CHAPTER 3
Inhibitors of Amino Acid Activation
Robert B. Loftfield
I. Introduction 107 A. Appropriateness of the Subject 107
B. The Dual Paths to Amino Acid Activation 108
C. Coverage H I I I . Noncompetitive Inhibitors H I
A. Antisulfhydryls H I B. Nucleophilic'Ions and Bases 112
C. Heat or Cold
i 1
4
III. Competitive Inhibitors 114 A. Against Adenosine Triphosphate 114
B. Amino Acids and Analogs 115
C. Transfer R N A ' s 118 IV. Conclusion 123
A. Stoichiometry of Inhibitors. 123 B. Use of Inhibitors to Determine Mechanism 124
C. Prospects for Therapy 126
References 127
I. INTRODUCTION
A. Appropriateness of the Subject
The title "Metabolic Inhibitors" implies that the reagents being con
sidered inhibit the complex series of reactions that collectively are known as metabolism. When the editors first requested a chapter on amino acid activation, I protested that the study of protein synthesis had been so highly dissected by biochemists that the inhibitors we know are in
hibitors of an isolated biochemical step rather than of metabolism. In fact, few of these inhibitors have any biological significance. However, the arguments of the editors and the proposed subject matter of the other
107
chapters have persuaded me that this discussion is appropriate in this volume.
1. INHIBITORS AS TOOLS IN ENZYME STUDIES
The use of inhibitors has been particularly helpful in elucidating the mechanism of action of the enzymes that activate amino acids. Applica
tion of the Hill equation (1) permits a determination of the number of interacting enzyme sites and has shown that even the polymeric en
zymes possess no interacting subunits (see Section IV,A). The ability to selectively inhibit one phase or another of the complex reaction be
tween ATP, amino acid, transfer RNA, and enzyme has been determina
tive in establishing reaction sequences [2-4). The use of substrate analogs has enabled investigators to establish which parts of a molecule are essential for binding to the enzyme (5-10). Still other inhibitors have been instrumental in discerning the nature of the "active site" of this class of enzymes.
2 . INHIBITORS IN THERAPY
The inhibition of amino acid activation and therefore of protein bio
synthesis is obviously a promising chemotherapeutic technique for mi
crobial infection or for cancer. In order to have a good therapeutic index, the inhibitory drug must be much more effective in the microbe or the tumor than in the host. None of the inhibitors presently known has therapeutic value, but analogous compounds might be found that are selectively concentrated or are otherwise selectively effective against tumors or microbes. It is necessary that the mechanism of action of these enzymes and the function of the various inhibitors be more com
pletely understood so that a rational pharmacology can be developed.
B. The Dual Paths to Amino Acid Activation
Since the discovery of transfer ribonucleic acid, it has been believed that the enzymes which activate amino acids (amino acid:tRNA ligases, aminoacyl-tRNA synthetases) catalyze the formation of enzyme-bound aminoacyl adenylates and subsequently transfer the aminoacyl residue to tRNA (11). Concerted mechanisms in which aminoacyl adenylate was not a discrete intermediate were considered by several workers (12-14), but these received little or no support even from their proponents. More recently, we felt that the evidence for a concerted reaction was totally
3. INHIBITORS OF AMINO ACID ACTIVATION 1 0 9
persuasive in many cases (3, 15). Now I am persuaded that these en
zymes catalyze two entirely different reactions, depending on whether tRNA is present (16). Of these only the esterification of tRNA is phy
siologically important, but both will be discussed. It should be empha
sized that the concerted mechanism has not been generally accepted as correct. Most often it is believed that the enzyme binds ATP, then amino acid to form Enz- (AA ~ AMP) and to release PPj; tRNA then reacts with the complex to form free AMP and Enz • (AA ~ tRNA), the AA
~ tRNA dissociating from the enzyme in the overall rate-limiting step (17-17e).
1. REACTION 1: T H E SYNTHESIS OF ENZYME-BOUND AMINOACYL ADENYLATE
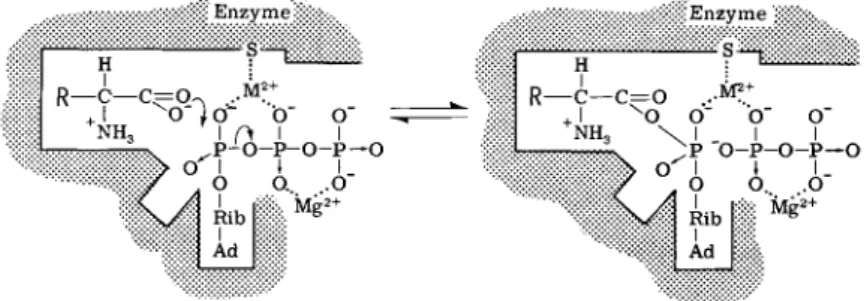
This reaction can be described by the formulation shown in Fig. 1.
A heavy-metal cation such as ferrous ion is associated tightly with the enzyme, possibly through a sulfhydryl residue. The amino acid car- boxylate ion makes a nucleophilic attack on the «-P of the M g A T P
2 - . The unknown cation is essential in dissipating the enormous negative charge associated with the transition state and, as will be noted below, any chelating agent that ties up the unknown metal is an inhibitor of this reaction. The reaction is easily and rapidly reversible such that
[ 3 2
P]PPi, can be incorporated into ATP at a rate that is usually deter-
:.- ENZYME •>
.M
2+
+ 1 O )o O" O" : NH, i
r>
I I.P-O-P-O-P— o O I \ I _
O Q. ..O Rib I
Ad I Mg
2
ENZYME
S^t | | o" '"or o"
i i i P "O-P-O-P—o O' I \ i _
o o, .o
Mg
;
2+
FIG. 1. Reaction path 1. A schematic representation of the reaction between enzyme, amino acid, and A T P in which enzyme-bound aminoacyl adenylate and pyrophosphate are formed. The enzyme is deliberately shown as distorted compared to the native conformation in Fig. 2 in order that the metal ion M
2+
can participate by helping to dissipate the intense negative charge in the transition state. A conventional nucleophilic attack by the carboxylate ion on the a-P of A T P displaces the 0- and 7- P as PPi, but the distorted conformation of the enzyme makes subsequent reaction with t R N A difficult. [From Loftfield and Eigner (3).]
mined only by the rate of synthesis of the aminoacyl adenylate. The aminoacyl adenylate can react with hydroxylamine to form hydroxamate or with tRNA to form aminoacyl ester, but the rate of this transfer is too slow to be physiologically significant (16); for instance, the half-life for transfer of leucine from adenylate to tRNA at 37°C is about 1 minute
(17) compared to an observed turnover of 40 moles/minute and a biologi
cal demand that is still greater. This first reaction does not take place with the enzymes specific for glutamine (IS), glutamic acid (13, 14), and arginine (18).
2. REACTION 2: T H E CONCERTED REACTION TO PRODUCE AMiNOACYL-tRNA (AND HYDROXAMATES)
Figure 2 schematically portrays the reaction between the terminal adenosine of tRNA, the amino acid, and ATP. The metal cation ( M
2 + )
FIG. 2. Reaction path 2. A schematic representation showing how t R N A , amino acid, and A T P react to form aminoacyl-tRNA, A M P , and PPi. It is proposed that a metal ion ( M
2 +
) bound tightly through a protein sulfur assists in orienting the substrate and delocalizing developing charges. The 2'-OH of the t R N A serves as a general base accepting the proton from the 3'-OH of adenosine. (If a stronger general base such as imidazole is present, it accepts the proton and accelerates the esterification.) The now very nucleophilic 3'-0~ of the adenosine attacks the amino acid carboxyl, displacing an oxygen to the a-P of A T P , in turn forcing the release of the /3,7-P's as M g P20 7
_
. The reaction is fully reversible so that exogenous [
3 2
P ] P P i may become incorporated into A T P . In the event that N H2O H
occupies t h e ^ M
2+
site the same series of reactions occurs, with the concerted for
mation of amino acid hydroxamate, A M P , and P P i ; however, in this case thermo
dynamic considerations make the reaction irreversible. [From Loftfield and Eigner (3).]
