 18.15 3.76 1667
n-CsHi2 309.23 19.44 4.10 1964
W-C6H14 341.90 20.78 4.46 2253
n-C?Hi6 371.59 22.48 4.95 2555
PHYSICAL CHEMISTRY OF FLUOROCARBONS 141 The vapor pressure approximated from the cell model gives*94) for the constant c in Table II,
- 2.303/ec = 0.678j*€i*. (9) For CF4, c = 730°K from Table II, and qz = z. A value for z is obtained
as approximately 16. The values of €i*/k for the fluorocarbon alkanes com
puted from the respective values of c and the above relationships with z = 16 are as follows:
r-mer C F4 C2F6 C3F8 C 4 F 1 0 C5F12 C6F i 4 C7F16
€l*jk, °K 155 156 157 158 159 159 159
qz 16 23 30 37 4 4 51 58
The constancy of the values indicates that this method holds some promise for treating fluorocarbons as successfully as it has treated hydrocarbons.
Extensions of this theory have been applied to mixtures containing only hydrocarbons. Mixtures of fluorocarbons with hydrocarbons have yet to be examined in this way.
The corresponding state behavior of fluorocarbons and other fluorine containing molecules demonstrates that they are farther from the behavior of spherically symmetrical molecules (the noble gases) than are the analogous hydrocarbon molecules*1 4 6*. Corresponding state treatments, however, preclude the investigation of details in molecular structure which are so important in comparing fluorocarbons with the more familiar hydrocarbon analogous.
III. Some Details of the Interaction of Polyatomic Molecules There is a theoretical basis in quantum mechanics for the exponent 6 in the attractive term for single-center, single-center interaction or more strictly for interactions between monotomic molecules. For such interactions a theoretical value for €* may be obtained from Slater and Kirkwood's expression for dispersion energy attraction together with the pair potential energy function, Eq. (2). The dispersion energy is the only attraction energy between nonpolar molecules. Neglecting second and higher order terms, the dispersion formula for the attraction energy €A between like molecules is given by Moelwyn-Hughes*1 1 1) as
€A= - 3v*Iy/Zlto*> (10)
where Z is the number of electrons in the outer shell, / is the ionization potential, and a is the polarizability at zero external field frequency, of
142 T. M. REED III
each of the molecules interacting, and r is the distance between molecular centers. From Eq. 2 when r = r* = 21 /6 a,
€A(r*)= - 2e*. (11)
From Eq. (10) with r = r*, and Eq. (11),
€* = 3a2/V^/16<A (12)
This relationship requires that the product of the experimental numbers e* and a6 divided by the molecular properties combined as 3 a2//16 should be equal to V'Z. In the Table III it is seen that Vz is approximately unity for H 2 and He. For somewhat larger atoms €*a6 is two to three times 3 a2//16, and for still larger atoms and molecules the ratio of e*cr6 to 3a2//16 increases with the size of the molecule. In the monatomic and diatomic molecules VZ ranges from 2 to 3. A value of 2.45 corresponds
3 2 0 0 1 1
0I . I 1 I I I I I I L_
0 4 8 12 o 16 2 0
P O L A R I Z A B I L I T Y , A3
F I G . 2 . Vapor pressure constant, c, of Eq. ( 4 ) .
