CHAPTER 19
Inhibitors of Catechol A m i n e Metabolism
T. L Sourkes and A. D'lorio
I. Introduction 79 II. Substances Interfering with the Biogenesis of Catechol Amines 80
through Inhibition of Dopa Decarboxylase
A. Inhibitors Acting on the Apodecarboxylase 80 B. Interference with the Function of Pyridoxal Phosphate 85
C. Other Inhibitors 87 III. Substances Interfering with the Metabolism of Catechol Amines . . 87
A. Inhibitors of Conjugation 88 B. Inhibitors of Oxidative Deamination 88
C. Inhibitors of O-Methyl Transfer 90 IV. Substances Acting a t Receptor or at Storage Sites 93
References 95
I. INTRODUCTION
Epinephrine was identified as a hormone and synthesized about 60 years ago, but it is only within the last 15 years that detailed biochemical knowledge of its formation and metabolism has become available. His- torically important findings have been the discovery of hydroxytyramine and norepinephrine in urine, in sympathetic nerves, and in the brain.
These compounds serve as precursors of epinephrine in the biosynthetic pathway leading from phenylalanine, through tyrosine, dihydroxyphenyl- alanine (dopa), dopamine, and norepinephrine, but they also have addi- tional functions. Other amino acids giving rise to catechol amines are ra-tyrosine and dihydroxyphenylserine. The development of sensitive fluorometric methods for the measurement of the catecholamines and the catechol amino acids in body fluids and tissues has revolutionized the study of sympathoadrenal physiology. Detailed reviews of the biochem- istry of the catechol amines have been prepared ( i , 2). Many useful
79
8 0 Τ. L. SOURKES AND A. D'lORIO papers on catechol amines will be found in the proceedings of a sym
posium held in 1958 (3).
In this chapter, inhibitors of catechol amine metabolism will be considered in the three categories: (a) biogenesis, (6) catabolism and
"detoxication," and (c) binding at receptor and storage sites. The enzymes concerned in biogenesis, beginning from dopa, are dopa decarboxylase, dopamine-/?-oxidase, and iV-transmethylase. Information on inhibitors of the latter two enzymes is lacking, and only the first is considered.
II. SUBSTANCES INTERFERING WITH THE BIOGENESIS OF CATECHOL AMINES THROUGH INHIBITION OF
DOPA DECARBOXYLASE
The amino acid precursors of the catechol amines undergo decarboxyla
tion through the action of a pyridoxal phosphate (PLP)-linked enzyme.
One of the earliest amino acid decarboxylases found in animal tissues was dopa decarboxylase, first described by Holtz, Heise, and Liidtke in 1938 (4). At present this enzyme is regarded as the catalyst for the decarboxy
lation of 5-hydroxytryptophan also ( 5 ) , and of some other aromatic amino acids. This section will review the literature primarily in relation to inhibitors of decarboxylase preparations which have been studied with dopa as the substrate. Hundreds of compounds have been tested for inhibition of dopa decarboxylase, but only a few of the active ones can be described here. The emphasis will be upon mechanisms of action. Further details will be found in references (6), ( 7 ) , and (8).
A. Inhibitors Acting on the A p o d e c a r b o x y l a s e
1. a-METHYLDOPA AND RELATED COMPOUNDS
a. Mechanism of Action in vitro. a-Methyldopa, a structural analogue of dopa in which the hydrogen atom on the α-carbon is replaced by a methyl group, exerts a biphasic action on dopa decarboxylase; in low concentrations it activates the enzyme, but above 1 0 ~5 Μ it displays the characteristics of a slowly reversible inhibitor competing with the sub
strate for sites on the enzyme surface. The mechanism of the activation has not been explained, although several experimental approaches have been tested {6). a-Methyl-3-hydroxyphenylalanine (a-methyl-meia- tyrosine) has similar properties. These compounds act as "pseudoirre-
19. INHIBITORS OF CATECHOL AMINE METABOLISM 81 versible" inhibitors, in the terminology of Ackermann and Potter (9);
that is, they are characterized by a relatively low apparent enzyme- inhibitor dissociation constant. The inhibitor can be removed from the enzyme by dialysis. The structural resemblance of these compounds to substrates of dopa decarboxylase suggests that they inhibit by establish
ing a stronger bond with the apodecarboxylase through their phenolic groups (10) than the substrate does. The meia-phenolic group is of special importance in this respect because inhibitory activity is greatly reduced in compounds lacking this group or bearing it masked by methylation.
Thus, α-methyl-m-tyrosine, at a concentration of 5 Χ 10~~3 Μ inhibits the dopa decarboxylase activity of an extract of pig kidney cortex 95%, whereas α-methyl-m-methoxyphenylalanine and α-methyl-p-tyrosine at the same concentration inhibit only 16-18%.
A series of α-methyl amino acids shows the following (descending) order of activity against kidney dopa decarboxylase: a-methyldopa, α-methyl-m-tyrosine, a-methyl-p-tyrosine, a-methylphenylalanine. These compounds also inhibit tyrosine decarboxylase of Streptococcus faecalis, but to a much smaller extent (6).
Like some other amino acids and amines, the α-methyl compounds are able to sequester the coenzyme, but the inhibition of the enzyme by this means occurs at concentrations of inhibitor where coenzyme binding is not significantly affected (see Section II, B, 2 6 ) .
b. Actions in vivo. After the demonstration of the inhibitory action of α-methyldopa in vitro (6), Dengler and Reichel (11) and Westermann et al. (12) showed that this compound is also active in intact animals.
This was confirmed by the direct measurement of catechol amines and of α-methyldopa in the tissues of rats injected with the inhibitor and with dopa (13). This study described two biochemical effects following the parenteral administration of α-methyldopa: (a) a decrease in the endoge
nous cerebral dopamine and norepinephrine, and (b) an inhibition of the decarboxylation of exogenous dopa. The action on endogenous cerebral amines is exemplified by the rapid fall in cerebral dopamine and norepine
phrine following the injection of the α-methyl amino acid. The concen
tration of α-methyldopa in the brain 3 hours after the injection of the compound is proportional to the dose administered. As soon as its con
centration falls below the levels which are inhibitory toward dopa decarboxylase, the resynthesis of the catechol amines begins; this is achieved in less than 24 hours for dopamine but requires 2 - 3 days for norepinephrine (14). Cerebral serotonin also declines in animals treated with α-methyldopa (15); the level is restored rapidly, as in the case of dopamine. The amine-depleting effect is not entirely specific to the
82 Τ. L. S 0 U R K E S AND A. D'lORIO
α-methyl amino acids, for other amino acids (m-tyrosine, o-tyrosine, tryptophan, 5-hydroxytryptophan) also cause a loss of cerebral catechol amines under the same conditions {16).