3. INHIBITORS OF AMINO ACID ACTIVATION 111 assists in binding the 2'- and 3'-hydroxyls. The 2'-OH serves as a general base to accept a proton from the 3'-OH. The 3'-0~ then, as a nucleophile, displaces the 0" from the amino acid to the a-P of the M g A T P
2 - , which in turn displaces PPi. If general bases stronger than the 2'-0H are able to complex with M
2 +
, the esterification is accelerated (19). The reaction is fully reversible so that this is also a route by which [
3 2
P]PPi can be incorporated into ATP. If NH2OH is in the site properly occupied by the adenosine hydroxyls, amino acid hyroxamate, AMP, and PPi are formed irreversibly. If M
2+
is removed or sequestered by chelating agents the reaction is inhibited.
C. Coverage
An effort has been made to review all the literature through December 1971. In some areas, such as inhibition by amino acid analogs or by mercurials, there have been so many studies that I have chosen to cite only those that appear to serve as general examples. The ligases that have been identified, purified, and studied during the last 15 years originate in every kind of microbe, plant, or animal. Although it will be occasion
ally necessary to define the source of the enzyme because it has unique properties, I shall usually confine myself to the more generally observed properties.
II. NONCOMPETITIVE INHIBITORS
The distinction between competitive and noncompetitive inhibitors is somewhat arbitrary, depending, in this chapter, partly on kinetics but partly on my interpretation of which function of the active site has been inactivated.
A. Antisulfhydryls
Almost every one of the hundreds of ligases tested has lost its activity in treatment with p-chloromercuribenzoate, p-hydroxymercuribenzoate, or p-chloromercuribenzene sulfonate, usually in the range of 10
-6 M [arginine (20), glutamic acid (14), leucine (21), and proline (22]. An
other inactivated by the mercurial but, in some cases, reactivated by
treatment with mercaptoethanol is the glycine enzyme (23, 24), which dissociates into four subunits but reassociates to an active enzyme in the presence of the mercaptoethanol (25).
Frequently, both the sulfhydryls and the enzymic activity are pro
tected from mercurials or oxidants by the simultaneous presence of sub
strates or substrate analogs [trytophan (26), valine (28), or phenyl
alanine (29) ]. Sometimes the mercurial destroys esterification activity much more rapidly than aminoacyl adenylate activity (i.e., ATP:PPi exchange) as in the case of the methionine enzyme (30) especially if the enzyme is being protected by methionyl adenylate (31). In another
case, the mercurial apparently distorts the threonine enzyme so as to forbid reaction with tRNA but to facilitate reaction with hydroxylamine (32). Similarly, the tryptophan enzyme loses esterification ability but gains hydroxamate-forming ability with 50 mM mercaptoethanol (33).
Two enzymes, the cysteine (34) and lysine (35, 36) enzymes, are resistant to p-chloromercuribenzoate and iodoacetate, but it should be noted that there are contrary reports and that resistance to mercurials is not always accompanied by resistance to dithiothreitol or iV-ethyl- maleimide. Thus, the lysine enzyme of E. coli loses both its lysyl adenylate synthetic ability and its esterification ability in parallel on treatment with dithiothreitol, which almost certainly acts on sulfhydryls (37). Stern and Peterkofsky particularly stress the need to establish that the mercurials are acting on the enzyme rather than on the substrate tRNA (37). However, in two well-studied cases, the effect of the anti- sulfhydryl has been established as being first on one of the SH groups of the enzyme, to destroy the esterification ability, and then on a second and third to inhibit aminoacyl adenylate synthesis (38, 39).
B. Nucleophilic Ions and Bases
The nucleophilic inhibitors are considered separately in two groups, monovalent and polyvalent, because the two groups have quite separate effects on the synthesis of aminoacyl adenylates and aminoacyl-tRNA.
1. MONOVALENT BASES
Where they have been examined, monovalent bases such as hy
droxylamine (40), tris, imidazole, ammonia (19), and collidine, lutidine, and pyridine (41) have all inhibited ATP:PPi exchange and, by in
ference, aminoacyl adenylate formation. The kinetics that provide infor-
3. INHIBITORS OF AMINO ACID ACTIVATION 1 1 3
mation on mechanism will be discussed in Section IV,B,1. On the other hand, each of these monovalent bases actually stimulates, with first-order kinetics, the production of aminoacyl-tRNA (19, 41). In the case of hydroxamate formation, this means that, over a limited range and with certain ligases, hydroxamate formation is proportional to the square of the concentration of NH2OH (19, 39, 42).
2. POLYVALENT BASES
From the earliest studies of these enzymes it was realized that pyro
phosphate was an inhibitor, the assumption being that it functioned only by reversal of the formation of aminoacyl adenylate. The low con
centrations required make it clear that such a simple interpretation is not accurate. Pyrophosphate is an extremely good chelating agent that binds to M
2+
in Figs. 1 and 2, thereby interfering with both types of reaction. Concentrations in the range of 1 mM are extremely effective in stopping hydroxamate formation or tRNA esterification [proline (43), valine, isoleucine, and leucine (3), arginine (20, 44), glutamic acid and glutamine (13), and lysine (49) ] . The inhibitory power is much greater in heterologous systems where 1 0
-8
M PPi inhibits the esterification of E.
coli t R N A V al
by Neurospora crassa phenylalanyl enzyme. The surprising fact that PPi inhibits the formation of aminoacyl adenylate and PPi, not by disturbing the equilibrium, but by interfering with the active site has been documented (3, 45).
Other compounds capable of sequestering a polyvalent cation in the reactive center are good inhibitors. Phosphate ion in concentrations near 0.01 M generally inhibits both types of reaction about 5 0 % or more [aspartic acid and tryptophan (45a), leucine and phenylalanine (46), asparagine (47), and serine (48)]. Phosphate competitively inhibits the binding of tRNA to valine ligase (27). Such inhibition is clearly of critical importance, in isolating and purifying the enzymes or in determining their kinetic constants since sodium phosphate or calcium phosphate is so often used in preparation or preservation of the enzymes.
Four other inhibitors of both kinds of reactions that probably function by sequestration of the central cation are an unidentified organic phos
phate (50), glyoxalate (51), phenanthroline (3), and methylene diphos- phonate (PcP) (4). The latter, an analog of PPt that cannot be hy- drolyzed by intracellular pyrophosphatase, ought to be a superb inhibitor of protein synthesis (and cell growth) because it reduces the esterification of lysine to less than 5 % of normal at a concentration of 2.0 mM.
It has been tested for its effect on the protein biosynthesis of intact
ascites cells and found to be noninhibitory, presumably because it failed to enter the cells (4). Recently (51a), PcP has been found to be an extremely cytotoxic agent in mammalian tissue cultures over a period of several hours of contact. This is certainly a good lead. Ethylenedi- aminetetraacetate and similar compounds are always inhibitory because they complex the M g
2+
essential to this type of reaction. However, excess M g
2+
is also inhibitory (28).
C. Heat or Cold
These enzymes are inactivated by heat, although for the most part they appear to be more tolerant of heat than the organisms from which they are derived. Not surprisingly, enzymes from thermophilic organisms resist thermal denaturation (52). Frequently, the presence of the sub
strates provides substantial protection against thermal denaturation. A more surprising observation in a special case is the cold inactivation of the proline enzyme (53) caused by dissociation of the subunits (54).
III. COMPETITIVE INHIBITORS
A. Against Adenosine Triphosphate
The enzymes are highly specific for the adenine residue and the reac
tion, of course, requires the triphosphate residue in order to generate AMP and PPi.
