Contents lists available atScienceDirect
Journal of Pharmaceutical and Biomedical Analysis
j o u r n a l h o m e p a g e :w w w . e l s e v i e r . c o m / l o c a t e / j p b a
Development, validation and application of LC–MS/MS method for quantification of amino acids, kynurenine and serotonin in human plasma
Dávid Virág
a, Márton Király
a, László Drahos
b, Andrea Edit Édes
c,d, Kinga Gecse
c,d, György Bagdy
d,e, Gabriella Juhász
c,d, István Antal
a, Imre Klebovich
a,
Borbála Dalmadi Kiss
a, Krisztina Ludányi
a,∗aDepartmentofPharmaceutics,SemmelweisUniversity,H ˝ogyesEndreutca7,BudapestH-1092,Hungary
bMSProteomicsResearchGroup,InstituteofOrganicChemistry,ResearchCentreforNaturalSciencesoftheHungarianAcademyofSciences,Magyar tudósokkörútja2,BudapestH-1117,Hungary
cSE-NAP2GeneticBrainImagingMigraineResearchGroup,SemmelweisUniversity,Nagyváradtér4,BudapestH-1089,Hungary
dDepartmentofPharmacodynamics,FacultyofPharmacy,SemmelweisUniversity,Nagyváradtér4,BudapestH-1089,Hungary
eMTA-SENeuropsychopharmacologyandNeurochemistryResearchGroup,HungarianAcademyofSciences,SemmelweisUniversity,Nagyváradtér4, BudapestH-1089,Hungary
a r t i c l e i n f o
Articlehistory:
Received16August2019
Receivedinrevisedform6November2019 Accepted27November2019
Availableonline28November2019
Keywords:
LC–MS/MS Aminoacid Serotonin Kynurenine
Fit-for-purposevalidation Surrogatematrix
a b s t r a c t
Alteredserotonergicneurotransmissionisakeyfactorinseveralneurologicandpsychiatricdisorders suchasmigraine.Humanandanimalstudiessuggestthatchronicallylowinterictalserotoninlevels ofplasmaandbrainmayfacilitateincreasedactivityofthetrigeminovascularpathway,andmaycon- tributetodevelopmentofrepeatedmigraineattacks.However,brainserotoninsynthesisisaffectedby theconcentrationoftryptophan,itsmetabolitesandanumberofaminoacids.Inthisworkasimpleand robustLC–MS/MSmethodforthequantitativedeterminationofvaline,leucine,isoleucine,phenylala- nine,tyrosine,tryptophan,serotoninandkynurenineinhumanplasmahasbeendevelopedandvalidated.
Samplepreparationwasachievedbyproteinprecipitation,usingtrifluoroaceticacid.Chromatographic separationwascarriedoutonaSupelcoAscentis®ExpressC18column(3.0mmi.d.×150mm,2.7m) equippedwithanAgilentZorbaxEclipseXDBC8guard-columnunderisocraticconditionsataflowrate of0.4mL/min,overa6.5minruntime.Mobilephasewas0.2%trifluoroaceticacid–acetonitrile(85:15, v/v).Theeightanalytesandtwointernalstandardswereionizedbypositiveelectrosprayionizationand detectedinmultiplereactionmonitoringmode.
A“fit-for-purpose”validationapproachwasadoptedusingsurrogatematrixforthepreparationof calibrationsamples.Thecalibrationcurvesofallanalytesshowedexcellentlinearitieswithacorrelation coefficient(r2)of0.998orbetter.Spikedsurrogatematrixsamplesandpooledhumanplasmawere usedasqualitycontrolsamples.Intra-dayandinter-dayprecisionswerelessthan11.8%and14.3%, andaccuracieswerewithintherangesof87.4–114.3%and87.7–113.3%,respectively.Stabilityofthe componentsinstandardsolutions,surrogatematrix,pooledplasmaandprocessedsampleswerefound tobeacceptableunderallrelevantconditions.Nosignificantcarryovereffectwasobserved.Thesurrogate matrixbehavedparalleltohumanplasmawhenassessedbystandardadditionmethodanddilutingthe authenticmatrixwithsurrogatematrix.Themethodwassuccessfullyappliedforanalysisof800human plasmasamplestosupportaclinicalstudy.
©2019TheAuthors.PublishedbyElsevierB.V.ThisisanopenaccessarticleundertheCCBYlicense (http://creativecommons.org/licenses/by/4.0/).
∗Correspondingauthor.
E-mailaddress:ludanyi.krisztina@pharma.semmelweis-univ.hu(K.Ludányi).
1. Introduction
Alteredserotonergicneurotransmissionisakeyfindinginsev- eralneurologicandpsychiatricdisordersincludingmigraine[1].
Migraineisacommondisablingprimaryheadachedisorderwith a globalprevalenceof 15–18%[2]. It is characterizedby throb- https://doi.org/10.1016/j.jpba.2019.113018
0731-7085/©2019TheAuthors.PublishedbyElsevierB.V.ThisisanopenaccessarticleundertheCCBYlicense(http://creativecommons.org/licenses/by/4.0/).
2 D.Virág,M.Király,L.Drahosetal./JournalofPharmaceuticalandBiomedicalAnalysis180(2020)113018
bingandusuallyunilateralmoderateorseverepainaccompanied byphono-andphotophobia,nauseaorvomiting,andworsening byroutinephysicalactivity[3].Humanandanimalstudiessug- gestthatchronicallylowinterictalserotoninlevelofplasmaand brainpredisposetoincreasedsensitivityofthetrigeminovascu- larpathwaythatmaycontributetothedevelopmentofrepeated migraineattacks[1,2,4,5].Inlinewiththeserotonergicdysfunc- tionhypothesis,the5-HT1B/1D(5-hydroxytryptaminereceptor1B and1Dsubtypes)agonisttriptansalleviatemigrainepain,while tryptophan depletion, which acutely decreases the brain sero- tonin concentration, increasesthe excitability of the brain and symptoms of the migraine attacks [1,5,6]. However, the brain serotoninconcentrationisaffectednotonlybytryptophanintake butalsobytheratiooftheplasmaconcentrationsoftryptophan andotherlargeneutralamino acids(LNAA)that arecompeting tooccupyatransporterattheblood brainbarrier,calledL-type amino acid transporter 1 (LAT1). Fernstrom defined a ratio of plasmatryptophanand LNAAs, namelytyrosine, phenylalanine, leucine,isoleucineand valine,which influenced thebraintryp- tophanconcentrations and serotoninsynthesis in experimental studies. We selected these LNAAs for this measurement based onhisstudy[7].Investigationoftheseaminoacidsisimportant becausedietarymanipulationoftryptophan/LNAAratiopromptly abletoelicitbehavioralalterations,althoughtheexactmechanism isnotfullyunderstood.Forexample,tryptophandepletionwith ingestionofatryptophanfreeaminoacidmixtureisacutelyable toincrease anxiety,lowermoodand contributetomoresevere courseofmigraineattacksinvulnerablesubjects[6–8].Thusour aimwastodevelop a methodtosimultaneouslymeasure tryp- tophan, LNAAs and the peripheral concentrations of serotonin and kynurenine (another compound synthesized from trypto- phanand have been implicatedin migraine) in humanplasma [9].
