Availableonlineatwww.sciencedirect.com
ScienceDirect
j ou rn a l h om ep a ge :w w w . i n t l . e l s e v i e r h e a l t h . c o m / j o u r n a l s / d e m a
Triethylene glycol dimethacrylate impairs
bioenergetic functions and induces oxidative stress in mitochondria via inhibiting respiratory
Complex I
Krisztina Mikulás
a,b, Péter Hermann
a, István Gera
c, Timea Komlódi
b, Gerg ˝o Horváth
b, Attila Ambrus
b, László Tretter
b,∗aDepartmentofProsthodontics,SemmelweisUniversity,Budapest,Hungary
bDepartmentofMedicalBiochemistry,MTA-SELaboratoryforNeurobiochemistry,SemmelweisUniversity, Budapest,Hungary
cDepartmentofPeriodontology,SemmelweisUniversity,Budapest,Hungary
a r t i c l e i n f o
Articlehistory:
Received13August2017 Receivedinrevisedform 22January2018 Accepted23March2018
Keywords:
DentalresinmonomerTEGDMA Reactiveoxygenspecies Cytotoxicity
Hydrogenperoxide Respiration
RespiratoryComplexI ATPproduction Oxidativestress
a bs t r a c t
Objectives.Earlierstudiesdemonstratedthatdentalresinmonomerslowercellularviability andprovokeoxidativestress.Reactiveoxygenspecies(ROS)formationhasakeyrolein triethyleneglycoldimethacrylate(TEGDMA)inducedadversereactions.Inthepresentstudy theeffectsofTEGDMAonmitochondrialfunctionswereinvestigatedtoidentifyadirect moleculartargetforcytotoxicity.
Methods.Mitochondriawereisolatedfromguineapigbrain.Themostimportantbioenergetic parameters,oxygenconsumption,membranepotential(m),andATPproductionwere assessed.MitochondrialH2O2productionandeliminationandtheNAD(P)Hlevelreported onredoxbalance.
Results.Mitochondriaweresupportedwithrespiratorysubstratestobeoxidizedbyeither ComplexI(CI)orComplexII(CII).mwasdepolarized,respirationandATPproduction wasgreatlydiminishedwhenapplyingCIsubstratesinthepresenceofTEGDMA.Thesame parametersremainedessentiallyunaffectedwhenCIIsubstrateplusTEGDMAwereapplied.
H2O2 productionbymitochondriawassignificantly stimulatedbyTEGDMAinthepres- enceofCIsubstrates.InthepresenceofTEGDMAmitochondrialeliminationofexogenous H2O2wasimpaired.WhenCIIsubstratesupportedthemitochondriaintheabsenceofADP theH2O2generationwasdecreased.NADHautofluorescenceresultsalsodemonstratedthe inhibitoryeffectofTEGDMAonCIactivity.
Abbreviations: AK,adenylatekinase;ANT,adenine-nucleotidetranslocator;␣-KG,alpha-ketoglutarate;␣-KGDH,alpha-ketoglutarate dehydrogenase;AP5,P1,P5-di(adenosine-5)pentaphosphate;BSA,bovineserumalbumin;CAT,carboxyatractylate;m,mitochondrial membranepotential;DMSO,dimethyl-sulfoxide;DTNB,5,5-dithiobis(2-nitrobenzoicacid);FCCP,carbonylcyanide-p-trifluoromethoxy- phenylhydrazone;GM,glutamateplusmalate;OXPHOS,oxidativephosphorylation;SLP,substrate-levelphosphorylation;Succ,succinate.
∗ Correspondingauthorat:DepartmentofMedicalBiochemistry,SemmelweisUniversity,T ˝uzoltóst.37-47,H-1094Budapest,Hungary.
E-mailaddress:tretter.laszlo@med.semmelweis-univ.hu(L.Tretter).
https://doi.org/10.1016/j.dental.2018.03.012
0109-5641/©2018PublishedbyElsevierInc.onbehalfofTheAcademyofDentalMaterials.
Significance.TEGDMAinhibitsCIintherespiratorychain,whichexplainseffectsinducedby TEGDMAonredoxhomeostasis,apoptoticandnecroticcelldeathsdescribedinprevious studies.IdentificationofthemoleculartargetofTEGDMAmayinfluencethedevelopment ofrelevantbiomaterialsandmayinducenewtherapeuticstrategiestocontroltheadverse effectsofresinmonomers.
©2018PublishedbyElsevierInc.onbehalfofTheAcademyofDentalMaterials.
1. Introduction
Dental composite resins are commonly used to restore structuraltooth damages. Thedevelopment ofresin-based composites(RBCs)hasresultedinestheticallypleasingand longlastingdentalrestorations[1].Recently,biocompatibil- ity ofthefilling materialshas becomeaclinically relevant issue[2].MostRBCscontainstronglyviscousmajormonomers andco-monomersoflowerviscositysuchastriethyleneglycol dimethacrylate(TEGDMA)[3–5].IncompleteRBCpolymeriza- tionmayleadtoanundesiredleachingofthemonomersinto theoraltissues[6,7].TEGDMAisofamphiphiliccharacterand themostcommonco-monomer releasedfrompolymerized composites [5,8–10]. Shortly after the polymerization pro- cesswas considered to becompleted, both 2-hydroxyethyl methacrylate (HEMA)and TEGDMA were detectable in the salivainmillimolarconcentrations[5,6,11,12].Aftermonths andyears,mechanicaldeteriorationandhydrolyticdisinte- grationbyunspecificsalivaryesterases may inducefurther releaseoftheresinmonomers,which areabletocrossthe dentin,enter the pulp,and reach concentrationsthat may indeedaffect cellularfunctions [8,13];the concentration of TEGDMA in the pulp was shown to reach levels of up to 4mM [11,14,15]. The systemic blood concentrations of the monomersareordersofmagnitudelower[6],thereforethe probabilityofthedevelopmentofTEGDMA-inducedadverse systemiceffects isnegligible [9]. TEGDMA and HEMA were reportedtoaffectvariouscellularfunctionsandtheviability ofthecellsintheoralcavity[12,16–18].Itwasreportedearlier thatTEGDMAcouldinitiatecellstressresponses,inducereac- tiveoxygenspecies(ROS)generation,andcauseglutathione depletioninawidevarietyofeukaryoticcellsinvitro[12,19,20].