The second action of α-methyldopa in vivo is its inhibition of decar
boxylation. This becomes evident when exogenous dopa or certain other amino acids are administered. The accumulation of dopamine under these conditions can be minimized by treatment of rats with α-methyldopa or α-methyl-m-tyrosine (but not a-methyl-3-methoxyphenylalanine) just before they receive a test load of L-dopa (10 m g / k g ) . The effect of various α-methyl amino acids upon the conversion of exogenous dopa to urinary
T A B L E I
EFFECT OF <X-METHYL AMINO ACIDS ON THE CONVERSION OF L-DOPA TO URINARY DOPAMINE
Urinary
Compound administered0 dopamine6
None 100 a-Methylphenylalanine 92
tt-Methyl-o-tyrosine 91 a-Methyl-ra-tyrosine 43 a-Methyl-3-methoxy-4-hydroxyphenylalanine 92
a-Methyl-2,5-dopa 40 α-Methyl tryptophan 91 a-Methyl-5-hydroxy tryptophan 10
a Injected intraperitoneally, in saline, 100 mg/kg, 0.5 hour before L-dopa (10 mg/kg) was given by the same route.
6 Twenty-four-hour output expressed as percentage of output in control animals receiving only dopa.
dopamine in the rat is shown in Table I (14, 17,18). The effect of a single injection of α-methyl-ra-tyrosine in this test lasts for a few hours (18).
c. Metabolism. Although the α-methyl amino acids tested manomet- rically in short-term incubations were reported as resistant to the action of tissue decarboxylase (6), more sensitive fluorometric methods have shown that they are slowly acted upon by enzymes (19), with the forma
tion of compounds bearing an isopropylamine side chain. "a-Methyl- dopamine" is 3,4-dihydroxyamphetamine. The decarboxylation of the α-methyl amino acids is in accord with the Westheimer model of the action of amino acid decarboxylases, which predicts that the α-hydrogen atom is not involved in this process (cf. 20).
1 9 . I N H I B I T O R S OF CATECHOL A M I N E M E T A B O L I S M 8 3
α-Methyl-ra-tyramine is a weak inhibitor of rabbit liver MAO, but α-methyldopamine is inactive (20a).
Subjects given 1 - 1 . 5 gm daily of α-methyldopa by mouth excrete about 1 0 % of the compound in the urine. Upon withdrawal of the drug α-methyldopa can be detected in the urine (in diminishing amounts) for 2 days. After intravenous administration, α-methyldopa is found in the plasma for at least 2 hours; it does not appear to enter the erythrocytes
{21).
d. Pharmacological and Clinical Effects. α-Methyldopa lowers blood pressure in hypertensive patients (22). Its hypotensive action has also been studied in the dog (23). It has a mild sedative action (22-24). Smith has compared its pharmacological actions in experimental animals to those of reserpine (25). It has now been tested widely in the treatment of hypertension (24), with success, and as a tranquillizer in schizophrenia, where it is ineffective (21). It exacerbates the tremor in Parkinson's dis
ease (25a).
2 . 3 - H Y D R O X Y C I N N A M O Y L D E R I V A T I V E S
a. Action in vitro. Many compounds based upon the structure shown in (I)
m-OH · CeH4— C = C — C — X
I I II ο
(I)
have been synthesized and tested as inhibitors of dopa decarboxylase.
Hartman et al. (7) have shown that the most active compounds possess an aryl group in the position designated by X in the above formula.
These are derivatives of chalcone (II).
C eH &—CH=CH—C—C eH s
II
(Π)
ο
In compounds with X — OH (cinnamic acid derivatives) less inhibitory activity is encountered [ ( 7 ) , cf. ( 6 ) ] . Intermediate potency is obtained with esters of cinnamic acid and with related ketones (X = an alkyl group). An additional phenolic group in the para position increases inhibi
tory activity, but not if the group is methylated. 3,4-Dihydroxycinnamic (caffeic) acid, for example, is more potent than either 3-hydroxycinnamic acid or 3-hydroxy-4-methoxycinnamic (isoferulic) acid.
8 4 Τ. L. SOTJRKES AND A. D'lORIO
Effective inhibition by members of this series is exerted upon contact with the enzyme. This contrasts with α-methyldopa, which requires a brief preincubation with the enzyme. 3-Hydroxycinnamic acid is effective in vitro at a concentration of 1 0 ~4 Μ, but other derivatives are much more potent. The most active compound in Hartman's series is 5 - ( 3 , 4 - d i h y - droxycinnamoyl)salicylic acid ( 7 , 26). I t is effective at 1 0- 6 M. These two compounds, as well as caffeic acid, act as competitive inhibitors of the enzyme ( 7 ) .
b. Studies in vivo (8). Some of the 3-hydroxycinnamoyl derivatives have been examined for inhibitory action in vivo. The compound most active in preventing the pressor response resulting from the intravenous injection of dopa in the cat or rat is 5-(3-hydroxycinnamoyl)salicylic acid. I t causes 5 0 % inhibition in vivo at a dose level of 6 . 6 mg/kg. Its catechol analogue, the most active compound in vitro, is less than half as active in vivo. Isoferulic and 3-hydroxyphenylpropionic acids both inhibit significantly in vivo (cat) despite the very weak action of the former compound on dopa decarboxylase in vitro and the inactivity of the latter. Discrepancies of this kind introduce some difficulties in predict
ing inhibitory activity in vivo from that in vitro. However, the most active compounds in both tests conform to the same structural features in that they contain the 3-hydroxycinnamoyl unit.
It appears that these compounds are readily metabolized, for the pressor response to injected dopa is affected only for about 3 0 minutes.
Injected caffeic acid disappears from blood in the same period of time.
A few of the compounds have a longer duration of action, and their action can be extended by repeated administration of the drug or injection by more than one route.
3 . F L A V O N E S
The flavones include derivatives of 2-phenylpyran-4-one and its 2 , 3 - dihydro analogue, 2-phenylchroman-4-one (2-phenylchromone). Among the well-known natural products in this group with antidecarboxylase activity are catechin (6, 7 ) , gossypin, and rutin ( 7 ) . They appear to be relatively more active in vivo than in vitro (8).