The methionyl enzyme does not use CTP, GTP, or UTP (40) and GTP, ITP, CTP, and TTP have been shown to be ineffective toward the proline, lysine, glutamic acid, isoleucine, and tyrosine enzymes (9).
Adenosine 5'-0-(l-thiotriphosphate) competitively inhibits the use of ATP by the phenylalanine enzyme (85). More to the point, all nucleo
side monophosphates tested have been shown to be competitive inhibitors of ATP (20, 23, 24, 40). In the most extensive survey to date, it has been shown that binding to the ATP site of the phenylalanine enzyme requires a 6-aminopurine, that the /?,y-diphosphate group plays no role in binding, and that the a-phosphate and the 5'-OH actually facilitate dissociation of the complex. Naturally all of these ATP fragments are competitive inhi
bitors of ATP in phenylalanine activation, the Ki for 5'-deoxyadenosine being 1 X 10"
6
M] the K{ for adenosine, 1.5 X 10"
5
M; and the Km or Kd for ATP, 1 X 10"
3
M (6). It seems entirely possible that 5'-deoxyadeno-
3. INHIBITORS OF AMINO ACID ACTIVATION 115 sine or some related, uncharged derivative should be able to diffuse through the cell membrane in sufficient quantity to severely disrupt protein synthesis, especially in rapidly growing cells.
The other compound that competes for the ATP site provides some of the evidence for two separate paths. 2-Deoxyadenosine triphosphate, although bound to the enzyme somewhat more poorly than ATP, is otherwise approximately as good as ATP in supporting the esterification of tRNA by the scheme of Fig. 2 [for glutamic acid (14)', lysine, proline, isoleucine (9); arginine (8); tyrosine (2); and phenylalanine (6)]. De- oxyadenosine triphosphate also participates in the formation of the unusual dinucleotide tetraphosphates AP4A, AP4G, dAP4dA, etc., as formed on the lysine enzyme (55). Nonetheless, there is absolutely no dATP: PPi exchange (i.e., no tyrosyl deoxyadenylate) in the absence of tRNA with the tyrosine enzyme (2) and very little exchange without tRNA in the case of the lysine enzyme (9). In fact, dATP is an excellent competitive inhibitor of ATP in the tyrosine-catalyzed ATP: PPi ex
change. Since dATP supports both esterification of t R N A p he
and phenyl- alanine-catalyzed dATP:PPi exchange, there can be no doubt that the thermodynamics are favorable to [
3 2
P]PPj incorporation into dATP if an aminoacyl deoxyadenylate forms. It should be noted, too, that in the absence of the cognate tRNA, the enzymes for glutamic acid (14), gluta
mine (13), and arginine (18) do not catalyze reaction 1; tRNA is essential to the exchange of [
3 2
P]PPi into'ATP.
B. Amino Acids and Analogs
1. AMINO ACIDS
The enzymes that activate amino acids show remarkable specificity.
In the case of the valine enzyme the addition or removal of a single CH2 group (i.e., valine —> a-aminobutyric acid, isoleucine, or norvaline) decreases the association of amino acid with enzyme by a factor of about 100 (28, 56). Nonetheless, close homologs do bind to the en
zyme and are "activated" to the level of an enzyme-bound amino
acyl adenylate. As such, they are able to tie up the enzyme. Thus, large excesses of aminobutyric acid or isoleucine competitively inhibit the formation of valine hydroxamate while a-aminobutyric hydroxamate or isoleucine hydroxamate is being formed (56). Fortunately, there is a still much greater specificity in the tRNA esterification reaction. Al- loisoleucine, which catalyzes ATP:PPi exchange and forms hydroxamate quite well with the isoleucine enzyme, hardly attaches to t R N A
I le (57)
and is, in fact, a competitive inhibitor of the isoleucine esterification of t R N A
I l e
. It appears that the presence of tRNA allosterically changes the affinity of the enzyme for competing substrates; in a sense, the tRNA produces a new enzyme that catalyzes a different reaction with different specificity (58). The D forms of a-aminobutyric acid, valine, and norva- line are all competitive inhibitors in the valine system, with Kh values between 10~
3
and 10~
2
M, not much greater than the corresponding Km values of the L acids, 10~
4 -10
-2
M (59). Many homologs that catalyze aminoacyl adenylate formation turn out to be competitive inhibitors of the corresponding tRNA esterification: 3-fluorotyrosine and hydroxypyri- dylalanine against tyrosine (60), canavanine against arginine (8), furano- mycin (I) against isoleucine minosine (II) and fluorophenylalanine
HO
/ Q _ > = CH
H3C—CH CH—CH—COO 0 = C N—CH\ / I \ / 2— CH—COOH
2
HC=CH NH3 I
+
HC=CH ^
(I) (II)
against phenylalanine (62), oxalysine and £rans-dehydrolysine against lysine (49).
Allo-4-hydroxyproline is activated by the proline enzyme without being transferred to t R N A
P ro
(22), while valine and leucine are activated by the isoleucine enzyme without being able to form A A ~ t R N A
I le
(67). In each of these cases the consequence is that the formation of the correct A A ^ t R N A is competitively inhibited by the incorrect amino acid. The arginine enzyme of E. coli is uniquely inhibited by several biosynthetic precursors of arginine like ornithine (69a). This inhibition can be inter
preted in terms of controlling the activity of several arginine enzymes under conditions of restricted or unrestricted arginine supply.
In other cases, the analog successfully fools the enzyme and becomes attached to tRNA, with the consequence that errors may be incorporated into protein: azetidinecarboxylic acid (III) (64),
H2C—CH—COOH
H2C—NH
(III)
thiazolidine-4-carboxylic acid, A^-ethylglycine and JV-methylglycine onto tRNA
P l
'° (22, 63, 63a), alloisoleucine onto t R N A I le
(57, 65), O-methyl- threonine onto t R N A i
l e
c o Zi (65, 66), canavanine onto t R N A A rg
(20), thiosine [ N H2C H2C H2S - C H2C H - ( N H2) C O O H ] onto t R N A
L ys
(35), and possibly 2-amino-4-methylhex-4-enoic acid onto t R N A
P he (67).
Thus, amino acids can be inhibitory in at least two ways: by compet-
3. INHIBITORS OF AMINO ACID ACTIVATION 1 1 7
ing for active sites and being at least partly converted to adenylates, as in reaction 1, or by successfully becoming attached to the appropriate tRNA, as in reaction 2 . In the first instance, protein synthesis may be slowed; in the second, an abnormal protein is formed. It is noteworthy that when these competitive inhibitors have been tried in vivo their cytotoxic effects have been reversed by an increase in the level of the one amino acid being competed against. Some of the competitor amino acids exist naturally in high concentration in certain tissues. Thus, Poly- gonatum multiflorum (Solomon's seal) contains much azetidinecarboxylic
acid, Canavalia ensiformis (jack bean) much canavanine, and Aesculus californica (California buckeye) much aminomethylhexenoic acid. In each instance, the corresponding proline- (68), arginine- (69), and phenylalanine-activating (67) enzyme systems are fully resistant to the bizarre amino acid analog.
2 . AMINO ACID DERIVATIVES
The carboxyl group of the amino acid appears to play no role in the binding of most amino acids to the activating enzymes. Hence, we find that tyramine (2, 70) against tyrosine, pyrrolidine and proline amide against proline (22), propyl- and sec-butylamine against valine (71), various phenylalanylamides against phenylalanine (5), the ethyl esters of phenylalanine tryptophan, glutamic acid, and leucine against the cor
responding amino acids (72), and tryptophan hydroxamate against tryp
tophan (73) are all competitive inhibitors of both reactions 1 and 2 and growth. The Ki for each of these is very near to the Km so that the esteri
fication reaction is approximately 5 0 % inhibited when an equal quantity of the derivative accompanies the amino acid. Amino acid derivatives in which the a-amino group is replaced by a hydroxyl cannot be activated and are excellent inhibitors of all reactions of the ligases, e.g., tyrosol
(IV) (70). [There is a report to the contrary (70a).]