The genotoxic [21] and mutagenic effects of TEGDMA are likelyconsequencesofROS-triggeredDNAdamages;thetoxic effects could apparently be eliminated applyingROS scav- engers[18,22–25].Itwasdemonstrated thatTEGDMA could induce,dependingontheconcentration,apoptotic[26,27]or necroticcelldeath[27–30].
These results inthe literatureraise the possibility of a common mechanism and perhaps molecular target which wouldberesponsiblefortheobservedcellularchanges,how- ever, the underlying mechanism behind these phenomena remains poorly understood. Recently the involvement of NADPHoxidase4(NOX4)inTEGDMA-inducedapoptosisand ROSgenerationwasdescribed[31].Earlieracollapsedmito- chondrialmembranepotentialandincreasedROSformation weredetectedingingivalfibroblastsafteratreatmentwith TEGDMA[32].Mitochondrialdysfunctioncertainlyinfluences cellviabilityasthedecreasedATPlevelcanleadtonecrotic
celldeath.Mitochondrialimpairmentcanincreasethelevel ofROS,provokeDNAdamages,andconsequentlyinduceapo- ptosis[33].Therefore,itappearedtobeplausibletoseekafter mitochondrialtargetsinordertounderstandthemechanism ofTEGDMA-inducedtoxicity.Takingintoaccountthatmito- chondrial impairment can either be primaryor secondary, andthatthedistinctionisnotalwaysclearinacellularsys- tem,isolatedmitochondriawerechosenastheobjectsofthis study.Accordingtoourhypothesis,mostofthetoxiceffectsof TEGDMAcouldpotentiallybeexplainedbythedevelopmentof bioenergeticinsufficiencyandincreasedROSgeneration.The hypothesisthatmitochondrialdysfunctionscouldberespon- siblefortheonsetofeithernecrotic[34,35]orapoptoticcell death [36]does notdemandany sophisticatedverification.
LowdosesofComplexI(CI)inhibitorsgenerallyinduceapo- ptosis,whilehigherdosesofthesamecompoundmayevoke necroticcelldeath[37].
TheroleofmitochondriaincellulargenerationofROSis alsowellknown[38].Variousmechanismsandmolecularenti- tiescanberesponsiblefortheenhancedROSgenerationof mitochondria[39,40].
In the present study, low millimolar concentrations of TEGDMAwereusedtochallengeisolatedbrainmitochondria and themostimportantbioenergeticfunctionswere evalu- atedinparallelwiththemeasurementofmitochondrialH2O2
homeostasis.Weproposethatinhibitionofthemitochondrial respiratory CImayberesponsibleformostofthe cytotoxic effectsofTEGDMA.
2. Materials and methods
2.1. PreparationofbrainmitochondriaAnimal experiments were performed in accordance with the relevant guidelines of the National Institute of Health (USA) and the Semmelweis University. Mitochondria were isolated fromguineapigbraincortexusingaPercollgradi- ent as detailed elsewhere [41]. Unless otherwise indicated, 0.1mg/mLmitochondrialproteinconcentrationwasapplied foralltheexperiments.
2.2. Incubationmedium
Measurementswerecarriedoutat37◦Cinanassaymedium containing the following components: 125mM KCl, 20mM HEPES,2mMK2HPO4,1mMMgCl2,and0.025(w/v)%bovine serumalbumin(BSA;freeoffattyacids).Mitochondriawere energizedbyeitheralpha-ketoglutarate(␣-KG),orglutamate
plusmalate(GM),orsuccinate(Succ),allin5mMconcentra- tion.
2.3. Measurementofmitochondrialoxygen consumption
Mitochondrial respiration was monitored using the high- resolutionrespirometrysystem Oxygraph-2k[42](Oroboros Instruments,Innsbruck,Austria).Oxygensensorswerecali- bratedatair-saturationandinoxygen-depletedmedia.
2.4. Measurementofmitochondrialmembrane potential( m)
Mitochondrial m wasdetected usingsafranine O (2M).
Thislipophiliccationicdyegetsdistributedamongmitochon- driaandthesurroundingmedium;thevalueofthepartition coefficientisthefunctionoftheactualmitochondrial m. MeasurementswereperformedusingaHitachiF-4500spec- trophotometer(HitachiHighTechnologies,Maidenhead,UK) at 495 and 585nm excitation and emission wavelengths, respectively,asdescribedpreviously[43].
2.5. ParallelmeasurementsforH2O2andNAD(P)H
H2O2formationbyisolatedmitochondriawasdetectedusing theAmplexUltraRedfluorescenceassay(forreview,seeRef.