4 . P H E N Y L P Y R U V I C A C I D
Phenylalanine and tyrosine are weak inhibitors of dopa decarboxylase (26, 27), but their keto acids are active ( 7 , 26, 27). Clark has shown (8) that 3-hydroxyphenylpyruvic and 3,4-dihydroxyphenylpyruvic acids are more effective. 4-Hydroxyphenyllactic acid has slight activity, but
1 9 . I N H I B I T O R S O F CATECHOL A M I N E M E T A B O L I S M 8 5
phenyllactic acid has none. He has also noted that 3-hydroxyphenylacetic acid has very weak activity, which is increased somewhat in its catechol analogue, dopacetic acid.
These compounds were tested using dopa decarboxylase prepared from kidney. Fellman, using extracts of beef adrenal medulla, obtained inhibi- tory effects with both phenylpyruvic and phenyllactic acids (28). He has suggested that inhibition of this enzyme by the excessive aromatic me- tabolites in the body fluids of phenylketonuric subjects plays a role in reducing endogenous catechol amine formation.
Martin et al. observed a fall in blood pressure following the injection of 4-hydroxyphenylpyruvic acid (26).
B. Interference with t h e Function of Pyridoxal P h o s p h a t e
1. P Y R I D O X I N E D E F I C I E N C Y
Deprivation of vitamin B6 results in diminished activity of hepatic dopa decarboxylase (29). In spite of this, young rats fed a diet deficient in this vitamin do not exhibit significant changes in the catechol amine content of the adrenal glands, liver, brain, spleen, or heart (17). The effects of the deficiency are reflected, however, in a reduced formation of urinary dopamine from injected dopa. The reduction is 3 5 - 5 0 % when the test is performed with L-dopa. This change represents the effect of the deficiency upon the activity of dopa decarboxylase in the tissues. Urinary dopamine is also formed from injected D-dopa, probably by a pathway involving oxidative deamination, followed by asymmetric transamination of the product to L-dopa and its decarboxylation. The transamination step has been demonstrated to occur in vivo (8). Pyridoxine deficiency causes a 7 0 - 8 0 % reduction in the rate of conversion of D-dopa to urinary dopamine in the rat. This effect sometimes appears earlier in the de- ficiency than the corresponding effect with L-dopa. The greater effect of the deficiency on the metabolism of D-dopa than on that of the natural isomer may signify that transaminase loses its coenzyme more readily under these conditions than the decarboxylase does (17).
Deoxypyridoxine has been tested as an antivitamin by adding it to a pyridoxine-deficient diet. After 1 3 days on this regime rats received D-dopa intraperitoneally as a test load. They then exhibited even greater inefficiency than the deficient controls in converting the amino acid to urinary dopamine. This may be a selective action upon transaminase, for when the rats were tested on the twenty-third day with an injection of L-dopa there was no difference in the deficient rats, whether they had been receiving deoxypyridoxine or not, although both groups excreted
86 Τ. L. SOTJRKES AND A. D'lORIO much less dopamine than their pyridoxine-supplemented controls (un
published, T. L. Sourkes).
2. A G E N T S R E A C T I N G W I T H T H E A L D E H Y D E F U N C T I O N
a. Carbonyl-Trapping Agents. The aldehyde group of codecarboxylase renders it susceptible to reaction with carbonyl-trapping agents such as hydroxylamine, semicarbazide, hydrazine and many of its derivatives.
Holtz et al. in their original paper on dopa decarboxylase reported its inhibition by cyanide (4), another aldehyde reagent. This phenomenon is common to all pyridoxal phosphate (PLP)-linked enzymes. Inasmuch as many hydrazides have a blood pressure-lowering action (30, 31), the possibility has been considered that they achieve this effect by coenzyme- binding with consequent decreased rate of formation of catechol amines.
At present such views are entirely speculative. Although isoniazid (iso- nicotinic acid hydrazide) inhibits dopa decarboxylase by forming a hydrazone with the coenzyme, this compound undergoes slow hydrolysis with the release, once again, of PLP. Addition of the hydrazide to an enzyme source permits the slow, but complete, decarboxylation of L-dopa
(32).
b. Amines and Amino Acids. The applicability of the nonenzymic re
action of amines with aldehydes to problems of PLP function has also received attention. Schott and Clark have shown that derivatives of phenylalanine and of phenylethylamine react readily with PLP only if (a) the amino group is free; (b) the ring bears a phenolic group meta to the side chain; and (c) there is an active hydrogen ortho to the side chain (33). These generalizations bear out the findings of Schoepf and Salzer, who studied the amine-aldehyde reaction (34). The reaction of an amine (in the phenylethylamine series) with pyridoxal or PLP consists of (a) formation of a Schiff base and (b) cyclization to form a tetra- hydroisoquinoline derivative (35). The examples provided in Table II show that norepinephrine and m-hydroxypropadrine react rapidly with PLP, but epinephrine and isoproterenol do so very slowly. Dopamine shows intermediate reactivity. Among the positional isomers of dopa, 3,4-dopa is most reactive. 2,4-Dopa, lacking a m-phenolie group, does not react measurably. 2,6-Dopa, which has neither m-hydroxyl nor free o-hydrogen, undergoes reaction with PLP but of an anomalous type (36).
The α-methyl amino acids react with the coenzyme, but do so very slowly.
The product of reaction of P L P with 3,4-dopa has slight inhibitory activity on dopa decarboxylase in vitro (6). A compound with the same Rf as this adduct is found in adrenal homogenates which have been incu
bated with labeled dopa (37). PLP products formed with norepinephrine
19. INHIBITORS OF CATECHOL AMINE METABOLISM 87
T A B L E II
NONENZYMIC REACTION OF PYRIDOXAL PHOSPHATE WITH AMINES AND AMINO ACIDS
Temperature (°C)
Substance 38° · 23° b
Norepinephrine 4 . 8
m-Hydroxypropadrine 3 . 2 —
m-Tyrosine 2 . 2 —
3,4-Dopa 3 . 2 0.83
Dopamine
—
0.24Epinephrine 0.13
—
2,5-Dopa 0.12 0.02
a-Methyl-m-tyrosine
—
0.07a-Methyldopa
—
0.06Isoproterenol 0.06
—
2,4-Dopa
—
No reactiona Reference (83).