In view of the special sensitivity of certain leukemic cells to depriva
tion of asparagine by asparaginase therapy, Harnden and Yellin (74) have synthesized a number of /?-aminoamides. When tested against the aspartic acid ligase in the esterification, all were excellent inhibitors of asparagine, the best being y-hydroxy-/?-aminobutyramide and as
paragine amide with Ki values in the range of 10~
5 M.
OH
(iv)
Salicylic acid competitively inhibits the conversion of glutamic acid to G l u ~ t R N A
G lu
without affecting the esterification of other tRNA's (74a). Several amino acid hydroxamates are excellent competitive inhib
itors of the parent amino acid, the Ki's being smaller than the correspond
ing Km's (74b). Phosphonate analogs of leucine, valine, and isoleucine are potent competitive inhibitors of ATP:PPi exchange catalyzed by the corresponding amino acids and ligases (74c).
3. AMINOALKYL ALCOHOLS R — C H ( N H2) — C H2O H
Reducing the carboxyl group of any amino acid to the corresponding carbinol results in an aminoalkyl alcohol that is extremely specific in competing with the related amino acid. Since they can form neither the adenylate nor the tRNA ester, these are inert competitive inhibitors with Ki values somewhat lower than the corresponding amino acid Km
[isoleucinol, leucinol, valinol, tyrosinol, phenylalaninol, prolinol, gly- cinol, lysinol, and methioninol (75)]. Each of these uniquely inhi
bits both reactions 1 and 2 with a Ki almost identical to that of the amino acid. Much more potent inhibitors are produced when the aminoalkyl alcohol is adenylated. These compounds react as competitive inhibitors of both amino acid and ATP with the sites for both the amino acid and ATP with Ki values of 8 X 1 0 ° M for methioninyl adenylate, 7 X 10~
9
M for isoleucinyl adenylate, 3 X 10"
8
M for valinyl adenylate, etc. In every case, the Ki is lower by a factor of 1000-10,000 than the Km for the amino acid. We shall comment briefly on the application of the Hill plot to this kind of inhibition in Section IV,A. Because the aminoalkyl adenylates are tightly bound to the ligase and quite unreac- tive, they have been useful for protecting the active site from denatura
tion or attack by antisulfhydryls (31).
C. Transfer RNA's
1. NATIVE tRNA
Many cells contain two or more tRNA's specific for a single amino acid. On occasion, these iso-accepting tRNA's may have quite different rates of association, dissociation, and reaction on the enzymes. Thus, Enz- (Leu~AMP) reacts rapidly with tRNA^
eu
but with a relatively large Km. The more tightly bound but more slowly reacting tRNA^
eu is a very specific competitive inhibitor of the tRNA^
eu
(75a). This experi
ment provides strong support for the argument that a single ligase
3. INHIBITORS OF AMINO ACID ACTIVATION 119 attaches a single amino acid to several tRNA's specific for that amino acid. In some cases native tRNA as well as periodate oxidized tRNA inhibits the ATPiPPi exchange (75b): this inhibition is consistent with two separate reactions leading to AA—tRNA (reaction 2) and to Enz- (AA—AMP) (reaction 1) respectively.
In general, noncognate tRNA from any source is not bound to the ligase and, except for their effect on chelating M g
2+
(76), such tRNA's or other RNA's have little or no inhibitory effect on either reaction 1 or 2. However, slightly modified cognate tRNA's bind to the "recogni
tion site" of the ligase almost as well as the substrate tRNA, are unreac- tive, and hence are powerful inhibitors. The simplest of these are the aminoacyl-tRNA's themselves, in other words the product. Product in- inhibition is observed; for instance, the Ki values for valyl-tRNA
V al and isoleucyl-tRNA
I le
are 3 X 10~
5
and 1 X 10"
5
M, respectively, each about 10 times greater than the Km of the tRNA for esterification (77). E. coli t R N A
I le
and He—tRNA 11 e
bind equally well to isoleucine:tRNA ligase, each Kd being about 10"
8
M (78). E. coli valine ligase binds free tRNA^
al and tRNA^
al
only slightly better than the corresponding valyl-tRNA's (27). Lagerkvist et al. (79) find that the valine enzyme forms isolatable complexes with Val-tRNA
V al
and with t R N A V a l
.
In many cases, ligases from one organism aminoacylate the cognate tRNA from another organism with quite respectable velocities (80, 81).
In other cases, where the rate of heterologous esterification is very low or zero, the heterologous tRNA is a competitive inhibitor of the amino acid-specific ligase and its homologous tRNA, the Ki being essentially identical to the Km for the homologous tRNA (77). Similar inhibitions are observed for the esterification of rat liver t R N A
G ln
by the t R N A G ln of yeast and of E. coli (82).
2. MODIFIED tRNA
The tRNA molecule is very large and appears to be bound to the ligase by forces that interact with almost the total surface (15, 16, 83). Hence, it is not surprising that minor modifications of the cognate tRNA that make it unable to accept an amino acid leave it a strong competitive in
hibitor. Gentle acetylation of t R N A V al
yields an acetylated t R N A V al whose Ki is only three times the Km of t R N A
V al
(84). Undermethylated
t R N A l
h e
C 0H has the same Km but a much reduced 7m ax for phenylalanine esterification and is, correspondingly, a competitive inhibitor (83a). Re
placement of one of the oxygens on the phosphorus joining the terminal
adenosine and cytosine by a sulfur produces a yeast t R N A that can accept phenylalanine about half as well as unmodified t R N A
p he
(85). The enzymic removal of seven nucleotides from the 3'-C-C-A end of yeast t R N A
V al
naturally destroys the acceptor ability, but the residue retains a great affinity for valine ligase and has a Kb only twice the Km of t R N A
V al
(86). It can even be shown that ATP is a partially competitive inhibitor of the binding of tRNA (87).
Gentle periodate oxidation of tRNA is limited to converting the 2',3'- vicinal diol residue of the terminal adenosine to a nonacceptor dialdehyde.
Such oxidation products are very specific and very effective competitive inhibitors of the corresponding undamaged tRNA's [valine and tyrosine (88), tyrosine (89), leucine (90), or other amino acids (91)]. Even more inhibitory is the oxidized tRNA treated with NH2OH (90).
One of the more surprising developments of recent years is the ability of fragments of tRNA to spontaneously reassociate and serve as accep
tors for activated amino acids (92). In one case, at least a quarter fragment from the 5'-G end of tRNAy^a
e
st combines with a two-fifths frag
ment from the 3'-C-C-A end of tRNAye h
a e
st to form a complex incapable of accepting phenylalanine but nonetheless a competitive inhibitor of the esterification of tRNA£e
h a e
8t (but not of tRNA*e y
a r
8t or t R N A ^ J by the respective amino acids (94). Similarly, oligo-G reacts with 3'-C-C-A quarter molecule from t R N A
T yr
to form a complex that specifically inhibits the esterification of t R N A
T yr (93).
3. SYNTHETIC OLIGONUCLEOTIDES
A number of synthetic oligonucleotides have been tested as inhibitors of aminoacyl-tRNA formation on the assumption that the oligonucleotide possesses fragments that mimic important parts of the tRNA molecules.
Thus, poly G and poly I competitively inhibit the formation of Glu- t R N A
G l u
, poly A and poly C inhibit the formation of Leu-tRNA L e u
, while neither poly A, poly I, poly U , or poly C affects esterification by serine
(95). In no case is the formation of the aminoacyl adenylate inhibited.
In another system, poly U is found to be a competitive inhibitor of t R N A
L ys
and t R N A V al
and indifferent to all other tRNA's (96). The lysine enzyme of yeast actually forms a catalytically inactive complex with poly U that can be isolated by Sephadex filtration or ultracentrifuga- tion. Several other enzymes and oligonucleotides do not form such stable inert complexes (96, 97). The esterification of t R N A
V al
is competitively inhibited by poly (A,C,U), but Holten and Jacobson (76) emphasize that all such experiments are difficult to interpret because of doubts about purity of polynucleotides, free cation concentrations, etc.