During a typical bioanalytical validation procedure, calibra- tion and quality control (QC) samplesare prepared by spiking the same sort of biological matrix as the study samples with knownamountsofanalyte.Thisensuresthattheanalyteofinterest facessameconditionswithrespecttomatrixeffectsandextrac- tion recoveries in the validation samples as well as the study samples.In thecase of endogenouscompounds, preparation of validationsamplesarehamperedbytheabsenceofanalyte-free (blank)matrix[10].Asaccuratequantitativeanalysisofendoge- nousanalytesiscrucial foranumber ofclinicalandnonclinical applications,increasinginterestisarisingforalternative,so-called
“fit-for-purpose” quantitation/validation approaches. There are fourcommonlyusedapproaches:standardaddition,background subtraction, surrogateanalyte in authentic matrix and authen- ticanalyteinsurrogatematrix[11–16].Applicability,advantages and drawbacksof these methods have recently been reviewed [17].
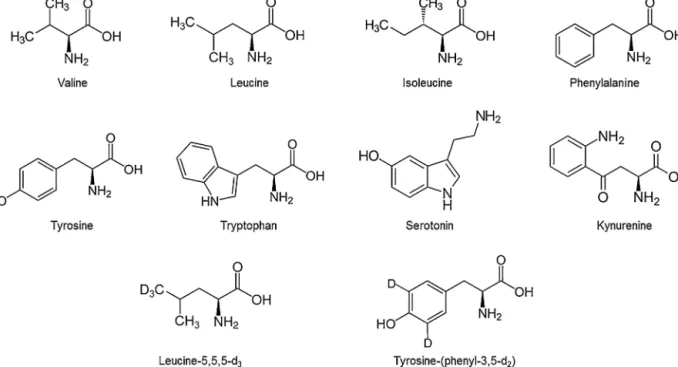
In the present article, we report a quick, accurate and reliable liquid chromatography-tandem mass spectrometry (LC–MS/MS) method for quantitative determination of valine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, sero- toninandkynureninein800humanplasmasamples.Twostable isotope-labeledinternalstandards(SIL-ISs)wereusedtoreduce assayvariation:leucine-5,5,5-d3(Leu-d3)foraliphaticaminoacids and tyrosine-(phenyl-3,5-d2) (Tyr-d2) for aromatic compounds, respectively.Duetothelargenumberofsamplesandcompounds tobeanalyzed,authentic analyte in surrogatematrix approach wasadoptedbyusingartificialplasmaasasurrogatematrixfor theconstructionofcalibrationstandardsand QCsamples.Some assayvalidationissuesofthemethodofchoicesuchasparallelism andlimitofquantitation(LOQ)arealsodiscussed.Fig.1showsthe eightanalytesandthetwoSIL-ISs.
2. Materialsandmethods
2.1. Chemicals
LC–MS grade acetonitrile waspurchased fromVWR Chemi- cals(Pool,England).LC–MSgradetrifluoroaceticacid(TFA)was purchased from Fisher Chemical (Loughborough, UK). Leucine (99.9%),isoleucine(99.5%),valine(99.3%),phenylalanine(99.0%), tyrosine (99.7%) were purchased from Dr. Ehrenstorfer GmbH (Augsburg,Germany).Tryptophan(≥99%)wasobtainedfromAlfa Aesar (Haverhill, MA, USA). Kynurenine (≥98%) was purchased fromCaymanChemicalCompany(AnnArbor,MI,USA).Serotonin (≥98%),l-leucine-5,5,5-d3 (99%Datom),l-tyrosine-(phenyl-3,5- d2) (98%D atom), phosphate bufferedsaline(PBS) and human serumalbumin(HSA)(≥96%)werepurchasedfromSigmaAldrich Inc.(St.Louis,MO,USA).K3EDTAtubeswereobtainedfromGreiner Bio-OneInternationalGmbH(Kremsmünster,Austria).Ultrapure waterwaspreparedwithanin-houseSimplicity®WaterPurifica- tionSystem(MerckMillipore,Burlington,MA,USA).
2.2. Standardsolutions
Stock solutions of the analytesand internal standards were madeupin0.2%trifluoroaceticacid–acetonitrile(85:15,v/v)and storedat−20◦C.Onthedayofanalysis,calibrationandQCstan- dardsolutions werepreparedbyserial dilutionoftwo separate primarystocksolutionswithwater.ISworkingsolutionwaspre- paredinwatertogiveaconcentrationof250g/mLforLeu-d3and 25g/mLforTyr-d2.Surrogatematrixwaspreparedbyadding4g ofHSAto100mLPBSsolution.
2.3. Plasmasamples
Humanbloodsampleswerecollectedinto3mLK3EDTAtubes frommigrainepatientswithoutauraandhealthyvolunteers(total numberofparticipantswas98,totalnumberofbloodsampleswas 800)byintravenouscannulation.Thesampleswereimmediately centrifuged,thenplasmasampleswerefrozenandkeptat−80◦C untiltheassay.Pooledplasmawaspreparedbymixingequalvol- umesofplasmasobtainedfrom6healthyindividuals.Thestudy protocolwasapprovedbytheScientificandResearchEthicsCom- mitteeoftheMedicalResearchCouncil,Budapest,Hungary.