6 Reference (6).
and dopamine have no effect on the cardiovascular system. Moreover, the pressor responses to norepinephrine are little affected by the intra- venous administration of massive doses of P L P or pyridoxal (8). Thus, sequestering of the coenzyme by substrate or products (or both) is a cause of inhibition of dopa decarboxylase in vitro, but it is not known whether this reaction plays a role under physiological conditions.
C. O t h e r Inhibitors
Other inhibitors of dopa decarboxylase have been described, but their mechanism of action is unknown. They include suramin (Naphuride) and trypan blue (38), 7-methylfolic acid and other folic acid analogues
(39, 40), and isoxazolidones like cycloserine (41). An extensive list of inhibitors is given by Clark (8).
III. SUBSTANCES INTERFERING WITH THE METABOLISM OF CATECHOL AMINES
The catabolism of catechol amines has been thoroughly investigated in recent years, and these experiments are summarized in several reviews (1, 42, 43). Three major pathways have been described which can act on catechol amines to produce inactive compounds. These reactions are
88 Τ. L. SOURKES AND A. D'lORIO conjugation, oxidative deamination, and O-methylation. One or more of these processes may act on a given compound.
A. Inhibitors of Conjugation
The first studies on the metabolism of catechol amines were mainly concerned with the excretion of conjugated amines (44)· It was found that the phenolic groups were substituted either with a glucuronide or a sulfate group, depending upon the species of animal. There is, however, no indication that this type of a reaction is an inactivating mechanism, for the biological activity of these conjugates has not been tested.
Some experiments have been conducted on the metabolism of epine
phrine in animals pretreated with iV-acetyl-p-aminophenol. The latter compound inhibits the conjugation of steroids in a competitive fashion (45). When it is injected simultaneously with epinephrine in rats, the excretion of conjugated metanephrine, which normally represents 3 9 % of the total metabolites of epinephrine, decreases to approximately 1 4 % of the total. This decreased conjugation is, however, not accompanied by an increase in toxicity as measured by the L D5 0 of epinephrine ( 0 . 3 7 mg/kg subcutaneously) (unpublished results, A. DTorio).
B. Inhibitors of Oxidative Deamination
1. N O M E N C L A T U R E A N D D E F I N I T I O N
Oxidative deamination of dopamine takes place according to the reaction shown in Eq. ( 1 ) .
(OH)2C6H8-CH2CH2NH2 + H20 + 02 (OH)2C6H8-CH2CHO + H202 + N H8 (1) The oxidation of 3-O-methyldopamine, norepinephrine, normetanephrine, epinephrine, and metanephrine proceeds analogously; the oxidation of the last two results in the formation of methylamine instead of ammonia.
About 3 0 years ago three enzymic activities of this type were described, but in 1 9 3 7 Blaschko et al. showed that "tyramine oxidase," "aliphatic amine oxidase," and "adrenaline oxidase" are identical (46). Later, Zeller gave the name "monoamine oxidase" (MAO) to the enzyme (or family of enzymes) with this unitary activity. B y contrast, the name "diamine oxidase" was reserved for the enzyme acting upon short chain diamines and histamine. More recent knowledge of the substrate specificity of these enzymes shows that this nomenclature is somewhat artificial (42, 47).
Nevertheless, the division into two groups is useful, as the following list of properties of the two enzymes shows.
19. I N H I B I T O R S OF CATECHOL A M I N E M E T A B O L I S M 89 Monoamine oxidase acts on the catechol amines, their O-methyl ana
logues, tyramine, serotonin, isoamylamine, and similar amines, as well as on long chain diamines; it is associated with the particulate material of the cell; it is inhibited by sulfhydryl reagents, octanol and iproniazid;
it is not inhibited by cyanide, hydroxylamine, and other carbonyl- trapping agents; no coenzyme has been detected.
Diamine oxidase acts on short chain diamines like putrescine and cadaverine, on polyamines like spermine and spermidine, and on hista
mine; it is easily extracted from tissues containing it; it is inhibited by cyanide, hydroxylamine, isoniazid, and other carbonyl reagents; it is not inhibited by iproniazid, octanol, or p-chloromercuribenzoic acid; it is said to require both flavin adenine dinucleotide and PLP for its activity.
2. I N H I B I T O R S OF M O N O A M I N E O X I D A S E ( M A O )
Inasmuch as inhibitors of the amine oxidases are described elsewhere in this book, the following functional classification is limited to those groups of compounds which have been tested as inhibitors of catechol amine metabolism, particularly in vivo. In some cases the inhibition under physiological conditions is primarily upon the me ία-methylated derivative of the catechol amine.
a. Inhibitors Affecting Sulfhydryl Groups. The presence of an essen
tial sulfhydryl group has been judged from the inhibition of M A O by p-chloromercuribenzoate (48, 49), iodoacetate (49), mercury salts (48- 50), arsenicals (49), and salts of cadmium and silver (50). The inhibition by mercuric ions can be reversed by glutathione and cysteine in the pres
ence of cyanide (48). M A O is also inhibited by an excess of certain sulfhydryl compounds (50), by methylene blue (51), and by tetrazolium salts (52). It is possible that these agents interfere with a reversible oxidation-reduction of the sulfhydryl group necessary for the reaction catalyzed by M A O .
b. Riboflavin Deficiency and Riboflavin Antagonists. Riboflavin- deficient rats have a decreased concentration of hepatic M A O (42, 53).
The vitamin does not seem to function in a coenzyme of M A O because neither flavin adenine dinucleotide nor flavin mononucleotide reactivate extracts of the enzyme made from livers of riboflavin-deficient rats and because enzymic activity is restored to normal levels only some days after supplying the deficient rats with riboflavin. Hawkins considers that ribo
flavin is involved in the biosynthesis of M A O itself (53).
It is nevertheless conceivable that a coenzyme does exist, in a tightly bound state within the tertiary structure of the apoenzyme.
Atabrine, a drug considered to be an inhibitor of yellow enzymes, in-
90 Τ. L. SOURKES AND A. D'lORIO hibits the action of MAO on epinephrine (54); this anti-MAO action has been confirmed (52). Galactoflavin, another antimetabolite of riboflavin, does not inhibit MAO in vitro (unpublished data). The action of these two compounds, Atabrine and galactoflavin, on amine metabolism in vivo
(studied with serotonin) is complex (55).