3. INHIBITORS OF AMINO ACID ACTIVATION 121 An enormous number of modifications of tRNA, cleavage of the di- hydrouridine residues, cyanoethylation of the pseudouridine residues, reaction of the guanines with kethoxal, etc., have resulted in tRNA's with negligible or reduced acceptor potential. Undoubtedly, many of these would competitively inhibit the esterification of the native tRNA and, by analogy, have little effect on aminoacyl adenylate formation.
However, most of these have not been tested for inhibitory activity.
4. DRUGS
Several alkaloids are believed to exert an effect on protein synthesis by interfering with the esterification of tRNA. It has been demonstrated that proflavin does not affect the synthesis of aminoacyl adenylate but inhibits severely the formation of isoleucyl- and leucyl-tRNA because it binds strongly to these tRNA's (98, 99). In the same way, kasugamy- cin changes the tertiary structure of t R N A
L ys
and t R N A A r g
, although its major effect is probably on the binding of esterified tRNA to mRNA and ribosomes (100). Pyrimethamine, hydroxystilbamidine, quinacrin, and acriflavin all inhibit the esterification of t R N A
V aI
at concentrations of 0.1 mM (101).
5. DIELECTRIC EFFECTS AND CATIONS
As noted above, studies with derivatives show that the electrically charged residues of ATP and amino acid apparently play little part in the binding of substrate to the enzyme. This is clear also from the independence of reaction 1 from salt effects (87, 102, 103), although there is one report that high concentrations of salt depress the rate of ATP:PPi exchange (104). On the other hand, the third substrate, tRNA, is an extremely large polyanionic molecule whose exterior is char
acterized by a unique shape and a monotonous surface consisting largely of alternating ribose units and charged phosphate residues. There
fore, it is not surprising that almost every worker has observed that the binding of the tRNA to the ligase (and the subsequent esterification) is enhanced by media of low dielectric constant (105-107) or inhibited by increasing concentrations of salt (24, 40, 4^a, 46, 76, 87, 107, 108, 114)- That the strongly inhibitory effect of salt is a consequence of the dielec
tric constant of the solution, almost regardless of the nature of the salt, is made clear by the Debye-Huckel relationship between binding con-
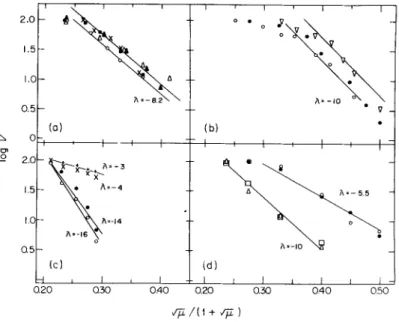
stant (as well as rate of esterification) and ionic constant. This was first demonstrated by Loftfield and Eigner (87) but is also made clear by the recalculation and replotting of other workers' data in Fig. 3.
In every one of the many plots shown, the effect of salt is to inhibit strongly the rate of esterification according to the relationship
/ V~v \ log V(esterification) = — 0.5z
+
z~ ( 7= )
where z +
and z~ are the numbers of interacting positive and negative charges in the enzyme-tRNA complex. In general, this product is between
2 . 0 I
- 4
' 1 ' 1 1 ' 1 i i ' 1
1.5 V » \ ^
—
1.0 -
0 . 5
A = - 8. 2 ^ A - - 1 0 \
V -
0 ( a )
Z| i 1 1 1
( b )
- i 1 1
•
I , I — o 2 . 0
' 1
1
1 A = - 3
i 1 1. —1 1
1
1
1.5 ~ \
• A = - 4
\ n A = - 5 . 5
1.0 A
V \ A = - I 4
= - 1 6 V •
0.5
(c)
1 , 1 ( d ) - 1
A =-10
] 1 i 1 I 1 -
0 . 2 0 0.30 0 . 4 0 0.20 0.30 0 . 4 0 0.50
FIG. 3. D e b y e - H u c k e l dependence of the rate of esterification of t R N A . (a) NaCl on t R N A |
e c
u
o H; O, NaCl on t R N A £
e
c o H; x, M g C l2 on t R N A ^ - ; A, M g S 04 on
t R N A ; • , KC1 on t R N A j
1
^ . [Data from(87) and unpublished work b y Loftfield and Eigner.] (b) O, N a C l ; # , N a2S 04; V, LiCl on t R N A ^
e c
u
o h, [Data recalculated from Fig. 2 of Smith (104).] (c) + , NaCl on t R N A ^
1
^ b y E. coli alanine enzyme; O, NaCl on t R N A ^oH b y Neurospora crassa phenylalanine enzyme; x, NaCl on t R N A j ^ b y E. coli enzyme; # , NaCl on t R N A j
8
^ b y N. crassa phenylalanine enzyme. [Data from Holten and Jacobson (114).] (d) • , KC1 on t R N A ^ L t hy yeast phenylalanine enzyme; O, N H4C 1 on tRNAyeLt b y yeast phenylalanine enzyme; • , N H4C 1 on
t R N A ™ ^ - b y yeast phenylalanine enzyme; A, KC1 on t R N A j
8
^ - b y yeast phenyl
alanine enzyme. [Data recalculated from Figs. 7B and C of Taglang et al. (103).]
All maximal uninhibited rates are normalized to 100. For orientation note that intra
cellular ionic strength is about 0.2 M, corresponding to \/~^i/(l + = 0.31.
3. INHIBITORS OF AMINO ACID ACTIVATION 123 5 and 20, with the greater ionic interaction found in heterologous reac
tions. A similar highly salt-sensitive reaction is the DNA-dependent RNA polymerase reaction, where z
+
z~ is about 40 for the interaction of the DNA primer with the polymerase (109).
A different sort of effect is noted with the prolyl-tRNA ligase where the monomeric subunits (stable in the cold or in salt solutions) catalyze only reaction 1. Dimerization promoted by glycerol or warming is re
quired to bind tRNA and effect esterification (54) •
Enzymes and tRNA's derived from the extremely halophilic bacteria Halobacterium cutirubrum bind so tightly that high salt concentrations are required to promote dissociation of product from enzyme. In this case, low salt concentrations are inhibitory (15, 16, 110). Svensson (111) has reviewed the need for some metal ion participation in these reactions.
The aminoacylation of t R N A A la
and t R N A C ys
is exceptionally sensitive to very low concentrations of Ca
2+
(111a). Monovalent cations such as NH^~ inhibit the formation of L y s ~ t R N A from ATP, lysine and tRNA while NH^" facilitates the synthesis of L y s ^ t R N A from E n z - ( L y s ~ AMP) and tRNA. Hele and Barber (111b) interpret this to indicate two separate pathways to the synthesis of Lys~tRNA.
IV. CONCLUSION
A. Stoichiometry of Inhibitors
The amino acid:tRNA ligases have a wide variety of physical proper
ties. They exist as single-chain polypeptides of molecular weight 110,000, as dimers or tetramers of subunits of 50,000-100,000 molecular weight, and possibly even as supermacromolecular aggregates (for a tabular review, see Table I, 16). Inhibitors have been instrumental in establish
ing that there is no subunit interaction even in enzymes such as methio
nine that are believed to be tetrameric. Plotting according to the Hill analysis gives a curve whose slope corresponds to the number of interact
ing subunits. Figure 4 (16) shows all such slopes to be exactly 1.0.
Because the aminoalkyl adenylates are stable and are firmly bound to the active site, these inhibitors have been useful in determining the number of active sites per enzyme molecule. Thus, there appears to be a single active site per monomeric subunit of the methionine enzyme
(four subunits), but only one active site per tetramer of the phenyl
alanine enzyme (31, 112, 112a).