P H Y S I C A L C H E M I S T R Y O F F L U O R O C A R B O N S 143
V Z F R O M E Q . (12)
Molecule e*lk
( ° K )
a ( A )
I (ev)
a ( A3)
VZ
H e (Q) 10.22 2.556 24.46 0.2036 1.22
H e (cl) 6.03 2.63 0.90
H2 (Q) 37.0 2.928 15.43 0.2023 0.93
H2( c l ) 33.3 2.968 1.05
H2( c l ) 38.0 2.915 1.08
H2( c l ) 29.6 2.87 0.76
N e 35.7 2.789 21.47 0.3925 2.33
29.5 2.858 2.23
35.6 2.749 2.14
34.9 2.78 2.22
A 124 3.418 15.68 1.626 2.19
116 3.465 2.23
119.8 3.405 2.07
122 3.40 2.09
K r 190 3.61 13.93 2.456 2.30
171 3.60 2.04
158 3.597 1.87
X e 229 4.055 12.08 3.9989 2.42
221 4.100 2.50
217 3.963 1.99
N2 91.5 3.681 15.51 1.734 2.24
79.8 3.749 2.18
95.05 3.698 2.40
95.9 3.71 2.46
o2 113 3.433 12.3 1.561 2.84
88.0 3.541 2.66
117.5 3.58 3.78
118 3.46 3.10
F2 112 3.37 17.8 1.26 2.66
15.7 3.02
C l2 357 4.115 13.2 4.60 2.85
257 4.400 3.07
C H4 137 3.882 13.16 2.58 2.46
144 3.796 2.26
148.2 3.187 2.40
(Continued on following page) T A B L E I I I
144 T. M . REED III T A B L E I I I (Continued)
Molecule e*jk a / a V z
(°K) (A) (ev) ( A3)
C F4 152.3 4.70 17.81 4.02 2.62
S F6 200.9 5.51 19.3 6.55 3.12
C2H6 230 4.418 11.6 4.48 3.39
243 3.954 2.36
C C k 327 5.881 11.0 11.11 4.58
to Z = 6, which indicates that approximately 6 electrons determine the interaction parameter. Neither CH4 or C F 4 show any unusual anomally with respect to dispersion interaction between like molecules on the basis of these considerations, since Z computed from experimental values of
€* and G in Table III are essentially the same. When, however, values of c from Table II are plotted versus molecular polarizability, as in Fig. 2, for the two homologous series, fluorocarbon alkanes and hydrocarbon alkanes, c v s a for the former is a straight line while that for the latter is a curve of decreasing slope. The hydrocarbons have a greater lattice energy for a given total molecular polarizability than do the fluorocarbons, when the number of carbon atoms per molecule is greater than one. We can attribute this difference to effects of molecular structure, which are not the same in the two series, in spite of their similarity in carbon skeleton.
A study has been made by Hamann and Lambert*6 4' 65> 9 2) of a mole
cular potential energy function obtained by summing the potential energies between the atom pairs of the interacting molecules. This work demon
strates that the single-center Lennard-Jones (12, 6) potential function is inadequate even for molecules such as C F 4 , SF6, and UF6, which are approximately spherically symmetrical (called quasi-spherical molecules).
A single-center potential function of the form of the Lennard-Jones function [Eq. (1)] is adequate for equilibrium properties of these mole
cules, when the exponents on the distance between molecular centers are m = 7 and n = 28 for the attractive and repulsive terms, respectively.
This empirical (28, 7) potential function is shown to give the same total mutual potential energy between pairs of molecules as that obtained from the sum of potential energies of the various atom centers of a pair of mole
cules. (63-65, 92) However, the application of the (28, 7) potential function to the quasi-spherical molecules, such as CF4, SF6, SiF4, and C(CHs)4, ' 'does not give a completely satisfactory correlation of the transport and
P H Y S I C A L C H E M I S T R Y O F F L U O R O C A R B O N S 145 equilibrium properties of these molecules/' as stated by McCoubrey and SinghG-05).
If we consider centers of interaction as existing at the center of each atom in a molecule, it appears that not only H,H interactions are import
ant but also, because of the small size of the H atom, C,H, and C,C inter
actions between centers on adjacent molecules are significant in the hydrocarbon series; whereas, because of the larger size of the F atom, only F,F interactions and perhaps C,F interactions contribute significantly to the intermolecular potential in fluorocarbons. The C,C and C,H attractions are stronger than the H,H attraction so that, as the ratio of C to H increases with increase in molecular size, the lattice energy increases above that which would be expected from the lattice energy of CH4 or of C2H6.
Because the C atoms are buried somewhat within the molecule by the peripheral H atoms, the contribution of C,C and C,H interactions to the net attractive energy (which varies as r~6) is a relatively greater fraction of the total attraction energy than is the contribution of these interactions to the net repulsive energy (which varies as r~12 or r~2s). The inner C atoms contribute to the attractive energy but not to the repulsive energy to the same extent. Consequently hydrocarbons enjoy a large attractive energy without the usual accompanying repulsive energy. Thus, they have unusually large lattice energies compared to fluorocarbons.