The recovery of MAO activity from inhibition by iproniazid is slower in the riboflavin-deficient rat (55a).
c. Inhibition by Substrate Analogues. Amphetamine and ephedrine are both competitive inhibitors of MAO (56), but they do not affect catechol amine metabolism in vivo (57, 58). α-Ethyltryptamine inhibits MAO in vitro and in vivo (59). All these compounds have an isoalkylamine side chain; they are not attacked by MAO.
Some amidines inhibit MAO (60). Propamidine has been tested in the cat and found to be without effect upon the concentration of endogenous catechol amines in various organs (58).
The derivatives of hydrazine include many potent inhibitors of MAO.
Iproniazid, the exemplar of this series, is an irreversible inhibitor of MAO in vitro and in vivo; this property was first described by Zeller and his colleagues (61). The kinetics of the inhibition have been further studied by Davison (62). Iproniazid inhibits the metabolism of epinephrine, norepinephrine (63-65), and dopamine (66) in vivo. Its biological actions and those of its congeners have been extensively studied (31, 67) because of the significant role of MAO in intermediary metabolism and because of the therapeutic benefits derived from the use of these drugs in mental depressions and in anginal pain.
Choline phenyl ethers are competitive inhibitors for the oxidation of epinephrine by guinea pig liver slices. Derivatives with an ortho or para substituent are more active than the parent compound; me ία-substituted derivatives decrease the activity. For example, choline p-tolyl ether
(TM6) is more active than the meta isomer (TM7) in vitro. A chlorine substituent confers greater inhibitory activity than a methyl group does (68). Choline p-tolyl ether has been studied by several investigators for possible effects in vivo. Schayer and his colleagues reported it to affect the metabolism of epinephrine (57) and norepinephrine (64) in vivo, but Corne and Graham could not confirm this (65).
C. Inhibitors of O-Methyl Transfer
In recent years the natural occurrence of the 3-O-methyl derivatives of catechols has been reported (69). The enzyme responsible for the O-methylation has been studied (70), and it has been shown to be
19. INHIBITORS OF CATECHOL AMINE METABOLISM
91
very active for all types of catechol derivatives. It does not, however, methylate phenolic rings with only one hydroxy 1 in the meta position.
A para substituent is apparently essential for the enzyme to react.
The enzyme is found in high concentration in liver, kidney, and brain;
some activity is also present in spleen, muscle, testis, and heart (71, 72).
Because of this wide distribution and the relatively poor pharmacological activity of the "methoxy" catechol amines (73) it is generally believed that the O-methylation process is one of the most active systems for the biological inactivation of catechol amines. Several inhibitors have been studied, but few kinetic studies on enzyme inhibition have been reported.
All the inhibitors presently reported in the literature are substances competing for the methyl group. These compounds are listed in Table III with a few comments on their activity. Most of them are known poten
tiators of the effects of catechol amines; this indicates that O-methyl- transferase is important in terminating the action of these hormones in the organism. It should also be pointed out that the various substances used at present are effective only at dose levels of the order of 25-50 m g / k g in vivo. More recently, Belleau and Burba (77a) reported that tropolones act as strong inhibitors of O-methylation. Musacchio and Goldstein (77b) injected 4-isopropyltropolone in rats and found it to pre
vent O-methylation in vivo. The 4-methyl derivative has been tested by Mavrides et al. (77c) in vivo and in vitro, and it was found to be a com
petitive inhibitor with a K% value of 1.5 Χ ΙΟ- 5 M.
The known synergism between epinephrine and thyroxine has been studied in some detail by Thibault (78). Spinks and Burn (79) and, more recently, Zile and Lardy (80) have shown that thyroxine inhibits amine oxidase, and they have suggested that this impairment is the cause of the potentiation. The results presented in Table III indicate, however, that O-methyltransferase is also blocked by thyroxine. It should be pointed out that thyroxine is the only substance listed which has no effect in vitro on either O-methyltransferase or amine oxidase; pretreatment of animals with it is required to obtain inhibition. Since thyroxine also interferes with methionine formation (81) it may be assumed that the action of this hormone is related to general transmethylation processes rather than to the more specific O-methylation reaction. However, recent studies (81a, b, c) indicate that several iodophenolic compounds and acidic derivatives of thyroxine, triiodo- and diiodothyronine all act as inhibitors of catechol- O-methyltransferase in vitro. As can be seen, thyroxine can decrease the activity of both monoamine oxidase and O-methyltransferase so that these inhibitions could be partly responsible for the thyroxine potentia
tion of epinephrine.
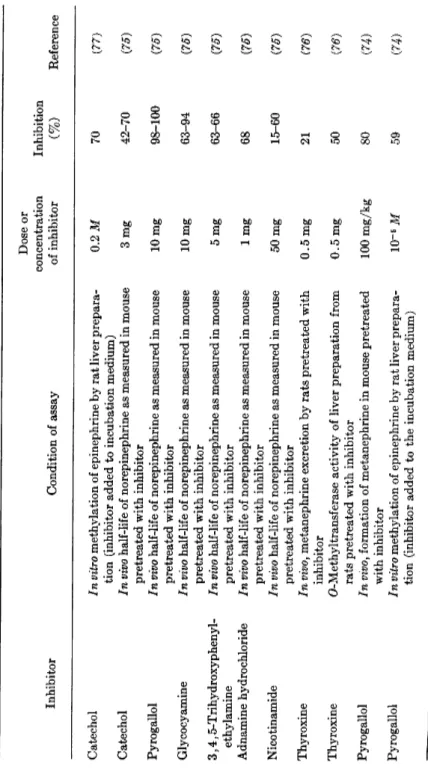
TABLE III INHIBITION OF O-METHYLTRANSFERASE Dose or concentration Inhibition Inhibitor Condition of assay of inhibitor (%) Reference Catechol In vitro methylation of epinephrine by rat liver prepara tion (inhibitor added to incubation medium) 0.2 Μ 70 (77) Catechol In vivo half-life of norepinephrine as measured in mouse pretreated with inhibitor 3mg 42-70 (75) Pyrogallol In vivo half-life of norepinephrine as measured in mouse 10 mg 98-100 (75) pretreated with inhibitor 10 mg (75) Glycocyamine In vivo half-life of norepinephrine as measured in mouse 10 mg 63-94 (75) pretreated with inhibitor (75) 3,4,5-Trihydroxyphenyl-In vivo half-life of norepinephrine as measured in mouse 5 mg 63-66 (75) ethylamine pretreated with inhibitor (75) Adnamine hydrochloride In vivo half-life of norepinephrine as measured in mouse 1 mg 68 (75) pretreated with inhibitor (75) Nicotinamide In vivo half-life of norepinephrine as measured in mouse 50 mg 15-60 (75) pretreated with inhibitor (75) Thyroxine In vivo, metanephrine excretion by rats pretreated with 0.5 mg 21 (76) inhibitor (76) Thyroxine O-Methyltransferase activity of liver preparation from rats pretreated with inhibitor 0.5 mg 50 (76) Pyrogallol In vivof formation of metanephrine in mouse pretreated with inhibitor 100 mg/kg 80 (74) Pyrogallol In vitro methylation of epinephrine by rat liver prepara tion (inhibitor added to the incubation medium) 10~5 Μ 59 (74)
92 Τ. L. SOURKES AND A. D'IORIO
19. INHIBITORS OF CATECHOL AMINE METABOLISM 93
IV. SUBSTANCES ACTING AT RECEPTOR OR AT STORAGE SITES
The substances mentioned previously in this chapter have known meta
bolic effects in the sense that they block enzymes involved in the bio
genesis or the catabolism of catechol amines. There are, however, many compounds which will block or enhance the effects of catechol amines without affecting their metabolic pathway. It is generally believed that such compounds act at receptor or at storage sites.