T A B L E 1
BALANCE SHEET FOR THE E X T E N T OF INHIBITION OF A T P : P P I EXCHANGE AND APPEARANCE OF HYDROXAMATE AND A M P AS CATALYZED B Y E. coli
V A L I N E E N Z Y M E "
N H2O H Imidazole Phenanthroline Reaction (2.5 M ) (0.74 M ) (0.015 M )
PPi: A T P exchange
N o inhibitor 375
&
375
6
375*
Inhibitor present 12. 5
C
202*
Difference = amount of inhibition 362. 5 306" 173"
;
1 4
C ] A M P formed from [
1 4
C ] A T P
N o PPi present 3. 0 / 1.0 1.
0.2 m M PPi present 1. 5
1 4
C]Valine converted to hydroxamate
N o PPi present 3. 0*
0.2 m M PPi present 1. 9
0
The data are presented in terms of micromoles converted per minute X milliliter of enzyme solution. 6
With 5 m M A T P , 0.1 m M [
3 2
P]PPI, 0.5 m M valine.
c
Same as a, but with 2.5 M N H2O H . d
Same as a, but with 0.74 M imidazole and 3 m M PPI.
e
Same as a, but with 15 m M phenanthroline and 1.25 m M PPI.
' 1.0 m M [
1 4
C ] A T P and 0.8 m M valine, 2.5 M N H2O H .
0 1.0 m M A T P and 0.8 m M [
1 4
C]valine, 2.5 M N H2O H .
B. Use of Inhibitors to Determine Mechanism
1. NH2OH AGAINST THE ATP:PPi EXCHANGE
If only reaction 1 were functioning, one might expect that NH2OH would react with enzyme-bound aminoacyl adenylate and thus reduce the amount of this intermediate available for reaction with [
3 2
P]PPi to form [
3 2
P ] A T P . This would of course mean that an amount of hydrox
amate and AMP would form in proportion to the amount of PPi that is prevented from reacting. Similarly, other bases such as imidazole or phenanthroline would cause the production of AMP and PPi in propor
tion to their effectiveness in reacting with the aminoacyl adenylate.
Table I (adapted from Loftfield and Eigner, 3) shows that the mechan
ism of inhibition of ATP:PPi exchange cannot be ascribed to competi
tion at the level of aminoacyl adenylate, that the competition is at some point earlier in the sequence.
3. I N H I B I T O R S OF A M I N O A C I D A C T I V A T I O N 1 2 5
1 I I i I I i 1 1 1 1 1— r
- 8 - 7 - 6 - 5 - 4 - 9 - 8 - 7 - 6 - 5 - 4 - 3 - 2 - I l o g ( / )
FIG. 4. (a) The Hill plot for the inhibition b y m e t h i o n y l - A M P ( O ) , isoleucinyl- A M P (A), and v a l y l - A M P ( • ) of A T P : P P i exchange catalyzed b y the correspond
ing enzymes of E. coli. (b) The Hill plot for the same inhibitors on the corresponding esterifications of E. coli t R N A . (c) The Hill plot for the PPi inhibition of A T P : P P}
exchange catalyzed b y isoleucine and the isoleucine:tRNA ligase of E. coli. All curves are recalculated from original data of (a) Table I V of Cassio et al. (75), (b) Table V of Cassio et al. (75), and Fig. 12 of Loftfield and Eigner (3). The straight lines of slope 1.00, pass through all points, establishing that there is neither positive nor nega
tive interaction between subunits. For orientation note that an ordinate value of 0.0 corresponds to 5 0 % inhibition, an ordinate value of + 1 . 0 corresponds to about 9 0 % inhibition, and + 2 . 0 is approximately 9 9 % inhibition. [From Loftfield (16).]
2. PPj AGAINST HYDROXAMATE OR AMiNOACYL-tRNA FORMATION
As in the previous section, one might have thought that PPi is inhibi
tory because it reduces the amount of aminoacyl-tRNA available for reaction with tRNA or NH2OH. Not only does this fail to account for the extremely effective inhibition by PPi (10~
8
M in some cases) but analysis as in Table II (from Loftfield and Eigner, 3) shows that PPi is not competitively reacting with any intermediate that otherwise forms a hydroxamate.
3. DEOXYADENOSINE TRIPHOSPHATE
As noted above, dATP substitutes for ATP with many amino acids in the synthesis of aminoacyl-tRNA ( 2 , 5, 9 ) . In the case of the phenyl alanine ligase, dATP supports a dATP:PPi exchange, showing that reaction 1 is operative and that PPi can react with phenylalanyl deoxy- adenylate. However, dATP is completely unable to support tyrosine- catalyzed dATP : PPi exchange in the absence of t R N A
T y r
, showing that another path (i.e., reaction 2 ) is operative.
T A B L E I I
B A L A N C E SHEET FOR PPi INHIBITION OF ISOLEUCINE HYDROXAMATE F O R M A T I O N
0
[
1 4
C]Isoleucine hydroxamate formation
N o PPi (vessel 1) 5 . 4 5 Plus 0.10 m M PPi (vessel 2) 1.20
Difference = extent of inhibition 4 . 2 5 A T P : P P i conversion with 0.1 m M [
3 2
P]PPi (vessel 3) 0 . 1 2
"Conditions: Each reaction vessel contained 0.10 m M [
1 4
C]isoleucine, 2.5 m M A T P ( M g
2
+ ) , 5.6 M N H2O H ( p H 7.5), 0.1 M tris ( p H 7.5), and 6 Mliters of the E. coli isoleucine enzyme. The second and third reaction vessels contained 0.1 m M PPi.
The third reaction mixture was identical to the second except that the PPi was radio
active and conversion to [
3 2
P ] A T P was determined. The reactions were carried out at 25°C and aliquots were removed for assay at 0, 5, 10, and 15 minutes. Units are micromoles converted per minute X milliliter of isoleucine enzyme solution.
4. SPERMINE
As noted above, M g 2+
is generally required for reaction 1 and has been considered as necessary for the overall esterification of tRNA. Re
cently it has been found that, although reaction 1 cannot use spermine in place of Mg
2 +
, reaction 2 can use spermine (113). Thus, although spermine is not actually an inhibitor of these enzymes, its failure to promote ATP:PPi exchange is helpful in establishing the mechanism by which these critical reactions proceed (113a).
5. AMINO ACID ANALOGS
Kinetic analysis of the effects of several inhibitors such as salicylate versus glutamate (74a) or several tyrosine derivatives versus tyrosine (2) have led several workers to conclude that the formation of amino
acyl-tRNA involves a completely random order of addition of the three substrates to the enzyme, followed by a reaction which could be concerted as in Fig. 2. A highly ordered addition and reaction sequence
A A
ATP + Enz - » Enz • ATP^»
Enz-ATP-AA -> E n z - A A ~ A M P + p P j ^ E n z - A A - A M P - t R N A ->
Enz • A A ~ t R N A + AMP -> Enz + A A ~ t R N A is advocated by others (17-17d).
C. Prospects for Therapy
The synthesis of protein is one of the most critical operations of the living cell. Anything that interferes with the accuracy of the synthesis
3. INHIBITORS OF AMINO ACID ACTIVATION 127 or the rate of synthesis is likely to damage the cell, especially if it is rapidly growing. Substances like puromycin, streptomycin, and actino- mycin are examples of how effective drugs can be at other points in protein biosynthesis. Only a few crude shotgun efforts have gone into developing compounds that specifically inhibit the esterification of tRNA but it is clear that this is an area where therapeutic applications are possible. A quick glance at the references will show that almost all the work on mechanism and on inhibition is the product of only the last three years. Surely as we learn more about the mechanism we can tailor compounds to specific purposes and specific tissues. The thought of introducing into specific cells nonmetabolizable potent inhibitors, such as derivatives of 5'-deoxyadenosine, of methylene diphosphonate, or aminoalkyl adenylates, is a product of only the last few years' under
standing. Much more can be expected.