2.4. Samplepreparation
Samplepreparationwasachievedbysimpleproteinprecipita- tion.ForthepreparationofcalibrationsamplesandQCsamplesin surrogatematrix,100Lofspikingstandardsolutionsand20Lof ISworkingsolutionweremixedwith900Lofsurrogatematrix.
Incaseofstudysamples,100Lofwaterand20LofISworking solutionweremixedwith900Lofhumanplasma.Forproteinpre- cipitation,200Loftrifluoroaceticacidwasaddedtoeachsample andvortexmixed.Aftercentrifugationat4000gfor10minat4◦C, aliquots(150L)ofthesupernatantweretransferredtoautosam- plervials.5Loftheresultingsolutionswereinjectedintothe LC–MS/MSsystem.
2.5. Liquidchromatography-tandemmassspectrometry
ThechromatographicseparationswereperformedonanAgilent 1260InfinityLCsystem(AgilentTechnologies,CA,USA).Theana- lyteswereseparatedonaSupelcoAscentis®ExpressC18column (3.0mmi.d.×150mm,2.7m)equippedwithanAgilentZorbax EclipseXDBC8guard-column(4.6mmi.d.×12.5mm,5m).The columnandtheautosamplerweremaintainedat25◦Cand4◦C, respectively.Thealiquotsofsampleswereelutedunderisocratic
Fig.1. Chemicalstructureoftheeightanalytesandtwointernalstandards.
conditionsover6.5minataflowrateof0.4mL/min.Themobile phase was composed of 0.2% trifluoroacetic acid– acetonitrile (85:15,v/v).Theneedlewaswashedfor10sintheflushportbefore everyinjectioninordertominimizecarryovereffect.
SampleswereanalyzedbyanAgilent6460triple-quadrupole massspectrometer(AgilentTechnologies, SantaClara, CA,USA) usingpositive electrospray ionization(ESI)and scheduledmul- tiple reaction monitoring (MRM) mode. Agilent MassHunter DataAcquisitionsoftware(versionB.04.01)wasusedtocontrol theequipment,MassHunterQuantitative QQQAnalysis(version B.05.01)andQualitativeAnalysissoftware(versionB.05.00)were appliedforquantitationanddataprocessing.Settingswereasfol- lows:capillaryvoltage,+3.5kV;nozzlevoltage,+400V.Nitrogen wasappliedasanebulizergasof45psi,acarriergasof10L/min at350◦C,andasheathgasof11L/minat350◦C.MRMtransitions, collisionenergies(CE)andfragmentorvoltagesforallcompounds wereautooptimizedbyOptimizerofAgilentMassHunterworksta- tion.
2.6. Validation
2.6.1. Calibrationcurves,LOQ
Thespikingstandardsolutions ofcalibrationstandardswere dilutedfromthestocksolutiontoobtaineightcalibrationlevels, andraninduplicateatthebeginningandtheendofeachbatch.The lowestandhighestpointsofthecalibrationcurvecoincidedwith thelowerlimitofquantitation(LLOQ)aswellastheupperlimitof quantitation(ULOQ).Thecalibrationcurvewasderivedbyplotting theconcentrationofthestandardsversustheanalytetoISpeak arearatiosusing1/xweightedleast-squareslinearregressions.
2.6.2. Accuracyandprecision
FourconcentrationlevelsofQCsampleswereusedforamino acids:LLOQ,lowQC,mediumQC,highQC,andthreeconcentra- tionlevelsforserotoninandkynurenine(LLOQ,lowQC,highQC) inordertocoverthecalibrationrange.Concentrationswereset accordingtotheEMAGuidelineonbioanalyticalmethodvalida- tion[18].Furthermore,pooledplasmawasappliedasanadditional levelofQCtoevaluatepotentialerrorscausedbymatrixdiffer-
ences[10].Endogenouslevelsofthesesamplesweredetermined bythemethodofstandardaddition.Calibrationrangesandcon- centrationsoftheQCsamplesaresummarizedinTable1.Intra-day accuracyandprecisionwereassessedbyevaluatingfivereplicates ofeachQCsamplesdescribedabove.Theinter-dayaccuracyand precisionwereestablishedbytherepetitionoftheintra-dayvali- dationprocedureonfiveconsecutivedays.Accuracywasexpressed aspercentageofthenominalconcentrationandprecisionwascal- culatedasthecoefficientvariation(%CV).Theacceptancecriteria forbothparametersweresetat±15%exceptfortheLLOQforwhich itshouldbewithin±20%[18].
2.6.3. Extractionrecovery,parallelism,dilutionintegrity
Extractionrecoverieswereevaluatedbycomparingtheanalyte responsesobservedin pre-spikedandpost-spiked samples.The experimentwasperformedinsixreplicates.Pre-spikedsamples werepreparedbyadding100LofhighQCstandardsolutionand 20LofISstandardworkingsolutionto900Lofsurrogatematrix, thenprecipitatedwithTFAandcentrifuged.Inthecase ofpost- spikedsamples,1000Lofsurrogatematrix(blanksample)was processedinthesameway.90Loftheresultingsupernatantwere mixedwith10LofhighQCstandardsolutionand2LofISstan- dardworkingsolution.Extractionrecovery(%)wascalculatedas peakareaofpre-spikedsamples/peakareaofpost-spikedsamples x100.IS-normalizedextractionrecovery(%)wasalsoexpressed fortheanalytesasextractionrecoveryoftheanalyte/extraction recoveryoftheISx100.
Thetermparallelismisgenerallyunderstoodtomeanhowwell asetofcalibrationstandardstracktheresponseoftheanalyteof interestinthebiologicalmatrix[19].Evaluationofparallelismis ofcriticalrelevancewhenusingsurrogatematrix,ashighdegree ofsuchparameterindicatesthatthereisnosignificantdifference betweentheauthenticandthesurrogatematrixwithrespectto extractionrecoveriesandmatrixeffects.Determinationofparal- lelismwascarriedoutbasedupon theexperiment proposedby Houghton etal.[20]. Endogenousconcentrationsof sixindivid- ualplasma samplesweredeterminedin triplicates bystandard addition.ThesamesampleswerespikedwithmediumQCstan- dardsolutionandwereseriallydilutedwithsurrogatematrixtwo,
4 D.Virág,M.Király,L.Drahosetal./JournalofPharmaceuticalandBiomedicalAnalysis180(2020)113018
Table1
CalibrationrangesandQClevelsoftheanalytes.