The storage of amines has been studied by many authors (82, 88), and it is well known that the catechol amines of the adrenal medulla or of nerve cells are localized in granules rich in protein and ATP. The catechol amines, ATP, and proteins presumably form a complex in these granules.
One can visualize the protein as a template for the catechol ring, with the A T P molecules attached to the protein by a phosphate bond and the other phosphates linked to the amino group of the catechol amines.
The chemical morphology of receptor sites is, however, more complex than this. The existence of different types of receptor site has been postu
lated on the basis of the various types of activity of catechol amines and the inhibition produced by adrenergic blocking drugs. There is at present no general agreement on the diversity of these sites (84, 85). In his classic work with epinephrine and ergotamine, Dale distinguished two types of receptor (86). Ahlquist (85, 87) has attempted to classify these receptors as α and β, according to whether the response is blocked by adrenergic blocking agents or not. Furchgott (88) has recently suggested a modifica
tion of this classification: α-receptors, for contraction of smooth muscle;
β-receptors, for relaxation of smooth muscle other than that of intes
tine, and also for increase in rate and strength of cardiac contraction;
y-receptors, for glycogenolysis; 8-receptors, for inhibition of intestinal smooth muscle.
In considering possible metabolic inhibitors the a- and γ-receptors are the only ones that are of interest. In the context of this chapter only α-receptors will be discussed. A certain number of compounds will compete with catechol amines for the reactive sites of these structures, thus pre
venting the effects of catechol amines on the receptors. Numerous studies in this field have been carried out on the β-haloalkylamine group of sub
stances (84, 89). It is believed (89) that a carboxylate or phosphate anion constitutes the active center of the excitatory α-receptors with which /?-haloalkylamines react to block the action of epinephrine.
Other groups of substances have also been used as adrenolytics, and it is assumed that they react in a similar fashion with the receptor sites.
94 Τ. L. SOURKES AND A. D'lORIO The most important members of the various groups are reported in Table IV. The only common feature of the various molecules presented there is the tertiary amine function. This may indicate that the reactive center of the receptors has the character assigned to it by Belleau (89).
T A B L E IV
ADRENERGIC BLOCKING SUBSTANCES
Group Prototypes Activity
Ergot alkaloids Ergotamine Prevents action of injected epinephrine or of nerve stimulation
Imidazolines Phentolamine Tolazoline
Same
β-Haloalkylamines Dibenamine
Dibenzyline
Prevents action of injected epinephrine and norepinephrine or of nerve stimula
tion (long-acting) Same
Dibenzazepine Azapetine Same, but short-acting Benzodioxanes Piperoxan
(Benzodioxane)
Prevents action of injected epinephrine
Receptor sites and storage sites have been discussed together in this chapter because they present some obvious chemical analogy to one another. The release of catechol amines from storage can be effected by three possible mechanisms: (a) direct or central stimulation of adrenergic nerves or adrenal medulla; (b) disruption of catechol amine-containing granules by lytic agents (examples: digitonin, lecithin, Tween 8 0 ) ; and (c) competition by analogous substances for the catechol amine binding sites within the granules.
In the present context the only compounds of interest are those corre
sponding to group (c), and of the many substances reported in the literature (83) at present only tyramine may be classified in this group.
It has, in effect, been reported that tyramine (90) will liberate catechol amines both in vivo and in vitro from storage granules. Since tyramine acts directly on the granules and has obvious structural analogies with catechol amines, it may be assumed that it is actually displacing catechol amines from their binding site.
19. INHIBITORS OF CATECHOL AMINE METABOLISM 95 Similarity of receptor and storage sites can be deduced from the action of certain adrenolytic agents (Dibenzyline, phentolamine, piperoxan, and Dibenamine) in releasing catechol amines from adrenal chromaffin gran
ules in vitro (91).
REFERENCES
1. J. Pellerin, J. Leduc, and A. D'lorio, Rev. can. biol. 17, 267 (1958).
2. T. L. Sourkes, in "Neurochemistry" (K. A. C. Elliott, I. H. Page, and J.
H. Quastel, eds.), 2nd ed., Chapter 24. Thomas, Springfield, Illinois, 1962.
3. 0 . Krayer, ed., "Symposium on Catecholamines." Williams & Wilkins, Baltimore, Maryland, 1959; also in Pharmacol. Revs. 11, 233 (1959).
4. P. Holtz, R. Heise, and K. Ludtke, Arch, exptl. Pathol. Pharmdkol.
Naunyn-Schmiedeberg's 191, 87 (1938).
5. J. H. Fellman, Enzymologia 20, 366 ( 1 9 5 9 ) ; E . Werle and D. Aures, Z.
physiol. Chem. Hoppe-Seyler's 316, 45 ( 1 9 5 9 ) ; A. Yuwiler, E . Geller, and S. Eiduson, Arch. Biochem. Biophys. 80, 162, (1959).