[44]). In this assay, Amplex Ultra Red (3M) is converted tothe highlyfluorescentcompoundresorufin byH2O2 and horseradishperoxidase(5U/2mL).Fluorescencewasdetected usingaPTIDeltascanfluorescencespectrophotometer(Pho- ton Technology International, Lawrenceville, NJ, USA); for excitation and emission, the 550 and 585nm wavelengths wereapplied,respectively.Eachmeasurementwascalibrated by100pmol H2O2attheendoftheexperiment.Inaparal- lelexperiment,usingthedouble-excitation/double-emission mode ofthe spectrofluorimeter, NAD(P)H fluorescencewas alsomeasured at340 and 466nm excitationand emission wavelengths,respectively.
2.6. MeasurementofmitochondrialH2O2elimination
MitochondrialeliminationofH2O2 wasmeasured asprevi- ouslydescribed[45].Briefly:glutamateplusmalatesupported mitochondria (0.1mg/mL) were preincubated in either the presenceorabsenceofTEGDMA (5mM)orDMSOfor5min.
At5minH2O2 (10M)wasgiven, then 50Lsampleswere takenat30sintervalsfor2min,andtheresidualH2O2was determinedbyAmplexUltraRedfluorimetry,asdescribedin thepreviousSection2.5.Measurementswerecalibratedwith knownamountsofH2O2.
2.7. Measurementofreducedglutathioneconsumption
The measurement of reduced glutathione (GSH) is based onthe reaction ofthe thiolSH group with5,5-dithiobis(2- nitrobenzoicacid) (DTNB). Freshly diluted GSH(2mM)was incubatedinthereactionmediumat37◦Cforvarioustime intervals.Aliquots were taken and the generatedTNBwas measuredviaspectrofotometryat412nm[46].
2.8. MeasurementofATPproduction
ATPproductionbyisolatedmitochondriawasdetectedusing acoupledenzymeassaythatapplieshexokinaseandglucose- 6-phosphatedehydrogenase,asdescribedpreviously[47].The assay mediumwas supplementedwith 3mM NADP+, 1.5U hexokinase,0.5Uglucose-6-Pdehydrogenase,5mMglucose, and 200M2-amino-phosphono-phentanoate(anadenylate kinase inhibitor). Mitochondria (0.05mg/mL), ADP (2mM), glutamate plus malate (5–5mM) or succinate (5mM), and TEGDMA(5mM)wereaddedasindicatedinthecorresponding figures.ADPwasconvertedtoATP,whichleftthemitochon- drion viathe adenine nucleotide translocase (ANT). In the medium,ATPphosphorylatedglucoseviahexokinaseandthe produced glucose-6-phosphate got further converted to 6- phosphogluconate via glucose-6-phosphate dehydrogenase;
withthe concomitantreductionofNADP+ toNADPH.Light absorbance of NADPH was recorded at 37◦C and 340nm (ε=6220M−1cm−1)usingaJASCOspectrophotometer(ABL&E- JASCOV-650,Tokyo,Japan).
2.9. DetectionofmitochondrialrespiratoryComplexI activity
FortheComplexIassaythemethoddescribedbyRaganand Hatefi [48] was used. The assay measures the rate of the conversionofNADHtoNAD+ whiletheelectronacceptoris Coenzyme Q1 (CoQ1). Mitochondria were incubated in the standardmediumandsubsequentlyfrozenandthawedthree times. Thereafter,200gwastransferredintothefollowing medium:100mMKH2PO4,40MCoenzymeQ1,1mMKCN,pH 7.4.50MNADHinitiatedthereaction,whichwasrepeatedin thepresenceofeither5mMTEDGMAor1Mrotenone.The initialrateofthechangeinabsorbanceat340nmwasdetected at37◦CusingaJASCOspectrophotometer.
2.10. Chemicals
TEGDMA was obtained from VOCO GmbH (Cuxhaven, Germany)anddissolvedindimethylsulfoxide(DMSO);the1M stocksolutionwasstoredat−20◦C.Thepurityoftriethylene glycoldimethacrylatewas97%;theremaining3%wastriethy- leneglycolmonomethacrylate.ThecontentofUVinhibitoris 150ppmHQME (hydroquinonemonomethylether)(0,015%), whichisusualforamethacrylatemonomer.
StandardlaboratorychemicalswerepurchasedfromSigma (St.Louis,MO,USA).TheAmplexUltraRedreagentwasfrom MolecularProbes(Eugene,OR,USA).
2.11. Statistics
PairwisecomparisonswereevaluatedbyStudent’st-test.Sta- tisticaldifferencesformultiplecomparisonswereevaluated usingtheone-wayANOVAmethod,orfordatanotfollowing normaldistributionANOVAonranksKruskarWallistestwas applied.Valuesofp<0.05wereconsideredtobestatistically significant.