QCtype Surrogatematrix Pooledplasma
Analyte Calibrationrange(g/ml) QClevel(g/ml)
LLOQ Low Medium High
Valine 1–100 1 3 20 80 24.05
Leucine 1–100 1 3 20 80 13.09
Isoleucine 0.5–50 0.5 1.5 10 40 7.82
Phenylalanine 1–100 1 3 20 80 6.98
Tyrosine 0.5–50 0.5 1.5 10 40 11.88
Tryptophan 0.5–50 0.5 1.5 10 40 7.81
Serotonin 0.1–2.5 0.1 0.3 – 2 0.20
Kynurenine 0.1–2.5 0.1 0.3 – 2 0.44
fiveandtentimes.Thenthedilutedandundilutedsampleswere measuredagainstsurrogatecalibrators.The%REand%CVofthe back-calculatedconcentrationsweremaximizedin±15%[20].By trackingtheeffectofdilutingtheplasmawithsurrogatematrix,the methodcanalsobeconsideredasadilutionintegritytest.
2.6.4. Stability
Stability of thestock solutions and working solutions were assessed atroom temperature (20◦C) for 24h and at 10◦C for tendays.Working solutionsof theeight analyteswerestudied usingbracketingapproach:onlythelowestandthehighestcal- ibrationstandardsweremeasured.Thelatterwasalsothestock solution.Stabilityoftheanalytesinmatrixwereevaluatedusing lowandhighQCsamplespreparedinsurrogateplasmaandpooled humanplasmasamples.Eachlevelswereanalyzedintriplicates after beingexposed to different conditions:room temperature for 24h, −20◦C for two months and three freeze-thaw cycles.
Stability of processed samples were determined at room tem- perature for 12hand autosampler temperature (4◦C) for 24h.
Sampleswereconsideredstablewhen the%RE waswithin15%
[18].
2.6.5. Carryover
Carryoverwasanalyzedbyassayingblanksamples(processed surrogatematrixsamples)injectedaftertheanalysisofapooled plasmasamplespikedwiththehighestcalibrationstandard.The acceptancecriteriaweresetat20%analyteresponseoftheLLOQ fortheanalytesofinterestand5%fortheISs[18].
2.7. Applicationofthemethodtoclinicalhumanplasmasamples Thevalidated methodwasappliedto800samplescollected duringtheclinicalstudydescribedearlier.Thesamplesweremea- suredintenseparateanalyticalruns.Surrogatecalibratorswere runatthebeginningandtheendofeachbatch.Low,medium,high QCsandpooledplasma sampleswereanalyzed induplicatesat eachbatchtokeeptrackofaccuracyandprecision.Reproducibil- ityofthedescribedmethodforincurredstudysampleswastested byreanalysisof10%ofthesamples(n=80).Reanalyzedsamples wereconsideredacceptablewhenthedifferencebetweenthepairs oftheresultswerewithin20%foratleast66.7%ofthesamples [18].
3. Resultsanddiscussion
3.1. Liquidchromatography-tandemmassspectrometry
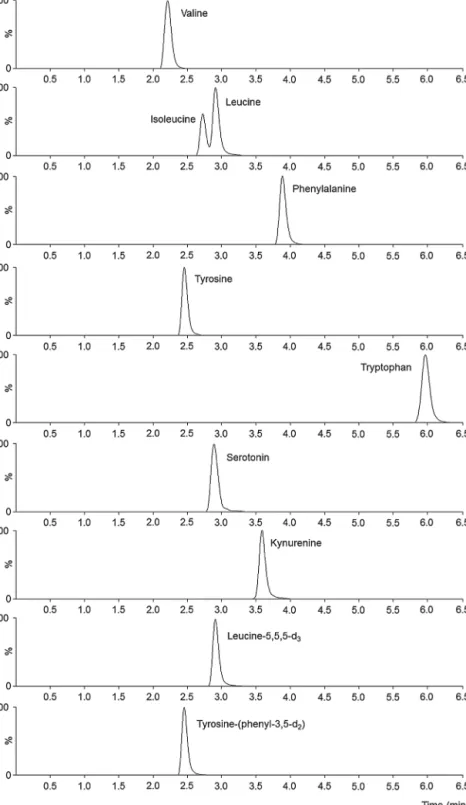
Thereisavastamountofliteratureonthequantitativedeter- minationof amino acids in plants, animal and humansamples [21–23].However,tothebestofourknowledge,nobioanalytical LC–MS/MSmethodhasbeenreportedforthesimultaneousanalysis
Table2
MRMparametersoftheanalytesandinternalstandards.
Compound Transition(m/z) Fragmentorvoltage(V) Collisionenergy(eV)
Valine 118→72 10 8
Leucine 132→86 2 8
Isoleucine 132→86 2 8
Phenylalanine 166→120 2 8
Tyrosine 182→136 30 12
Tryptophan 205→188 40 4
Serotonin 177→160 45 4
Kynurenine 209→192 45 4
Leu-d3 135→89 45 4
Tyr-d2 184→138 45 10
ofaminoacids,serotoninandkynurenineinhumanplasma.Amino acidsareveryoftenderivatizedinorder toenhancefluorescent detection selectivity, sensitivity or chromatographic resolution [23,24].However,thederivatizationagentcanbringanadditional sourceofinterferencesand errorsintothesystemcomplicating methodvalidation[24].Itshouldbealsotakenintoaccountthat derivatizationcansignificantlyincreasethelaborintensityofthe method,which can becrucial when thenumber of samplesto beanalyzedislarge. Inthis particularcase,where theaimwas themeasurementof800(withvalidationsamplesfarover1000) plasmasamples,weprimarilyfocusedonspeed,simplicityandhigh throughputduringmethoddevelopment.MSconditionsincluding MRMtransitions,collisionenergies(CE)andfragmentorvoltages wereindividuallyoptimizedforallanalytesandSIL-ISsinpositive ionmodebyOptimizerofAgilentMassHunterworkstationinorder toenhanceselectivityandsensitivityofthemethod.Asshownin Table2,theoptimizationprocedureresultedinsameMRMtransi- tionsforisomericaminoacidsleucineandisoleucine.Becauseof theinsufficientmassspectrometricselectivity,properchromato- graphic resolution wasnecessary to separatesuch compounds withoutcompromisingpeakshape orareasonablyshortanalyt- icalrun time. As representative MRMchromatograms in Fig.2 demonstrates,aSupelcoAscentis® ExpressC18column(3.0mm i.d.×150mm,2.7m)providedsatisfactoryseparationoftheana- lyteswithina6.5minrun.Anisocraticconditionconsistingof0.2%
TFA-acetonitrile(85:15,v/v)atflowrateof0.4mL/minwascho- seninordertoavoidtheneedforcolumnreequilibrationbetween consecutiveinjections.AZorbaxEclipseXDBC8guard-columnwas usedtoprotecttheanalyticalcolumn,therefore,performanceof theseparationintermsofpeakshapesandretentiontimeswas consistentthroughoutthestudy.