6. T. L. Sourkes, Arch. Biochem. Biophys. 51, 444 (1954).
7. W. J. Hartman, R. I. Akawie, and W. G. Clark, J. Biol. Chem. 216, 507 (1955).
8. W. G. Clark, Pharmacol. Revs. 11, 330 (1959).
9. W. W. Ackermann and V. R. Potter, Proc. Soc. Exptl. Biol. Med. 72, 1 (1949).
10. H. Blaschko, Biochim. et Biophys. Acta 4, 130 ( 1 9 5 0 ) ; T. L. Sourkes, P.
Heneage, and Y. Trano, Arch. Biochem. Biophys. 40, 185 (1952).
11. H. Dengler and G. Reichel, Arch, exptl. Pathol. Pharmakol. Naunyn- Schmiedeberg's 232, 324 ( 1 9 5 7 ) ; 234, 275 (1958).
12. E. Westermann, H. Balzer, and J. Knell, Arch, exptl. Pathol. Pharmakol.
Naunyn-Schmiedebergfs 234,194 (1958).
13. G. F. Murphy and T. L. Sourkes, Rev. can. biol. 18, 379 (1959).
14. T. L. Sourkes, "The Extrapyramidal System and Neuroleptics," Proceed
ings of the International Symposium, Universite de Montreal, Novem
ber 1960. Editions Psychiatriques, Montreal, 1961; also in Rev. can. biol.
20, 187 (1961).
15. S. M. Hess, R. H. Connamacher, M. Ozaki, and S. Udenfriend, Pharmacol.
Exptl. Therap. 134, 129 (1961) ; C. C. Porter, J. A. Totaro, and C. M.
Leiby, ibid., p. 139.
16. T. L. Sourkes, G. F. Murphy, B. Chavez, and M. Zielinska, J. Neurochem.
8,109 (1961).
17. T. L. Sourkes, G. F. Murphy, and V. R. Woodford, Jr., J. Nutrition 72, 145 (1960).
18. G. F. Murphy and T. L. Sourkes, Arch. Biochem. Biophys. 93, 338 (1961).
19. H. Weissbach, W. Lovenberg, and S. Udenfriend, Biochem. Biophys. Re
search Communs. 3, 225 (1960).
20. S. Mandeles, R. Koppelman, and Μ. E. Hanke, J. Biol. Chem. 209, 327 (1954).
20a. Ε. V. Heegaard and G. A. Alles, J. Biol. Chem. 147, 505 (1943).
96 Τ. L. SOURKES AND A. D'lORIO 21. T. L. Sourkes, G. F. Murphy, and B. Chavez, Proc. 3rd World Congr.
Psychiatry, Montreal, June 1961. Univ. of Toronto Press, Toronto and McGill Univ. Press, Montreal, 1962, p. 649; J. Med. Pharm. Chem. 5, 204 (1962).
22. J. A. Oates, L. Gillespie, S. Udenfriend, and A. Sjoerdsma, Science 131, 1890 (1960).
23. L. I. Goldberg, F. M. DaCosta, and M. Ozaki, Nature 188, 502, (1960).
24. Anonymous, Brit. Med. J. 1,1676 (1962).
25. S. E . Smith, Brit. J. Pharmacol. 15, 319 (1960).
25a. A. Barbeau, G. F. Murphy, and T. L. Sourkes, in "Monoamines et Sys- teme nerveux central (J. de Ajuriaguerra, ed.). Georg, Geneva, and Masson, Paris, 1962.
26. G. J. Martin, R. Brendel, and J. M. Beiler, Exptl. Med. Surg. 8, 5 (1950).
27. P. Gonnard, Bull. soc. chim. biol. 32, 535 (1950).
28. J. H. Fellman, Proc. Soc. Exptl. Biol. Med. 93, 413 (1956).
29. H. Blaschko, C. W. Carter, J. R. P. O'Brien, and G. H. Sloane-Stanley, J.Physiol. (London) 107,18P (1948).
30. W. von Schuler and R. Meier, Helv. Physiol, et Pharmacol. Acta 13, 106 (1955).
31. G. Zbinden, L. O. Randall, and R. A. Moe, Diseases Nervous System 21, P t 2 , 89 (1960).
32. D. Palm, Arch, exptl. Pathol. Pharmacol. 234, 206 (1958).
33. H. F. Schott and W. G. Clark, J. Biol. Chem. 196, 449 (1952).
34. C. Schoepf and W. Salzer, Ann. Chem. 544,1 (1940).
35. D. Heyl, E. Luz, S. A. Harris, and K. Folkers, J. Am. Chem. Soc. 74, 414 (1952).
36. T. L. Sourkes, Rev. can. biol. 14, 49 (1955).
37. J. Pellerin and A. D'lorio, Can. J. Biochem. Physiol. 35, 151 (1957).
38. H. Blaschko, J. Physiol. (London) 101, 337 (1942).
39. O. Schales and S. S. Schales, Arch. Biochem. 24, 83 (1949).
40. G. J. Martin and J. M. Beiler, Arch. Biochem. 15, 201 ( 1 9 4 7 ) ; P. Gonnard, Bull. soc. chim. 3 3 , 1 4 (1951).
41. H. J. Dengler, E. Rauchs, and W. Rummel, Arch, exptl. Pathol. Pharmakol.
Naunyn-Schmeideberg's 243, 366 (1962).
42. T. L. Sourkes, Rev. can. biol. 17, 328 (1958).
43. J. Axelrod, Physiol. Revs. 39, 751 (1959).
44. D. Richter and F. C. Macintosh, Am. J. Physiol. 135, 1 (1941).
45. G. Corte and W. Johnson, Proc. Soc. Exptl. Biol. Med. 97, 751 (1958).
46. H. Blaschko, D. Richter, and H. Schlossman, Biochem. J. 31, 2187 (1937).
47. J. R. Fouts, L. A. Blanksma, J. A. Carbon, and E. A. Zeller, J. Biol. Chem.
225, 1025 ( 1 9 5 7 ) ; P. Hagen and N. Weiner, Federation Proc. 18, 1005 (1959).
48. J. S. Friedenwald and H. Herrmann, J. Biol. Chem. 146, 411 (1942).
49. T. P. Singer and E. S. G. Barron, J. Biol. Chem. 157, 241 (1945).
50. J. R. Lagnado and T. L. Sourkes, Can. J. Biochem. and Physiol. 34, 1185 (1956).
51. F. J. Philpot, Biochem. J. 31, 856 (1937).
52. J. R. Lagnado and T. L. Sourkes, Can. J. Biochem. and Physiol. 34, 1095 (1956).
19. INHIBITORS OF CATECHOL AMINE METABOLISM 97 53. J. Hawkins, Biochem. J. 51, 399 (1952).
54. N. Allegretti and D. Vukadinovic, Arhiv kem. 22, 191 ( 1 9 5 0 ) ; in Chem.
Abstr. 46,10449a (1952).