ACKNOWLEDGMENTS
The preparation of this essay has been, in part, supported b y research grants from the American Cancer Society (BC-11B) and the United States Public Health Service (CA-08000). I am grateful to colleagues for advice and criticism, to my secretaries, Mrs. Samaha and Miss Clenney, for their painstaking assistance and to m y wife, Ella, for patient understanding.
REFERENCES
1. R. B. Loftfield and E. A. Eigner, Science 1 6 4 , 305 (1969).
2. D . V . Santi and V. A. Pena, FEBS Lett 1 3 , 157 (1971).
3. R . B. Loftfield and E. A. Eigner, J. Biol. Chem. 2 4 4 , 1746 (1969).
4. P. C. Zamecnik and M . L. Stephenson, "Proceedings of the Alfred Benson Foundation Symposium I," p. 276. Academic Press, New York, 1969.
5. D . V . Santi and P. V . Danenberg, Biochemistry 1 0 , 4813 (1971).
6. D . V. Santi, P. V. Danenberg, and K. A. Montgomery, Biochemistry 1 0 , 4821 (1971).
7. S. L. Owens and F. E. Bell, J. Biol. Chem. 2 4 5 , 5515 (1970).
8. I. Hirschfield and H . J. P. Bloemers, / . Biol. Chem. 2 4 4 , 2911 (1969).
9. S. K . Mitra and A . H . Mehler, Eur. J. Biochem. 9, 79 (1969).
10. K. Randerath, C. M . Janeway, M . L. Stephenson, and P. C. Zamecnik, Biochem.
Biophys. Res. Commun. 2 4 , 98 (1966).
11. P. Berg and E. J. Ofengand, Proc. Nat. Acad. Sci. U.S. 4 4 , 78 (1958).
12. M . P. Stulberg and G. D . Novelli, in "The Enzymes" (P. D . Boyer, H . Lardy, and M . Myrback, eds.), 2nd rev. ed., V o l . 6, p. 401. Academic Press, New York, 1962.
13. J. M . Ravel, S. F. Wang, C. Heinemeyer, and W . Shive, J. Biol. Chem. 2 4 0 , 432 (1965).
14. M . P. Deutscher, J. Biol. Chem. 2 4 2 , 1132 (1967).
15. R . B. Loftfield, in "Protein Biosynthesis" ( E . McConkey, ed.), Dekker, New York, 1971.
16. R. B. Loftfield, Progr. Nucl. Acid Res. Mol. Biol. 1 2 , 87 (1972).
17. P. Rouget and F. Chapeville, Eur. J. Biochem. 4 , 310 (1968).
17a. C. C. Allende, H. Chaimovich, M . Gatica, and J. E. Allende, J. Biol. Chem., 2 4 5 , 93 (1970).
17b. A. V . Parin, E. P. Savelyev and L. L. Kisselev, FEBS Lett. 9 , 163 (1970).
17c. S. A. Berry and M . Grunberg-Manago, Biochim. Biophys. Acta, 2 1 7 , 83 (1970).
17d. M . Y a m s and P. Berg, / . Mol. Biol, 4 2 , 171 (1969).
17e. E. W . Eldred and P. R. Schimmel, Biochemistry, 1 1 , 17 (1972).
18. A. H. Mehler and S. K . Mitra, J. Biol. Chem. 2 4 2 , 5495 (1967).
19. R . B. Loftfield and E. A. Eigner, Biochemistry 7 , 1100 (1968).
20. S. K . Mitra and A. H. Mehler, J. Biol. Chem. 2 4 2 , 5490 (1967).
21. J. Vanhumbeeck and P. Lurquin, Eur. J. Biochem. 1 0 , 213 (1969).
22. T. S. Papas and A. H . Mehler, J. Biol, Chem. 2 4 5 , 1588 (1970).
23. C. Bublitz, Biochim. Biophys. Acta 1 1 3 , 158 (1966).
24. B. Niyomporn, D . Dahl, and J. L. Strominger, / . Biol. Chem. 2 4 3 , 773 (1968).
25. D . L. Ostrem and P. Berg, Proc. Nat. Acad. Sci. U.S. 6 7 , 1967 (1970).
26. M . De Luca and W . D . McElroy, Arch. Biochem. Biophys. 1 1 6 , 103 (1966).
27. C. Helene, F. Brun, and M . Yaniv, / . Mol. Biol. 5 8 , 349 (1971).
28. M . Yaniv and F. Gros, J. Mol. Biol. 4 4 , 1 (1969).
29. F. Fasiolo, N . Befort, Y . Boulanger, and J. P. Ebel, Biochim. Biophys. Acta.
2 1 7 , 305 (1970).
30. F. J. Lawrence, Eur J. Biochem. 1 5 , 436 (1970).
31. D . Cassio, Eur. J. Biochem. 4 , 222 (1968).
32. D . I. Hirsh, J. Biol. Chem. 2 4 3 , 5731 (1968).
33. A. V. Parin and L. L. Kisselev, Mol. Biol. 3 , 636 (1969).
34. H . L. James and E . T . Bucovaz, J. Biol. Chem. 2 4 4 , 3210 (1969).
35. R . Stern and A. H . Mehler, Biochem. Z. 3 4 2 , 400 (1965).
36. J. Waldenstrom, Eur. J. Biochem. 3 , 483 (1968).
37. R. Stern and A. Peterkofsky, Biochemistry 8 , 4346 (1969).
38. T. K u o and M . DeLuca, Biochemistry 8, 4762 (1969).
39. M . Iaccarino and P. Berg, J. Mol. Biol. 4 2 , .151 (1969).
40. G. A. Hahn and J. W . Brown, Biochim. Biophys. Acta 1 4 6 , 259 (1963).
41. R. B. Loftfield and E. A. Eigner, unpublished observations (1969).
42. D . G. Knorre, Mol. Biol. 2 , 715 (1968).
43. C. Bublitz, Biochem. Biophys. Acta 1 2 8 , 165 (1966).
44. S. K . Mitra and A. H . Mehler, J. Biol. Chem. 2 4 1 , 5161 (1966).
45. F. X . Cole and P. Schimmel, Biochemistry 9 , 480 (1970).
45a. G. Keith, J. Gangloff, and G. Dirsheimer, Biochimie 5 3 , 661 (1971).
46. I. B. Rubin, A. D . Kelmers, and G. Goldstein, Anal Biochem. 2 0 , 533 (1967).
47. C. Hedgcoth, J. Ravel, and W . Shive, Biochem. Biophys. Res. Commun. 1 3 , 495 (1963).
48. M . H . Makman and G. L. Cantoni, Biochemistry 5 , 2246 (1966).
49. E. M . Lansford, N . M . Lee, and W . Shive, Archiv. Biochem. Biophys. 1 1 9 , 272 (1967).
50. T . Okamoto, Biochim. Biophys. Acta 1 3 8 , 198 (1967).
51. A. Vescia, M . Romano, and M . Cerra, Boll. Soc. Ital. Biol. Sper. 4 2 , 642 (1966).
3. INHIBITORS OF AMINO ACID ACTIVATION 129
51a. P. Zamecnik, personal communication (1971).
52. J. Vanhumbeeck and P. Lurquin, Biochem. Biophys. Res. Commun. 31, 908 (1968).
53. T . S. Papas and A. H . Mehler, / . Biol. Chem. 243, 3767 (1968).
54. M . L. Lee and K . H. Muench, / . Biol. Chem. 244, 223 (1969).
55. K. Randerath, M . L. Stephenson, C. M . Janeway, and P. C. Zamecnik, Biochem.
Biophys. Res. Commun. 24, 98 (1966).