3.2. Validation
3.2.1. Calibrationcurves,LLOQ
Themeanregressioncoefficients(r2)foralltheanalyteswere over0.998indicatingexcellentlinearities.Thecalibrationranges weresufficienttocoveranalyteconcentrationsinalmostallhuman
Fig.2.ExtractedMRMchromatogramsoftheanalytesandinternalstandards.
plasmasamples.LLOQ is describedas thelowestconcentration withacceptableaccuracyandprecisionwhichlargelydependson thesensitivityoftheinstrument[18].However,inthisstudy,only theLLOQofserotoninandkynurenineweredeterminedwiththis approach.In thecaseof aminoacids,measurement ofaccuracy andprecisionattheLLOQmaynotbeadequate,consideringthat suchconcentrationsareoutsidethephysiologicallyrelevantrange [25].Thusthelowestcalibrationlevels,followingamorepragmatic approach,weresettobeapproximatelyonetenthoftheconcen- trationsmeasuredinpooledplasma.Thismethodsimplifiesthe preparationand dilutionofcalibrationstandardswhenworking withalargenumberofanalytes,whilecoveringtherelevantcon-
centrationrange,evenifdownregulationoftheanalytesisexpected [20,25].
3.2.2. Accuracyandprecision
Intra-day and inter-day accuracies and precisions for surro- gate matrix andpooled plasma QC samplesaresummarized in Table3.Inthisstudy,theintra-dayandinter-dayprecisionswere lessthan11.8%and14.3%,andaccuracieswerewithintheranges of87.4–114.3%and87.7–113.3%,respectively.Theresultsindicate thatthemethodisaccurateandpreciseenoughforthemeasure- mentofplasmasamples.
6 D.Virág,M.Király,L.Drahosetal./JournalofPharmaceuticalandBiomedicalAnalysis180(2020)113018
Table3
Intra-dayandinter-dayaccuracyandprecisionoftheQCsamples.
QCtype Surrogatematrix Pooledplasma
QClevel LLOQ Low Medium High
Accuracy(%) CV(%) Accuracy(%) CV(%) Accuracy(%) CV(%) Accuracy(%) CV(%) Accuracy(%) CV(%)
Intra-day
Valine 111.7 7.4 93.0 11.8 100.4 11.3 110.8 8.8 92.4 3.7
Leucine 102.7 5.7 95.3 2.5 93.8 4.9 89.7 3.3 90.9 4.0
Isoleucine 114.3 7.2 104.8 3.3 108 3.2 113.3 7.0 101.3 3.9
Phenylalanine 100.2 5.2 102.1 5.6 99.8 6.4 102.9 2.8 102.3 6.4
Tyrosine 98.6 4.3 87.4 2.5 88.9 10.5 103.7 4.2 107.0 3.3
Tryptophan 106.8 6.8 106.0 6.5 95.8 10.2 108.0 4.6 97.2 6.7
Serotonin 101.0 8.1 92.7 2.9 – – 87.8 4.7 95.1 7.2
Kynurenine 110.7 8.2 114.3 10.1 – – 113.4 9.4 110.9 9.7
Inter-day
Valine 107.6 10.2 96.5 11.9 97.0 9.7 111.7 9.0 91.5 5.3
Leucine 102.5 6.4 95.6 7.8 91.5 5.6 92.4 7.1 95.8 8.2
Isoleucine 114.4 8.2 106.7 9.6 103.0 6.8 112.6 8.3 106.4 8.7
Phenylalanine 102.1 7,4 100.1 7.2 96.5 6.9 104.6 8.8 108.8 5.9
Tyrosine 111.2 14.3 102.3 13.3 102.9 10.7 109.5 7.6 104.5 3.5
Tryptophan 102.9 8.1 96.7 7.6 95.4 6.3 113.3 4.7 109.5 7.0
Serotonin 99.5 14.2 87.7 7.7 – – 91.1 7.4 89.2 9.1
Kynurenine 92.4 13.8 90.7 10.6 – – 95.0 12.8 91.2 11.4
3.2.3. Extractionrecovery,parallelism,dilutionintegrity
Extraction recovery of the method was evaluated by com- paringthemeanpeak areaof pre-extractedand post-extracted surrogatematrix samples. The sample preparation process did not influence considerably the concentrations of the analytes anISs(extraction recoveriesvariedbetween85.2–101.4%).Fur- thermore,theIS-normalizedextractionrecoveryvalueshighlight that the selected SIL-ISs are appropriate references of the analytes (IS-normalized extraction recoveries varied between 93.6–111.8%). Results of extraction efficiency evaluation are detailedinTable4.