In the series of fluorocarbon alkanes the F,F interactions predominate to give a uniform increase in lattice energy essentially independent of the ratio of the number of carbon atoms to the number fluorine atoms per molecule. Most of the intermolecular interaction in the fluorocarbon series arises from centers located in the peripheral F atoms, while in the hydrocarbon series some of the centers are centrally located in carbon atoms of the molecule and others are located in the peripheral hydrogen atoms. The usual repulsive energy associated with the attraction term is not diminished, as in hydrocarbons, by the presence of intervening struc
tures. The net interaction and lattice energy in fluorocarbons are thus smaller in magnitude than those for hydrocarbons of the same polariza
bility.
The intermolecular pair potential energy function for fluorocarbon molecules should thus exhibit a narrower potential well with steeper sides than that possessed by analogous hydrocarbon molecules (105a).
From these hypotheses we may explain the following comparative behaviors of hydrocarbons and fluorocarbons, all of which are substanti
ated in this chapter:
(1) The lattice energy for hydrocarbons versus the total molecular polarizabilities will be larger than that for fluorocarbons of the same total polarizability.
146 T . M . R E E D I I I
(2) Because there are more centers of significant influence in the hydrocarbons than in the fluorocarbons, the attraction interaction in hydrocarbons will reduce the volume of the assemblage, causing the com
pressibility of hydrocarbon to be less than that of fluorocarbons.
(3) The effect of isomerization or branching in the molecular structure in hydrocarbons will be greater than this effect in the fluorocarbons and in the opposite direction. For example, if a completely substituted carbon atom exists in a structure, such as it does in neopentane, this carbon atom is considerably removed from contributing its full attractive (as well as repulsive) interactions with adjacent molecules by the methyl groups attached to it. Such an isomerization of the C5F12 molecule should not alter to such a large extent the mutual interaction with neighbors, since there is little interference with F,F interactions produced by altering the structure from normal pentforane to neopentforane.
Chain branching of a hydrocarbon thus reduces the number of inter
molecular C,C interactions by covering carbon atoms with methyl groups.
The van der Waals forces are thus diminished by isomerization in the hydrocarbon series, and boiling points are consequently lowered by branching. In the fluorocarbon series branching produces a small increase in the van der Waals forces between molecules, which may be attributed to a greater exposure (greater number of neighbors) of a fluorine atom to surrounding molecules, when it is attached to a methforyl group than when it is attached to a methforene group.
The above hypotheses are a more concrete statement of the idea of
"interpenetration" in hydrocarbons discussed by Simons and Dunlap(1 5 5>.
This "interpenetration" here appears explicitly as an overlapping of multicenters of molecular interactions in hydrocarbon substances. Such an overlapping is small in the fluorocarbon series, and in this sense (negligible overlap) the fluorocarbons are more analogous to the single- center molecules, the inert gases. In other respects, in their macroscopic behavior, and in particular the corresponding state for fluorocarbons which depend implicitly upon the detailed structural makeup of the mole
cule, the fluorocarbons may demonstrate greater or lesser deviations from the behavior of the inert gases than is shown by the behavior of hydrocarbons.
Further predictions of the above hypotheses are:
(1) The compressibilities of liquids composed of molecules of branched chain isomers in the hydrocarbon series should be greater than for the straight chain structure.
(2) The densities of the liquids composed of branched-chain isomers in the hydrocarbon series should be less than for the straight chain struc
ture. The densities of liquid fluorocarbon should be greater for the branched chain than for the straight chain isomers.
P H Y S I C A L C H E M I S T R Y O F F L U O R O C A R B O N S 147 IV. Virial Coefficients and Equations of State for Gases The second virial coefficient B has been reported for several fluoro
carbon molecules over limited temperature intervals. In Fig. 3 are plotted values of BPcjRTc versus the reduced temperature Tr = T/Tc. The experimental data for C2F6<122>177) is reproduced over the temperatures range 0.6 < Tr < 1.3 using the spherocylinder model of Kihara<75> and
+ . 2 5
- . 2 5
BPc R TC .5
- . 7 5
- 1.0
• - C F 4
® — C2F6
o — C4F1 0
•
— 5FI 2 C— C6F ,4
V - S 6 F
C5 'C6
C2 K I H A R A M O D E L
1.5 2 2.5 3
Tr
F I G . 3. S e c o n d virial coefficient vs temperature for fluorocarbons.
the three parameters a = 4.71 A, / = 1.48 A, and e*/& = 225°K. This value of a was obtained in the following way. The difference between 3.65 A (a for F2 molecule) and 1.44 A (F—F bond distance in F molecule) is the distance between F atom centers on adjacent molecules when the potential energy is zero, and includes the excess of cr over the van der Waals diameter for F,F interactions on adjacent molecules. This difference is 2.21 A.