55. Μ. H. Wiseman and T. L. Sourkes, Biochem. J. 78, 123 (1961).
55a. Μ. H. Wiseman-Distler and T. L. Sourkes, Can. J. Biochem. and Physiol.
41, 57 (1963).
56. P. J. G. Mann and J. H. Quastel, Biochem. J. 34, 414 (1940).
57. R. W. Schayer, Κ. Υ. T. Wu, R. L. Smiley, and Y. Kobayashi, J. Biol.
Chem. 210,259 (1954).
58. U. S. von Euler and S. Hellner-Bjorkman, Acta Physiol. Scand. 33, Suppl.
1 1 8 , 2 1 (1955).
59. Μ. E . Greig, R. A. Walk, and A. J. Gibbons, J. Pharmacol. Exptl. Therap.
127, 110 (1959).
60. H. Blaschko and R. Duthie, Biochem. J. 39, 347 (1945).
61. E. A. Zeller, J. Barsky, J. R. Fouts, W. F. Kirchheimer, and L. S. Van Orden, Experientia 8, 349 ( 1 9 5 2 ) ; E . A. Zeller and J. Barsky, Proc.
Soc. Exptl. Biol. Med. 81, 459 (1952).
62. A. N. Davison, Biochem. J. 67, 316 (1957).
63. R. W. Schayer and R. L. Smiley, J. Biol. Chem. 202, 425 ( 1 9 5 3 ) ; M. Good- all, Pharmacol. Revs. 11, 416 (1959).
64. R. W. Schayer, R. L. Smiley, K. J. Davis, and Y. Kobayashi, Am. J. Phys- iol. 182,285 (1955).
65. S. J. Corne and J. D. P. Graham, J. Physiol. (London) 135, 339 (1957).
66. M. Goldstein, A. J. Friedhoff, and C. Simmons, Biochim. et Biophys. Acta 33, 572 (1959).
67. A. N. Davison, Physiol. Revs. 38, 729 (1958).
68. B. G. Brown and P. Hey, Brit. J. Pharmacol. 11, 58 (1956).
69. M. D. Armstrong, A. McMillan, and Κ. N. F. Shaw, J. Biol. Chem. 218, 293 ( 1 9 5 6 ) ; F. De Eds, A. N. Booth, and F. T. Jones, ibid. 225, 615
( 1 9 5 7 ) ; J. Pellerin and A. D'lorio, Rev. can. biol. 16, 371 (1957).
70. J. Pellerin and A. D'lorio, Proc. 21st Meeting, Can. Physiol. Soc, Ottawa, 1957; J. Pellerin and A. D'lorio, Can. J. Biochem. and Physiol. 36, 491
( 1 9 3 8 ) ; J. Axelrod, Science 126, 400 (1957).
71. J. Axelrod, W. Albers, and C. D. Clemente, J. Neurochem. 5, 68 (1959).
72. A. D'lorio, in "Methods in Medical Research" (J. H. Quastel, ed.), Vol. 9, p. 208. Yearbook, Chicago, Illinois, 1961.
73. J. Champagne, A. D'lorio, and A. Beaulnes, Science 132, 419 (1960).
74. J. Axelrod and M. J. Laroche, Science 130, 800 (1959).
75. S. Udenfriend, C. R. Creveling, M. Ozaki, J. W. Daly, and B. Witkop, Arch. Biochem. Biophys. 84, 249 (1959).
76. A. D'lorio and J. Leduc, Arch. Biochem. Biophys. 87, 224 (1960).
77. A. M. Bacq, L. Gosselin, A. Dresse, and J. Renson, Science 130, 453 (1959).
77a. B. Belleau and J. Burba, Biochem. et Biophys. Acta 54, 195 (1961).
77b. J. Musacchio and M. Goldstein, Federation Proc. 21, 334 (1962).
77c. C. Mavrides, K. Missala, and A. D'lorio, Can. J. Biochem. and Physiol. 41, 1581 (1963).
78. O. Thibault, Compt. rend. soc. biol. 142, 499 (1948).
79. A. Spinks and J. H. Burn, Brit. J. Pharmacol. 7, 93 (1952).
80. M. Zile and H. A. Lardy, Arch. Biochem. Biophys. 82, 411 (1959).
98 Τ. L. SOURKES AND A. D'lORIO 81. E . L. Oginsky, Arch. Biochem. 26, 327, (1950).
81a. A. D'lorio and C. Mavrides, Can. J. Biochem. and Physiol. 40, 1454 (1962).
81b. A. D'lorio and C. Mavrides, Can. J. Biochem. and Physiol. 41, 1779 (1963).
81c. A. D'lorio and C. Mavrides, in "Symposium on Regulation of Enzyme Activity and Synthesis in Normal and Neoplastic Liver," Oct. 1962, Indianapolis (G. Weber, ed.) Pergamon Press, N e w York (1963).
82. H. Blaschko, G. V. R. Born, A. D'lorio, and N. R. Eade, J. Physiol. (Lon
don) 133, 548 (1956) ; B. Falck, N. A. Hillarp, and B. Hbgberg, Acta Physiol. Scand. 36, 360 (1956).
83. N. R. Eade, Rev. can. biol. 17, 299 (1958).
84. M. Nickerson, Pharmacol. Revs. 1, 27 (1949).
85. R. P. Ahlquist, Pharmacol. Revs. 11, 441 (1959).
86. Η. H. Dale, J. Physiol. (London) 34, 163 (1906).
87. R. P. Ahlquist, Am. J. Physiol. 153, 586 (1948).
88. R. F. Furchgott, Pharmacol. Revs. 11, 429 (1959).
89. B. Belleau, Can. J. Biochem. and Physiol. 36, 731 (1958).
90. H. J. Schumann, Arch, exptl. Pathol. Pharmakol. Naunyn-Schmiedeberg's 238, 41 (1960) ; U. S. von Euler and F. Lishajko, Experientia 16, 376
(1960).
91. A. D'lorio and J. G. Lague, Can. J. Biochem. and Physiol. 41, 121 (1963).