3. Results
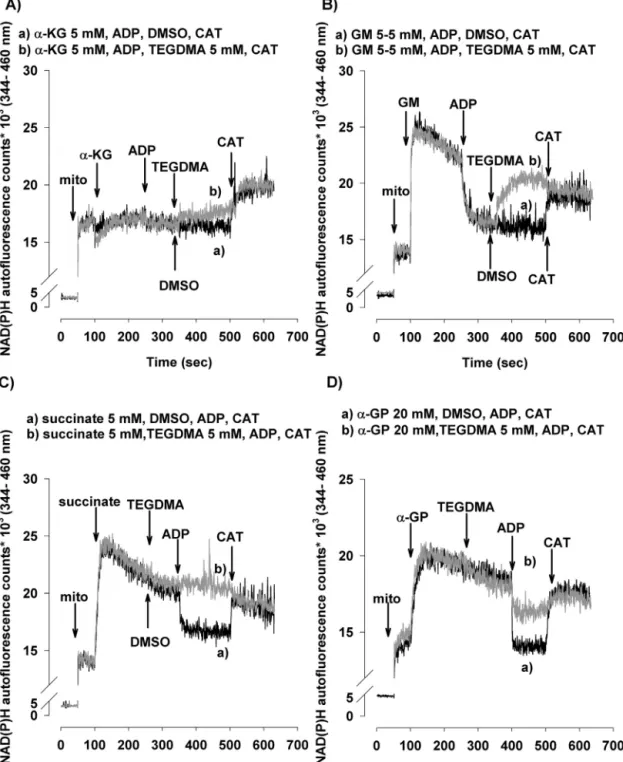
3.1. TheeffectofTEGDMAonmitochondrial respiration
Mitochondria were energized with by either alpha- ketoglutarate (␣-KG), or glutamate plus malate (GM) or succinate (Succ) as respiratory substrate. NADH is formed wheneither␣-KGorGMgetsoxidizedinsidethemitochon- drion. Since the produced NADH is oxidized by CI, ␣-KG and GM are both considered to be Complex I substrates;
whereasoxidationofsuccinateispredominantlyassociated withComplex II. Oxidativephosphorylation (OXPHOS) was initiated by ADP (2mM). Afterwards, mitochondria were challenged with TEGDMA. Experiments were terminated withcarboxyatractylate(CAT)(2M)whichinhibitsADP-ATP exchange[49].
3.1.1. OxidationofComplexIsubstrates
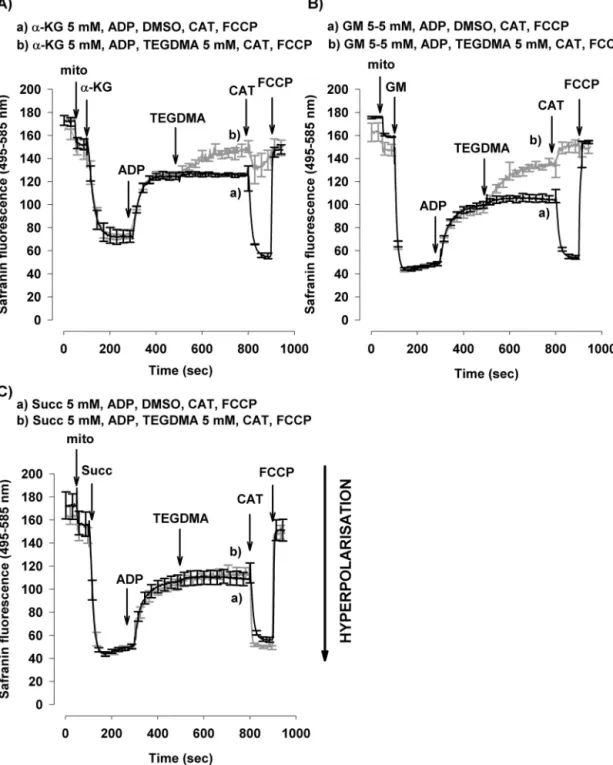
InthepresenceofADPoxygenconsumptionwasincreased from12±1to98±2nmol/min/mgproteinin␣-KG-supported mitochondria (Fig. 1A fororiginal traces and Fig. 1B). The high respiratory control ratio (RCR: oxidation rate in the presenceof ADP vs oxidation rate in the absence of ADP) indicated the high quality of mitochondrial preparations usedinthisstudy(RCR>7).Asubsequentadministrationof TEGDMA(5mM)resultedin69%decreaseinoxygenconsump- tion (from98±2to30±2nmol/min/mgprotein) (Fig. 1A,B).
ChallengingmitochondriawithanANTinhibitordecreased the ␣-KG-supported respiration both in the absence and presence of TEGDMA (Fig. 1A,B). In GM-energised mito- chondria,respiration wasstimulatedbyADPfrom25±2to 200±9nmol/min/mgprotein.Similarlytotheeffectobserved with␣-KG,addition ofTEGDMA (5mM) decreasedthe rate of the ADP-stimulated respiration by 88% (from 200±9 to 24±3nmol/min/mg protein). Significant effect of TEGDMA onADP-stimulatedO2consumptionwasdetectedfrom2mM concentration (Fig. 1C). CAT lowered the rate of oxygen consumptionbothinTEGDMA-treatedandnon-treatedmito- chondria.
3.1.2. OxidationsupportedbyaComplexIIsubstrate Unlike with the NADH-linked substrates, in succinate- supported mitochondria TEGDMA given after ADP did not influencesignificantlytherateofoxygenconsumption(from 166±10nmol/min/mg protein to 159±5nmol/min/mg pro- tein)(Fig.1D).TheseresultsunequivocallyimplythatTEGDMA doesnotinhibitComplexIIfunction.
3.2. EffectofTEGDMAonthemitochondrial membranepotential( m)
The misanimportantparameterofmitochondrialbioen- ergeticcompetence,whosebuildingupisaprerequisitefor efficientOXPHOS-dependentATPsynthesis. m wasmea- sured in isolated mitochondria supported by either ␣-KG or GM or Succ. The experimental protocol was identical to that of the oxygen consumption measurement (Sec- tion 3.1). In the presence of respiratory substrates, the
inner membrane became hyperpolarized (Fig. 2), indicated bya decreaseofsafraninfluorescence. m was higherin GM-supported mitochondria than in mitochondria respir- ing on␣-KG, reflecting the higher rate ofoxidation ofGM (Figs. 2A,B and 1B,C). Subsequent addition of ADP caused depolarization with both substrates. Both in ␣-KG and in GM supported mitochondria, TEGDMA induced further depolarization(Fig.2A,Btraceb).AdditionofCATtoTEGDMA- treatedmitochondriaresultedinacompletedepolarization, as opposed to control mitochondria where CAT induced membrane hyperpolarization. The total depolarization of mitochondriawasachievedusinguncouplercarbonylcyanide- p-trifluoromethoxy-phenylhydrazone (FCCP) (250nM). Con- trary to that observed with CI substrates in succinate supported mitochondriaTEGDMA didnot depolarize mito- chondria in the presence of ADP and subsequent addi- tion of CAT resulted in an elevation of m both in TEGDMA-treated mitochondria and in control condition (Fig.2C).Consequentlymmeasurementsalsosupportthe notion that Complex I activity is selectively inhibited by TEGDMA.