For C2F6 in the Kihara model this excess, 2.21 A, was added to the product
148 T. M. REED III
of the sine of 180° minus the tetrahedral FCF angle (110°) and twice the C—F bond distance (2 x 1.33 A) to give cr for C2F6 as 4.73 A. The core length / of the Kihara sphero-cylinder for C2F6 is the C—C bond distance in this molecule (1.48 A)*98>. Using these values for cr and /, e*jk was obtained from the experimental second virial coefficients. Other com
binations of values for cr, /, and e*/k (for example a = 4.38 A, / = 1.48 A, a*Ik = 245°K) fit the data as well, but the value of 4.7 A for cr is the same as that found by MacCormack and Schneider*96* by fitting the Lennard- Jones (12, 6) spherical model to their data on C F4. The points obtained experimentally for CF4 lie on the same curve and its extension in Fig. 3, when a value of 31 atm is used for the critical pressure of CF4 (see Section V). This critical pressure has not been determined experimentally. This curve on Fig. 3 for CF4 and C2F6 gives a second virial coefficient for CF4 at its normal boiling point of —495 ml per mole. The convex core model of Kihara*8 3) fitted to the high temperature data (Tr > 1) for CF4 predicts a second virial coefficient of approximately — 520 ml per mole at the nor
mal boiling point. These values agree fairly well. No experimental value is available for CF4 in this low temperature region.
Second virial coefficients have been reported for W- C 5 F 1 2 and for U-CQFU^ in the low temperature region (TR ~ 0.5 to 1.0). The reduced coefficients BPcjRTc for these molecules lie slightly below the curve for C2F6. As the number of centers of intermolecular interaction in a molecule increases, it is expected*66) that the reduced virial coefficient should de
crease at a given reduced temperature.
For the molecule SF6 experimental*2 7'6 7'9 6) second virial coefficients give values of BPCIRTC which coincide with those for the fluorocarbons.
MacCormack and Schneider*96) obtained a value of 200.9°K for e*/& and 5.51 A for cr by fitting the Lennard-Jones (12, 6) spherical model to their data. This value for e*jk is not too different from the value 225°K obtained above for C2F6, which also contains six fluorine atoms. When we add the value 2.21 A (distance between F atom centers when the molecular pair potential energy is zero obtained above) to twice the S—F bond distance (2 x 1.68 A) the value of cr for SF6 is calculated as 5.57 A, which is essentially the value obtained by MacCormack and Schneider. Hamann et al.W) obtained cr = 5.90 A and e*/& = 188.7°K for the (12, 6) fit to their data on SF6.
The second virial coefficients for SiF4 give (12, 6) parameters, calcu
lated by Hamann et a/.*6 7), as a = 5.57 A, e*/& = 148.7°K. The latter is close to e*jk = 152.5°K for CF4, which also contains four F atoms. The value 2.21 A plus twice the Si—F bond distance (2 x 1.81 A) is 5.83 A, about 0.2 A higher than the (12, 6) potential cr value.
Hamann and Lambert*6 4' 6 5) have discussed the virial coefficients of
P H Y S I C A L C H E M I S T R Y O F F L U O R O C A R B O N S 149 CF4, SiF4, and SFQ in relationship to the (12, 6) single-center potential function and the (28, 7) empirical approximation to the multicenter potential energy function for quasi-spherical molecules. Tables for calcu
lating B from the parameter e*jk and r* for the (28, 7) potential are given in reference (64). Values of the parameters are given in Table IV. From a comparison of the values of the critical constants*64) and of the entropies of vaporization*6 3'6 5) for quasi-spherical molecules these authors conclude that the (28, 7) potential function is more accurate than is the (12, 6) potential for such polyatomic molecules. The inert gases, and N 2 , O2, CO, and C H 4 are better represented by the (12, 6) potential function, while CF4, SiF4, SF6, and C ( C H 3) 4 are better described by the (28, 7) potential function.
Computations of the second virial coefficients for the fluorocarbons CF4 through C 6 F 1 4 have been made by the author using the corresponding state method of Pitzer*1 2 6 - 1 2 9) and the vapor pressure data of Table I.