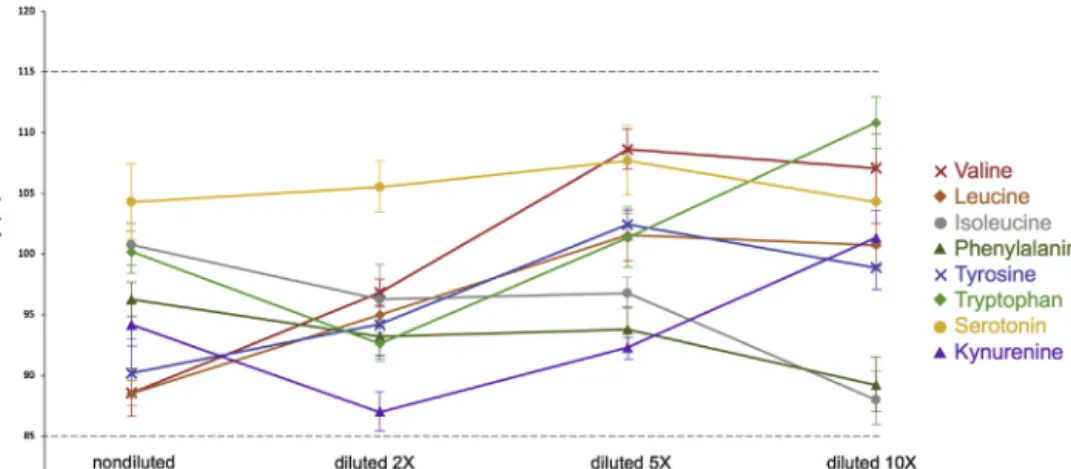
Fortheparallelismexperiment,backgroundconcentrationsof theindividualplasmasamplesweredeterminedbystandardaddi- tion.Evaluationofthesamesampleswithsurrogatecalibrators resulted very similar concentrations for all theanalytes (RE is lessthan10%).Fig.3illustrates theuseof standardadditionto assessparallelismwithsurrogatematrix methodin thecase of isoleucine.Moreover,themeasuredanalyteconcentrationsinthe spikedsamples at each dilution levels were within the accep- tancecriteria,whichindicatesthatthesurrogatematrixbehaves similarlytotheauthenticbiologicalmatrix.Fig.4illustratesthe relatively balanced accuracy profile of a representative plasma samplewhendilutedzero-two-,five-,ten-timeswithsurrogate matrix.
3.2.4. Stability
Thestocksolutionsandworkingsolutionsoftheanalytesand internalstandardswerestable foratleast24hat20◦C and for 10daysat10◦C,respectively.Humanplasmasamplesfrozento
−20◦Candthawedtoroomtemperatureforthreecyclesdidnot changeconsiderablytheconcentrationsof theanalytes.Human plasmaandsurrogatematrixspikedwithQCstandardswerestable atroomtemperaturefor24hand−20◦Cfor2months.Noapparent changewasobservedintheconcentrationsoftheprocessedsam- plesafterbeing24hatautosamplertemperature(4◦C)and12hat roomtemperature.
3.2.5. Carryover
Carryoverwasanalyzedbyinjectingblanksamplesafterpooled plasmaspikedwiththehighest calibrationstandard.AsTable5 demonstrates, responses of the analytes were 0.7–17.2% and 0.8–1.0%fortheinternalstandards,respectively.
3.3. Applicationofthemethodtoclinicalhumanplasmasamples Thevalidatedmethodwassuccessfullyappliedinsupportof aclinicalstudytomeasuretheplasmaconcentrationoftheeight analytesin 800humanplasmasamplesobtainedfrommigraine patientsand healthyindividuals.Valine,isoleucineandtyrosine concentrationswereabovetheULOQinlessthan1%ofthesam- ples.Insuchcases,anotheraliquotofthesampleweredilutedtwo timeswithsurrogatematrix,thenprocessedandmeasuredagain.
AccuracyandprecisionoftheQCsamplesdidnotchangeconsid- erablycomparedtotheresultsofthemethodvalidation,incurred samplereanalysiswasalsowithintheacceptancecriteria.Table6 summarizestheconcentrationrangesin800studysamples.
3.4. Strategiesforquantitativeanalysisofendogenous compounds
Regulatoryguidancesare designedprimarilytoaddressvali- dationofdrugmoleculesaswellastheirmetabolites.However, reflecting on the challenges of endogenous analytes, the draft version ofthe ICHguideline of bioanalytical methodvalidation dedicatesasectionforthequantitativedeterminationofthesecom- pounds,inwhichtheabove-mentionedquantitationstrategiesare detailed[26].Consideringthatbothmethodshaveadvantagesand limitations,theirapplicabilityisdeterminedbythespecificanalyt- icalchallenge.Inthepresentcase,whenthenumberofsamplesto beanalyzedislargeandtheamountofeachsampleisrelatively low(insomecases1mLorless),theuseofstandardadditionis narrowedbytherequirementformultiplemeasurementsofevery studysamples.Comparedtohealthyindividuals,downregulation ofcertainanalytesisexpectedinmigrainepatients,whichmeans that sampleswithlowerconcentration thanthecontrol matrix shouldbetakenintoaccount.Therefore, theuseofbackground subtractionis not recommendeddue tothepotentialmeasure- menterrors introducedbyextrapolationbeyondthecalibration range[20]. Thesituationisfurthercomplicatedwhen thereare multipleanalyteswithvariableendogenouslevels[17].Surrogate analyteinauthenticmatrixtypicallyrequirestwoversionsofSIL- ISsoftheanalyteofinterest[10].Despitebeingawell-accepted approach,itsapplicationislimitedbytheavailabilityandhighprice ofstableisotope-labeledanalogs,especiallywhentheanalysisof numerousanalytesisneeded.Undersuchcircumstances,surro- gatematrixapproachmaybethemethodofchoice,whichenables reliablequantitationofmultipleanalyteswithrelativelylowlabor
Table4
Meanextractionrecoveriesoftheanalytesandinternalstandardsinsurrogatematrix.
Valine Leucine Isoleucine Phenylalanine Tyrosine Tryptophan Serotonin Kynurenine Leu-d3 Tyr-d2
Extractionrecovery(%) 87.5 101.3 88.6 101.4 95.0 95.1 85.2 97.2 90.6 91.0
IS-normalizedextractionrecovery(%) 96.6 111.8 97.8 111.3 104.4 104.5 93.6 106.7 – –
Fig.3.Illustrationoftheuseofstandardadditiontoassessparallelism.Endogenousconcentrationsweredeterminedbysurrogatecalibrationandbyextrapolatingtothe negativex-interceptfromthestandardadditioncalibrationcurve.Similarityoftheresultsindicatingparallelismbetweenhumanplasmaandtheartificialmatrix.