3.3. TheeffectofTEGDMAonH2O2generationand elimination
Theprominentroleofmitochondriainmodulationofredox homeostasisiswidelyaccepted[40,50].Mitochondriapartici- patenotonlyinROSgeneration,butalsoinROSdetoxication (for review see Ref. [44]). In the following experiments, mitochondrial H2O2 production and elimination were both measuredinthepresenceofCIsubstratesandH2O2produc- tionwasalsomeasuredwithnon-CIsubstrateslikeSuccand alpha-glycerophosphate(␣-GP).
3.3.1. H2O2formationinmitochondriasupportedby NADH-linkedsubstrates
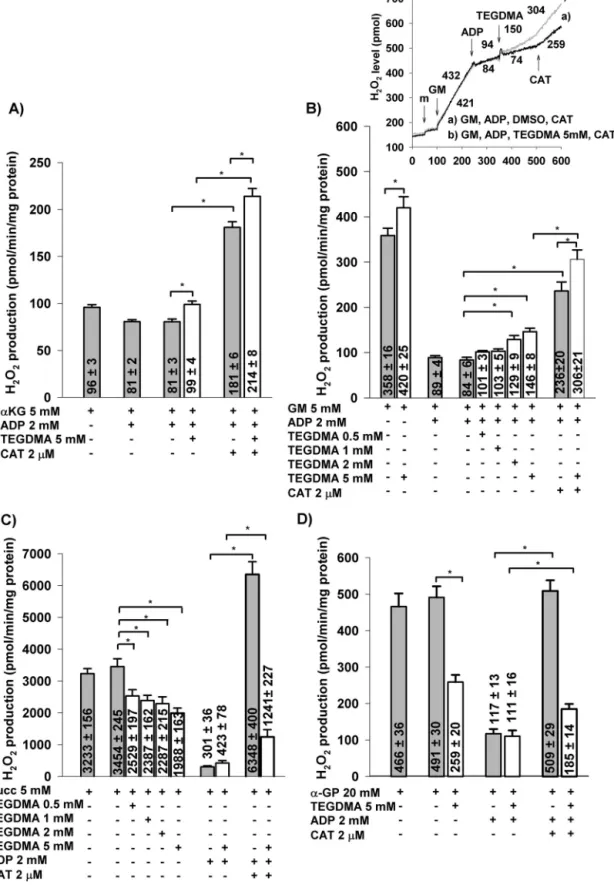
GM and ␣-KG both initiated H2O2 production in mito- chondria, as demonstrated in Fig. 3A,B. Addition of TEGDMA elevated ROS production by 17% (from 358±16 to420±25pmol/min/mg protein) (Fig. 3B).AdditionofADP resulted in a reduction in the rate of H2O2 production [41,51] in mitochondria energized by either ␣-KG or GM, due to depolarization of the inner mitochondrial mem- brane. TEGDMA augmented the rate of H2O2 production by 18% (from 81±2 to 99±4pmol/min/mg protein) in ␣- KG-supported mitochondria (Fig. 3A), and by 74% (from 84±6 to 146±8pmol/min/mg protein) in GM-supported mitochondria (Fig. 3B and B inset). Significant stimulation of H2O2 production has already been observed at 2mM TEGDMA (Fig. 3B).The addition ofCATcaused remarkable stimulation of H2O2 formation both in the presence and absence ofTEGDMA.Itisknown that thehorseradishper- oxidase (HRP)-Amplex Ultra Red system not only detects H2O2 formation, butit alsoeliminatesH2O2 [44]mitigating the H2O2-mediated oxidative stress.In order toassess the effect of TEGMA without this interference, mitochondria were pretreated with TEGDMA for 10min in the presence of glutamate plus malateand the absence ofAmplex plus HRP.
Fig.1–EffectsofTEGDMAontheO2consumptionrateoftheisolatedbrainmitochondriainthepresenceofvarious respiratorysubstrates:␣-ketoglutarate(␣-KG;A,B),glutamateplusmalate(GM;C),orsuccinate(Succ;D).Mitochondriawere incubatedinthe‘standardmedium’describedunderSection2.ADP,TEGDMA(graycurve),DMSO(blackcurve),and
carboxyatractylate(CAT)weregivenasindicatedontheoriginaltraces(1A).TheeffectsofTEGDMA(whitebars)(withvarious concentrationsinGM-supportedmitochondriaaswell1C)arecomparedtocontrols(graybars,noTEGDMAonlyDMSOwas added).Oxygenconsumption,writtenonthebars,isexpressedinnmol/min/mgprotein(mean±SEM).Significant differencesareindicated*p<0.05.
Fig.2–TheeffectofTEGDMAonmin␣-ketoglutarate(A),glutamateplusmalate(B)andsuccinate(C)respiringbrain mitochondria.Mitochondria(0.1mg/mL)wereincubatedinstandardmediumasdescribedinSection2.TEGDMA(5mM),
␣-ketoglutarate(␣-KG;5mM),glutamateplusmalate(GM;5–5mM),succinate(Succ;5mM),ADP(2mM),carboxyatractylate (CAT;2M)andFCCP(250nM)wereaddedasindicated.TheeffectofTEGDMA(darkgrey;traceb)iscomparedwithcontrols (withoutTEGDMAblack;tracea).Tracesrepresenttheaverage±SEMoffourindependentexperiments.