56. R . B. Loftfield and E. A. Eigner, Biochim. Biophys. Acta 130, 426 (1966).
57. R. B. Loftfield, L. Hecht, and E. A. Eigner, Biochim. Biophys. Acta 72, 383 (1963).
58. R . B. Loftfield and E . A. Eigner, / . Biol. Chem. 240, 1482 (1965).
59. S. L. Owens and F. E. Bell, / . Mol. Biol. 38, 145 (1968).
60. J. M . Ravel, M . N . White, and W . Shive, Biochem. Biophys. Res. Commun.
20, 352 (1965).
61. K . Tanaka, M . Tamaki, and S. Watanabe, Biochim. Biophys. Acta 195, 244 (1969).
62. I. K . Smith and L. Fowden, Phy to chemistry 7, 1065 (1968).
63. M . K o n d o and C. R . Woese, Biochemistry 8, 4177 (1969).
63a. L. Fowden and M . H. Richmond, Biochim. Biophys. Acta 71, 459 (1963).
64. A. G. Atherly and F. E. Bell, Biochim. Biophys. Acta 80, 510 (1964).
65. M . Smulson and M . Rabinowitz, Arch. Biochem. Biophys. 124, 306 (1968).
66. M . E. Smulson, M . Rabinowitz, and T . R . Breitman, J. Bacteriol. 94, 1890 (1967).
67. J. W . Anderson and L. Fowden, Biochem. J. 119, 677 (1970).
68. P. J. Peterson and L. Fowden, Biochem. J. 97, 112 (1965).
69. L. Fowden and J. B. Frankton, Phy to chemistry 7, 1077 (1968).
69a. D . W . Y e m and L. S. Williams, / . Bacteriol. 107, 589 (1971).
70. S. Kijima, T. Ohta, and K. Imahori, / . Biochem. (Tokyo) 63, 434 (1968).
71. S. L. Owens and F. E. Bell, / . Biol. Chem. 245, 5515 (1970).
72. I. Owens and J. J. Blum, / . Biol. Chem. 242, 2893 (1967).
73. G. Lemaire, M . Dorizzi, and B. Labouesse, Biochim. Biophys. Acta 132, 155 (1967).
74. M . R . Harnden and T . O. Yellin, / . Med. Chem. 13, 1095 (1970).
74a. M . Burleigh and M . J. H. Smith, J. Pharm. Pharmacol, 23, 590 (1971).
74b. T. Tosa and L. I. Pizer, J. Bacteriol, 106, 972 (1971).
74c. S. Neale, Chem. Biol. Interact., 2, 349 (1970).
75. D . Cassio, F. Lemoine, J. P. Waller, E. Sandrin, and R . A. Boissonas, Biochem
istry 6, 827 (1967).
75a. M . K o n d o , Biochem. J., 121, 567 (1971).
75b. D . V. Santi, P. V. Dannenberg and P. Satterly, Biochemistry, 10, 4804 (1971).
76. V. Z . Holten and K . B. Jacobson, Biochemistry 6, 1293 (1967).
77. R . B. Loftfield and E . A . Eigner, Acta Chem. Scand. 17, S117 (1963).
78. M . Yarus and P. Berg, J. Mol. Biol 28, 479 (1967).
79. U. Lagerkvist, L. R y m o , and J. Waldenstrom, / . Biol. Chem. 241, 5391 (1966).
80. R . B. Loftfield, E. A. Eigner, and J. Nobel, Biol. Bull. 135, 181 (1968).
81. K. B. Jacobson, Progr. Nucl. Acid Res. Mol. Biol. 11, 461-488 (1970).
82. M . P. Deutscher, Arch. Biochem. Biophys. 125, 758 (1968).
83. F. Cramer, Progr. Nucl. Acid Res. Mol. Biol. 11, 391-422 (1970).
83a. M . P. Stulberg and L. R . Shugart, Cancer Res. 31, 67 (1971).
84. D . G. Knorre, Mol. Biol. 3 , 579-583 (1969)..
85. E. Schlimme, F. von der Haar, F. Eckstein, and F. Cramer, Eur. J. Biochem, 1 4 , 351 (1970).
86. U. Lagerkvist and L. R y m o , / . Biol. Chem. 2 4 5 , 435 (1970).
87. R . B. Loftfield and E. A. Eigner, / . Biol. Chem. 2 4 2 , 5355 (1967).
88. J. Torres-Gallardo and M . Kern, Proc. Nat. Acad. Sci. U.S. 5 3 , 91 (1965).
89. H. Hayashi and K. Miura, Cold Spring Harbor Symp. Quant. Biol. 3 1 , 63 (1966).
90. G. M . deVries, Biochim. Biophys. Acta 1 4 5 , 169 (1967).
91. V . A . Korzhov and L. S. Sandakhchiev, Biokhimiya 3 1 , 71 (1966).
92. R . W . Chambers, Progr. Nucl. Acid Res. Mol. Biol. 1 1 , 489-524 (1970).
93. B. Beltchev and M . Grunberg-Manago, FEBS Lett. 1 2 , 27 (1970).
94. B. Beltchev and M . Grunberg-Manago, FEBS Lett. 1 2 , 24 (1970).
95. M . Deutscher, Biochem. Biophys. Res. Commun. 1 9 , 283 (1965).
96. C. H. Letendre, J. M . Humphreys, and M . Grunberg-Manago, Biochim. Biophys.
Acta 1 8 6 , 46 (1969).
97. S. A. Berry and M . Grunberg-Manago, Biochim. Biophys. Acta 2 1 7 , 83 (1970).
98. H. Grosjean, J. Werenne, and H. Chantrenne, Biochim. Biophys. Acta 166, 616 (1968).
99. J. Werenne, H. Grosjean and H . Chantrenne, Biochim. Biophys. Acta 1 2 9 , 585 (1966).
100. N . Tanaka, H . Yamaguchi, and H . Umezawa, J. Biochem. (Tokyo) 6 0 , 429 (1966).
101. J. Han and J. Han, Science 1 6 4 , 560 (1969).
102. A. Peterkofsky, S. J. Gee, and C. Jesensky, Biochemistry 5 , 2789 (1966).
103. R . Taglang, J. P. Waller, N . Befort, and F. Fasiolo, Eur. J. Biochem. 1 2 , 550 (1970).
104. D . W . E. Smith, / . Biol. Chem. 2 4 4 , 896 (1969).
105. P. S. Sarin and P. C. Zamecnik, Biochem. Biophys. Res. Commun. 1 9 , 198 (1963).
106. P. 0 . Ritter, F. J. Kull, and K . B. Jacobson, / . Biol. Chem. 2 4 5 , 2114 (1970).
107. P. O. Ritter, F. J. Kull, and K . B. Jacobson, Biochim. Biophys. Acta 1 7 9 , 524 (1969).
108. Y . Takeda and K. Igarashi, Biochim. Biophys. Acta 2 0 4 , 406 (1970).
109. O. W . Jones and P. Berg, / . Mol. Biol. 2 2 , 199 (1966).
110. F. Griffiths and S. T . Bayley, Biochemistry 8 , 541 (1969).
111. I. Svensson, Biochim. Biophys. Acta 1 4 6 , 252 (1967).
111a. A. K. R o y , Biochim. Biophys. Acta. 2 4 6 , 349 (1971).
111b. P. Hele and R . Barber, Biochim. Biophys. Acta, 2 5 8 , 319 (1972).
112. C. J. Bruton and B. S. Hartley, / . Mol. Biol. 5 2 , 165 (1970).
112a. K. H. Muench and D . R. Joseph, Miami Winter Symp. 2 , 172 (1971).
113. K. Igarashi, K . Matsuzaki, and Y . Takeda, Biochim. Biophys. Acta 2 6 2 , 476 (1972).
113a. A. Pastuszyn and R . B. Loftfield, Biochem. Biophys. Res. Comm. 4 7 , 7 7 5 (1972).
114. V. Z. Holten and K. B. Jacobson, Arch. Biochem. Biophys. 1 2 9 , 283-290 (1969).