These values were found to be essentially identical with the experimental data in the ranges where it is available. This corresponding state method, however, does not reproduce the properties*36) of cyclobutforane. The virial coefficients of cyclobutforane have been fitted successfully by the models of Kihara and of Stockmayer*36). This molecule is a nonplanar ring.
Third virial coefficients are reported for CF4 and SFQ by MacCormack and Schneider*96) for SF6 by Clegg, Rowlinson, and Sutton*2 7) and for cyclobutforane by Douslin, Moore, and Waddington*3 6).
The only data on the compressibility factor (PVjRT) available for fluorocarbon alkanes*18), that for W- C 4 F 1 0 , can be reproduced for both the liquid phase and for the vapor phase by the correlations of Pitzer*1 2 6 - 1 2 9).
Constants in equations of state for various fluorine containing substances are given in Table V.
Second virial coefficients have been measured for mixtures of C H4 + SF6 by Hamann, Lambert, and Thomas*6 6). [They also report values f o r C H4 + C ( C H3)4a n d f o r C H4 + Si(CH3)4.] Garner and McCoubrey**3) determined the second virial coefficients for mixtures of W- C 5 H 1 2 + W- C 5 F 1 2 and for W- C 5 H 1 2 + W- C 6 F 1 4 . Tripp and Dunlap*1 6 8 a) have de
termined second virial coefficients for a mixture of W- C 4 H 1 0 + n - C 4 F i o ,
for a mixture of (CH^O + C 3 F 7 H , and for these four pure components.
These data are reproduced in Table VI as Bnh the second virial coefficient for the mixture and B12, the cross term coefficient in the relationship:
Bm = yi2Bn + 2 v i v2£ i 2 + v22£ 2 2 ,
where y is mole fraction in the gas phase and B\\ is the second virial co
efficient for pure component 1 and B22 is that for pure component 2.
The coefficient B12, and thus the second virial coefficient for the
150 T. M . REED III T A B L E I V
PARAMETERS I N S I N G L E - C E N T E R P O T E N T I A L E N E R G Y F U N C T I O N S , E Q . (1) A. From Second Virial Coefficients
n —
( ° K )
12, w = r*
( A ) 6
a (A)
Ref
erence
n =
( ° K )
28, m = r*
( A ) 7
a (A)
Ref
erence
C F4 152.5 5.28 4.70 75 315 4.63 4.33 64
W-C5F12 162 12.82 11.42 53
«-C6Fl4 163 14.50 12.91 53
F2 121 4.05 3.61 77
S i F4 148.7 6.28 5.59 67 331 5.03 4.71 64
S F6 188.7 6.63 5.90 67 414 5.37 5.03 64
200.9 6.19 5.51 96
C H4 148.2 4.28 3.81 75
« - C5H i 2 176 9.91 8.82 53
C ( C H3)4 236 8.25 7.35 64 581 6.09 5.70 64
c y c l o - ( C H2) 3A 210 6.8 6.1 34 610 4.5 4.2 34
c y c l o - ( C H2) 6 590 7.3 6.8 34
C6H6 570 7.2 6.7 34
a Better potential: n = 26, m = 6.5, e*jk = 4 6 0 ° K , r* = 5.2 A . T A B L E I V (Continued)
PARAMETERS I N S I N G L E - C E N T E R P O T E N T I A L E N E R G Y F U N C T I O N S , E Q . (1) B. From Gas Viscosities
n = 12, m = 6 n = 28, m—1
€*/k r* a Ref e*/k r* a Ref
( ° K ) (A) (A) erence ( ° K ) (A) (A) erence
CF4 288 4.49 4.20 105
C2F6 163 6.24 5.56 105a 334 535 5.01 105a
«-C5Fi2 195 8.26 7.36 105a
«-C6Fl4 160 9.04 8.05 105a W-C7F16 356 8.99 8.01 112a
285 9.51 8.47 112a
C2F4 152 5.75 5.12 105a 289 5.01 4.69 105a
F2 112 3.78 3.37 75
B F3 178 4.74 4.22 4 4
178 4.74 4.22 104a
S i F4 147 5.57 4.96 4 4 286 4.85 4.54 105
148 5.56 4.95 104a
S F6 259 5.63 5.01 4 4 439 5.00 4.68 105
155 6.13 5.46 104a
U F6 255 6.60 5.88 104a
C ( C H3)4 4 7 0 6.09 5.70 105