3.3.2. EffectofpretreatmentwithTEGDMAonROS production
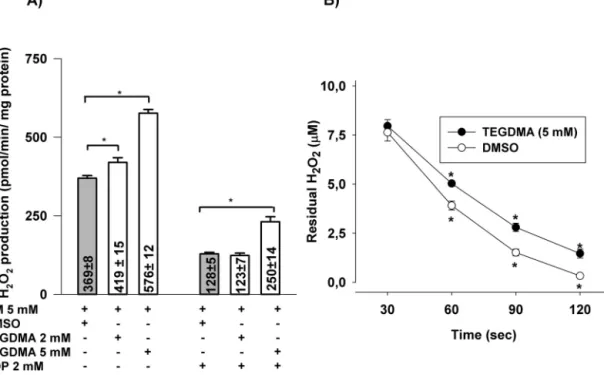
Afterthe10minpreincubation,H2O2 productionwasmea- sured by Amplex plus HRP. In the presence of TEGDMA (5mM)H2O2productionwasstimulatedby56%(from369±8 to576±12pmol/min/mg protein) (Fig. 4A).Addition ofADP decreasedtherateofH2O2formation,butinthepresenceof TEGDMA95%stimulationwasdetected.
3.3.3. H2O2eliminationinmitochondriasupportedby NADH-linkedsubstrates
Mitochondriawere challengedwithexogenous H2O2 inthe presenceorabsenceofTEGDMA(5mM)andthedisappearance ofH2O2wastestedasdescribedinSection2.Inthepresence ofTEGDMA asignificant decreaseinH2O2 elimination was observed(Fig.4B).
Fig.3–EffectsofTEGDMAonH2O2productionintheisolatedbrainmitochondriasupportedby␣-ketoglutarate(␣-KG;A), glutamateplusmalate(GM;B),succinate(Succ;C),or␣-glycerophosphate(20mM)(␣-GP;D).IntheinsetofB,originaltraces andnumbersthatrepresenttheratesofH2O2production(inpmol/min/mgprotein)aredisplayed.Carboxyatractylate(CAT) wasaddedasindicatedtothemitochondriaincubatedwithGM,ADP,TEGDMA(graycurve),orDMSO(blackcurve)(InsetB).
ForA–D,theratesofH2O2productionwerealsodemonstratedonabarchart(mean±SEM;whitebars:TEGDMAwaspresent, greybars:noTEGDMAwaspresent).Statisticalanalysiswasappliedasabove(seeFig.1).
Fig.4–EffectsofpreincubationofmitochondriawithTEGDMAonH2O2production(A),andH2O2elimination(B).
Mitochondriawerepreincubatedfor10minwithComplexIsubstrateglutamateplusmalate(5–5mM)intheincubation mediumintheabsence(greybars)orpresenceofTEGDMA(whitebars).Afterthis10min,horseradishperoxidaseand AmplexUltrawereadded(asdescribedinSection2)andH2O2productionwasmeasured.Dataareexpressedas mean±SEM(n≥7)(A).RemovalofexogenouslyaddedH2O2byglutamateplusmalatesupportedmitochondriainthe presenceorabsenceofTEGDMA(5mM).Mitochondria(0.1mg/ml)wereincubatedfor10min,thenchallengedwithH2O2
(10M).TheresidualH2O2wasmeasuredfromaliquotstakenafterdifferenttimeintervals,asdescribedinSection2.Data areexpressedasmean±SEM(n=9)(B).Significantresults(p<0.05)areindicatedbyasterisks(AandB).
3.3.4. TheinteractionofTEGDMAwithGSH
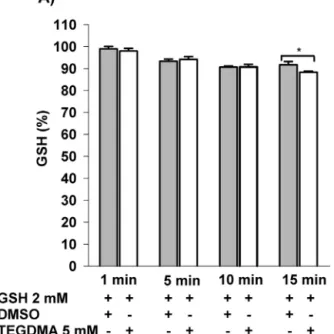
Similarlyto the experiments described elsewhere [52], the reactionofGSHwithTEGDMA(5mM)wasinvestigatedina mitochondrion-freereactionmedium.At15minofincubation timea3.7%decreaseinGSHconcentrationwasdetectedinthe presenceofTEGDMA(5mM)(Fig.5).
3.3.5. H2O2formationinthecoursesofoxidationof non-ComplexIsubstrates
3.3.5.1. H2O2 generation in mitochondria supported by suc- cinate. TEGDMA or DMSO were applied to mitochondria energized with succinate. TEGDMA (5mM) decreased the rate of H2O2 production by 42% (from 3454±245 to 1988±163pmol/min/mg protein). Significant decrease of H2O2 formation has been observed already at 0.5mM of TEGDMA. Addition of ADP further decreased H2O2 pro- duction to 423±78pmol/min/mg protein in the presence of TEGDMA (5mM) and to 301±36pmol/min/mg protein in DMSO-treated mitochondria(Fig. 3C). Inhibition of ANT by CAT stimulated again the ROS production. H2O2 gen- eration in TEGDMA-treated group (1241±227pmol/min/mg protein) was significantlylower than that ofcontrolgroup (6348±400pmol/min/mg protein) (Fig. 3C) owing to the CI inhibition.