Fig.4.Assessmentofparallelismusingdilutionlinearitytest.Spikedhumanplasmasamplesweremeasuredagainstsurrogatecalibratorswhendilutedtwo-,five-,ten-times andnondiluted.Thedatawasthemeanoftriplicates.Theanalyteconcentrationsweremultipliedbythedilutionfactors.
Table5
Carryoveroftheanalytesandinternalstandards.
Valine Leucine Isoleucine Phenylalanine Tyrosine Tryptophan Serotonin Kynurenine Leu-d3 Tyr-d2
Carryover(%) 17.2 4.8 8.3 2.1 8.7 0.7 2.6 2.7 1.0 0.8
intensity,evenwhendownregulationoftheanalytesisexpected [1,7,9].Apotentialdrawbackofthemethodisthesystematicerror thatmay beintroducedbythelack ofparallelismbetweenthe biofluidandthesurrogatematrix[19,20].However,parallelismcan beensuredbythepropercompositionofthesurrogatematrixand monitoredbydilutionlinearitytestingorstandardaddition[19].
Usingstandardadditiontoinvestigateparallelismclearlyindicates thatfit-for-purposestrategiesshouldbeconsideredascomponents ofananalyticaltoolbox,thosecanbeappliedindividuallyandin combinationtomakesurethatnoneofthevalidationcriteriaare compromised.
Table6
Concentrationrangesoftheanalytesin800studysamples.
Analyte Concentrationrange(g/ml)
Valine 4.63–115.58
Leucine 6.31–71.42
Isoleucine 1.68–97.66
Phenylalanine 1.65–36.76
Tyrosine 1.16–82.20
Tryptophan 1.17–39.35
Serotonin 0.27–1.70
Kynurenine 0.10–2.04
8 D.Virág,M.Király,L.Drahosetal./JournalofPharmaceuticalandBiomedicalAnalysis180(2020)113018
4. Conclusion
A simple, fast and reproducible LC–MS/MSbased bioanalyt- icalmethod hasbeen developed for simultaneous quantitation ofvaline,leucine,isoleucine,phenylalanine,tyrosine,tryptophan, serotoninandkynurenineinhumanplasma.Thechromatographic method wascapable to separate isomeric amino acids leucine andisoleucine.Afit-for-purposevalidationapproachwasadopted byemployingsurrogatematrixforcalibration,andpartlyforthe constructionofQC samples.Thesurrogatematrix wasfoundto beparallelwithhumanplasma,when investigatedbystandard additionanddilutionintegritytesting.Somequantitationandval- idationissues of endogenous compounds have been discussed.
Themethodwasabletomeetthecriteriaimposedbytheregu- latoryguidances ofEMAand FDA[18,27],and demonstratedits valueintheanalysisofatotalnumber800humanplasmasam- ples.
Funding
This work wassupported by the Hungarian Brain Research Program–GrantNo.KTIANAP13-2-2015-0001(MTA-SE-NAPB GeneticBrain ImagingMigraine Research Group), by the Hun- garianAcademyofSciences(MTA-SENeuropsychopharmacology and Neurochemistry Research Group), and by the Hungarian Brain Research Program – Grant No. 2017-1.2.1- NKP-2017- 00002. GK was supported by the New National Excellence ProgramoftheMinistryofHuman Capacities(ÚNKP-18-2-I-SE- 86).
ThestudywasalsofinancedbytheHigherEducationInstitu- tionalExcellenceProgrammeoftheMinistryofHumanCapacities inHungary,withintheframeworkofthemolecularbiologythe- maticprogrammeofSemmelweisUniversity.
DeclarationofCompetingInterest Authorsdeclarenoconflictofinterest.
References
[1]M.Deen,C.E.Christensen,A.Hougaard,H.D.Hansen,G.M.Knudsen,M.
Ashina,Serotonergicmechanismsinthemigrainebrain—asystematicreview, Cephalalgia37(2017)251–264,http://dx.doi.org/10.1177/
0333102416640501.
[2]P.J.Goadsby,P.R.Holland,M.Martins-Oliveira,J.Hoffmann,C.Schankin,S.
Akerman,Pathophysiologyofmigraine:adisorderofsensoryprocessing, Physiol.Rev.97(2017)553–622,http://dx.doi.org/10.1152/physrev.00034.
2015.
[3]HeadacheClassificationCommitteeoftheInternationalHeadacheSociety (IHS),Theinternationalclassificationofheadachedisorders,3rdedition, Cephalalgia38(2018)1–211,http://dx.doi.org/10.1177/0333102417738202.
[4]G.Juhasz,T.Zsombok,E.A.Modos,S.Olajos,B.Jakab,J.Nemeth,J.Szolcsanyi,J.
Vitrai,G.Bagdy,NO-inducedmigraineattack:strongincreaseinplasma calcitoningene-relatedpeptide(CGRP)concentrationandnegative correlationwithplateletserotoninrelease,Pain106(2003)461–470.
[5]R.C.A.Guedes,M.dasG.R.deAraújo,T.C.Verc¸osa,F.M.Bion,A.L.deSá,A.
Pereira,R.Abadie-Guedes,Evidenceofaninversecorrelationbetween serotonergicactivityandspreadingdepressionpropagationintheratcortex, BrainRes.1672(2017)29–34,http://dx.doi.org/10.1016/j.brainres.2017.07.
011.
[6]P.D.Drummond,Tryptophandepletionincreasesnausea,headacheand photophobiainmigrainesufferers,Cephalalgia26(2006)1225–1233,http://
dx.doi.org/10.1111/j.1468-2982.2006.01212.x.
[7]J.D.Fernstrom,Largeneutralaminoacids:dietaryeffectsonbrain
neurochemistryandfunction,AminoAcids45(2013)419–430,http://dx.doi.
org/10.1007/s00726-012-1330-y.
[8]S.N.Young,Acutetryptophandepletioninhumans:areviewoftheoretical, practicalandethicalaspects,J.PsychiatryNeurosci.38(2013)294–305, http://dx.doi.org/10.1503/jpn.120209.
[9]L.Vécsei,L.Szalárdy,F.Fülöp,J.Toldi,KynureninesintheCNS:recent advancesandnewquestions,Nat.Rev.DrugDiscov.12(2013)64–82,http://
dx.doi.org/10.1038/nrd3793.