3.3.5.2. H2O2formationinmitochondriasupportedby˛-GP.In brainmitochondriaalpha-glycerophosphate dehydrogenase
(␣-GPDH)ishighlyactive[53].Thisenzyme(suchassuccinate dehydrogenase)providestherespiratorychainwithelectrons and canalsogenerateRET.In␣-GP-energizedmitochondria TEGDMA mediated a 47% suppression ofH2O2 production (from 491±30 to 259±20pmol/min/mg protein) (Fig. 3D).
AdditionofADPloweredtherateofH2O2generationinboth groups.InthepresenceofADPtherewasnoobservablediffer- encebetweenthetwotreatments(DMSOorTEGDMA).Under controlconditionsCATrestoredtherateofH2O2production to the level detected in the ADP-free state (from 117±13 to509±29pmol/min/mgprotein).InTEGDMA-treatedmito- chondriatherewasalsoaCAT-mediatedstimulationofH2O2
generation (from111±16to185±14pmol/min/mgprotein), but theeffectwasmuchsmallerthan theonemeasuredin solvent-treatedmitochondria(Fig.3D).
3.4. EffectsofTEGDMAontheNAD(P)Hlevel
TheapplicationofbothNADH-linked(␣-KG,GM)andFADH2- linkedsubstrates(Succand␣-GP)elevatedthemitochondrial NAD(P)Hlevel(Fig.6).TheelevationoftheNAD(P)Hlevelby Succ and/or␣-GP canlikelybeascribed totheRETmecha- nism[54–56].TheADP-inducedstimulationofrespirationwas accompaniedbyadecreasedsteady-statelevelofNAD(P)H.
InTEGDMA-treated mitochondriatheNAD(P)Hsteady-state levelwashigherbothwitheitherNADHorFADH2-linkedsub- strates(Fig.6tracesb).Inmitochondriarespiringon␣-KGthe
Fig.5–EffectofincubationwithTEGDMAontheGSHlevel inmitochondrion-freemedium.GSH(2mM)wasincubated withTEGDMA(5mM)inthestandardincubationmedium andatthetimeintervalsindicatedaliquotsweretakenand thereducedglutathioneconcentrationwasdeterminedby 5,5-dithiobis(2-nitrobenzoicacid)(DTNB),
spectrophotometrically(ε412=13600M−1cm−1).Resultsare expressedaspercentageoforiginalGSHconcentration;
mean±SEM(n≥7).Significance(p<0.05)isindicatedby asterisk.
TEGDMA-inducedelevationoftheNAD(P)Hlevelwasslowand gradual(Fig.6A),butinGM-supportedmitochondriaTEGDMA inducedafastelevationoftheNAD(P)Hlevel(Fig.6B).Inthe presenceofCATtherewasnodifferenceintheNAD(P)Hsteady state between control and TEGDMA challenged mitochon- dria(Fig.6).However,inTEGDMA-treatedsuccinate-energized mitochondriaADPdidnotdecreasetheNADP(H)levelatall, contrary to TEGDMA-treated ␣-GP-supported mitochondria whereamoderatedecreaseoftheNAD(P)Hlevelwasobserved (Fig.6C,Dtracesb).
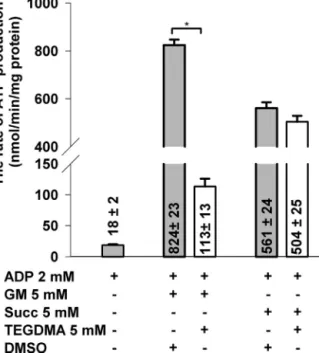
3.5. EffectofTEGDMAonATPsynthesis
For a bettercharacterization ofthe relevant mitochondrial bioenergetics,inadditiontooxygenconsumptionand m
therate ofATPproductionwasalsoassessed(Fig.7).After administeringADP,intheabsenceofrespiratorysubstrates, a compromised rate of the adenylate kinase (AK) coupled ATP synthesis (18±2nmol/min/mg protein) could still be detectedas200MAP5wasapparentlynotsufficienttofully inhibit the activity of AK. OXPHOS was initiated with the addition of respiratory substrates in the presence of ADP.
InGM-supportedTEGDMA-treatedmitochondriatherateof ATPproductionwasloweredby86%(decreasedfrom824±23 to113±13nmol/min/mgprotein)ascomparedwithcontrols.
However,additionofTEGDMA(5mM)tosuccinate-energized
mitochondriaresultedinadropof10.2%onlyintherateofATP synthesis(from561±24to504±25nmol/min/mgprotein).
3.6. TheeffectofTEGDMAonComplexIactivity
InordertoproveourhypothesisthatTEGDMAwasaninhibitor ofCI,theNADH-CoQreductaseactivitywasdirectlymeasured.
TheCIactivitywasdecreasedby70.6±3.3and61.8±3.2%in thepresenceof5and2mMTEGDMA,respectively(datanot shown).
4. Discussion
IthasbeenshownthatTEGDMAreleaseduetoincomplete polymerizationandbiodegradationinoralenvironmentcan impair various cellular functions, provoke oxidative stress (for review see Ref.[18]), apoptotic and necrotic cell death [28,30,57,58] and trigger genetic mutations [8,59]. Selected resin monomerswere demonstrated todeplete the poolof glutathione(GSH),anintracellularantioxidant[12].Inhibition ofcell proliferation, stimulationofapoptosis and impaired differentiationofosteoclasts[60]canbeallmediatedbythe overproductionofROS.Thebeneficialeffectsofantioxidants e.g. N-acetylcysteine (NAC), Trolox (vitamin E), or ascorbic acid (vitamin C) also suggest that the cytotoxic effects of TEGDMAwouldbetheconsequencesofincreasedROSgen- eration[20,57,61–65].Thecellularmechanismthatliesbehind theincreasedrateofROSproductionhasnotbeenelucidated yet[18].TheinvolvementofthemitochondrioninTEGDMA toxicityhasalreadybeenproposed[32].Arecentstudyempha- sizedtheimportanceoftheTEGDMA-mediatedactivationof NOX4intheROSgenerationandapoptosis[31].Sincemostof theATP,requiredforvariouscellularfunctions,isproduced bymitochondria,thisorganelleplaysapivotalroleincellular viabilityanddeath.Themitochondrionisalsoaprimarysite forROSproductionandelimination(forreview,seeRef.[38]).