[10]W.Jian,R.W.Edom,N.Weng,Importantconsiderationsforquantitationof small-moleculebiomarkersusingLC–MS,Bioanalysis4(2012)2431–2434, http://dx.doi.org/10.4155/bio.12.247.
[11]Y.Huang,A.Louie,Q.Yang,N.Massenkoff,C.Xu,P.W.Hunt,W.Gee,Asimple LC–MS/MSmethodfordeterminationofkynurenineandtryptophan concentrationsinhumanplasmafromHIV-infectedpatients,Bioanalysis5 (2013)1397–1407,http://dx.doi.org/10.4155/bio.13.74.
[12]J.Jiang,C.A.James,P.Wong,Bioanalyticalmethoddevelopmentand validationforthedeterminationofglycineinhumancerebrospinalfluidby ion-pairreversed-phaseliquidchromatography-tandemmassspectrometry, J.Pharm.Biomed.Anal.128(2016)132–140,http://dx.doi.org/10.1016/j.jpba.
2016.05.019.
[13]X.Lai,J.A.Kline,M.Wang,Development,validation,andcomparisonoffour methodstosimultaneouslyquantifyl-arginine,citrulline,andornithinein humanplasmausinghydrophilicinteractionliquidchromatographyand electrospraytandemmassspectrometry,J.Chromatogr.BAnal.Technol.
Biomed.LifeSci.1005(2015)47–55,http://dx.doi.org/10.1016/j.jchromb.
2015.10.001.
[14]N.C.vandeMerbel,K.J.Bronsema,M.W.J.vanHout,R.Nilsson,H.Sillén,A validatedliquidchromatography-tandemmassspectrometrymethodforthe quantitativedeterminationof4-hydroxycholesterolinhumanplasma,J.
Pharm.Biomed.Anal.55(2011)1089–1095,http://dx.doi.org/10.1016/j.jpba.
2011.03.017.
[15]Y.Shi,X.Xu,M.Fang,M.Zhang,Y.Li,B.Gillespie,S.Yorke,N.Yang,J.C.
McKew,W.A.Gahl,M.Huizing,N.Carrillo-Carrasco,A.Q.Wang,Quantitative hydrophilicinteractionchromatography-massspectrometryanalysisof N-acetylneuraminicacidandN-acetylmannosamineinhumanplasma,J.
Chromatogr.BAnal.Technol.Biomed.LifeSci.1000(2015)105–111,http://
dx.doi.org/10.1016/j.jchromb.2015.07.018.
[16]A.Q.Wang,B.P.Wei,Y.Zhang,Y.J.Wang,L.Xu,K.Lan,Anultra-highsensitive bioanalyticalmethodforplasmamelatoninbyliquid
chromatography-tandemmassspectrometryusingwaterascalibration matrix,J.Chromatogr.BAnal.Technol.Biomed.LifeSci.879(2011) 2259–2264,http://dx.doi.org/10.1016/j.jchromb.2011.06.010.
[17]R.Thakare,Y.S.Chhonker,N.Gautam,J.A.Alamoudi,Y.Alnouti,Quantitative analysisofendogenouscompounds,J.Pharm.Biomed.Anal.128(2016) 426–437,http://dx.doi.org/10.1016/j.jpba.2016.06.017.
[18]EuropeanMedicinesAgency,CommitteeforMedicinalProductsforHuman Use,GuidelineonBioanalyticalMethodValidation,2011,July2011https://
www.ema.europa.eu/en/documents/scientific-guideline/guideline- bioanalytical-method-validationen.pdf(Accessed4November2019).
[19]B.R.Jones,G.A.Schultz,J.A.Eckstein,B.L.Ackermann,Surrogatematrixand surrogateanalyteapproachesfordefinitivequantitationofendogenous biomolecules,Bioanalysis4(2012)2343–2356,http://dx.doi.org/10.4155/bio.
12.200.
[20]R.Houghton,C.HorroPita,I.Ward,R.Macarthur,Genericapproachto validationofsmall-moleculeLC–MS/MSbiomarkerassays,Bioanalysis1 (2009)1365–1374,http://dx.doi.org/10.4155/bio.09.139.
[21]N.Zheng,H.Xiao,Z.Zhang,X.Gao,J.Zhao,Rapidandsensitivemethodfor determiningfreeaminoacidsinplanttissuebyhigh-performanceliquid chromatographywithfluorescencedetection,ActaGeochim.36(2017) 680–696,http://dx.doi.org/10.1007/s11631-017-0244-5.
[22]M.K.Dhillon,S.Kumar,G.T.Gujar,AcommonHPLC-PDAmethodforamino acidanalysisininsectsandplants,IndianJ.Exp.Biol.52(2014)73–79.
[23]G.Sharma,S.V.Attri,B.Behra,S.Bhisikar,P.Kumar,M.Tageja,S.Sharda,P.
Singhi,S.Singhi,Analysisof26aminoacidsinhumanplasmabyHPLCusing AQCasderivatizingagentanditsapplicationinmetaboliclaboratory,Amino Acids46(2014)1253–1263,http://dx.doi.org/10.1007/s00726-014-1682-6.
[24]J.Pawliszyn,HandbookofSolidPhaseMicroextraction,Elsevier,Amsterdam, 2012,http://dx.doi.org/10.1016/C2011-0-04297-7.
[25]N.C.vandeMerbel,Quantitativedeterminationofendogenouscompoundsin biologicalsamplesusingchromatographictechniques,TrACTrendsinAnal.
Chem.27(2008)924–933,http://dx.doi.org/10.1016/j.trac.2008.09.002.
[26]EuropeanMedicinesAgency,CommitteeforMedicinalProductforHuman Use,ICHGuidelineM10onBioanalyticalMethodValidation,DraftGuidance, 2019,March2019https://www.ema.europa.eu/en/documents/scientific- guideline/draft-ich-guideline-m10-bioanalytical-method-validation-step-2b en.pdf(Accessed4November2019).
[27]U.S.DepartmentofHealthandHumanServices,FoodandDrug
Administration,GuidanceforIndustry,BioanalyticalMethodValidation,2018, May2018https://www.fda.gov/files/drugs/published/Bioanalytical-Method- Validation-Guidance-for-Industry.pdf(Accessed4November2019).