Therefore,weinvestigatedtheresponsesofvariousmitochon- drialfunctionstoTEGDMA.
4.1. EffectsofTEGDMAonmitochondrialrespiration
Mitochondrialrespirationisasensitiveindicatorofmitochon- drialfunction[66].Inmitochondriawhichweresupportedby either␣-KGorGMTEGDMA(5mM)ledtoadecreaseinoxygen consumption(Fig.1A–C),whereasinFADH2-linked,succinate- supportedmitochondriatherateofoxygenconsumptionwas not affected bythis treatment(Fig. 1D). Our resultsare in contrast withanearlierobservation[67]wherethe respira- tion ofisolatedsuccinate-rotenone-supportedmitochondria was inhibitedbyTEGDMA;thiscontradictionmightbedue todifferent experimentalsetups.Inthe abovepaperHL-60 cellswerepreincubatedwithTEGDMAfor1h,andmitochon- driawereisolatedandinvestigatedthereafter.Inthecourse oftheincubationperiodmitochondriamighthaveundergone suchastructuraldamagesthatcouldpotentiallyalsoimpair thesuccinate-dependentrespiration.Inthepresentstudy,the reduced rates ofmitochondrial respiration observedin the presenceofCIsubstratescanpotentiallybeexplainedby:(i)a decreasedATPaseactivity [68–70], (ii)theinhibitionofANT
Fig.6–EffectsofTEGDMAonthesteady-statelevelofNAD(P)Hinthebrainmitochondriasupportedby␣-ketoglutarate(A), glutamateplusmalate(B),succinate(C),or␣-glycerophosphate(D).Mitochondria(0.1mg/mL)wereincubatedwithvarious respiratorysubstrates.TEGDMAorDMSO,ADP(2mM),andcarboxyatractylate(CAT,2M)wereaddedasindicated.
Representativeoriginaltracesofexperimentsaredisplayed(grey,traceb:TEGDMAwaspresent;black,tracea:noTEGDMA waspresent).
[71],(iii)areduced rateoftransportformetabolitesand/or inorganicphosphate[72],or(iv)thecompromisedactivities ofCIand/orselecteddehydrogenaseenzymes[73,74].How- ever,thenormalrateofrespirationwhichwasdetectedinthe presenceofsuccinateexcludedthereasonslistedunderi,ii, andpartiallyiii.Inordertoelucidatethemechanismthatlies behindthedepressionofrespirationtheeffectsofTEGDMA onmitochondrial mwereassessed.
4.2. EffectsofTEGDMAonmitochondrial m
Maintenance of the mitochondrial membrane potential is crucialfornormalmitochondrialphysiology.Theprotongra- dientisusedbymitochondriatofacilitatetransportprocesses for ions [75], metabolites [76] and proteins[77] across the mitochondrial inner membrane.Previously, m was mea- suredingingivalfibroblastsandmitochondrialdepolarization
Fig.7–EffectsofTEGDMAontherateofATPsynthesisin brainmitochondriarespiringoneitherglutamateplus malateorsuccinate.TEGDMA(whitebars)orDMSO(grey bars)wereappliedbesidesADPthroughoutallthe experiments.TherateofATPsynthesis,writtenonthe bars,isexpressedinnmol/min/mgprotein(mean±SEM) n>11.Significantdifferencesareindicatedby*p<0.05.
was detected[32]. Incellular systems, however, mitochon- dria are energized simultaneously withvarious respiratory substrates and hence it is difficult to determine the true target of a compound. In the present study, m mea- surementsalsoconfirmed thatTEGDMA inhibits theCI.In mitochondriasupportedwithNADH-linkedsubstrates,addi- tion of TEGDMA decreased m (Fig. 2A,B). Contrary to that, in succinate-energized mitochondria m was unaf- fectedbyTEGDMA(Fig.2C).Thesefindingsagainprovethat TEGDMA does not inhibit the ANT or the ATP-ase; when using selective inhibitors of these molecules m would have been hyperpolarized, independently of the respira- torysubstratesapplied[78].Theresultsareconsistentwith the proposed inhibitory effect ofTEGDMA on the CI func- tion.
4.3. EffectsofTEGDMAonROShomeostasis
As we discussed earlier, there is a general agreement that exposition of cells to TEGDMA provokes oxidative stress.However,thesourcesand mechanismsoftheredox imbalance have not been elucidated, yet. Very recently it was suggested that the activation of NADPH oxidase 4 has a key role in the TEGDMA-associated ROS generation [31].
Ourhypothesisthatmitochondrialtargetsarealsoinvolved inthecellulartoxicityofTEGDMAwouldbesubstantiatedif administrationofTEGDMAcouldstimulatethemitochondrial ROSproduction.
4.3.1. H2O2formationwithrespiratorysubstrateslinked toComplexI
Theadditionofrespiratorysubstratestomitochondriainiti- atedmitochondrial H2O2generation (Fig.3).Both␣-KGand GMstimulatedtheH2O2 production,buttodifferentextent (Fig.3A,B).ThehigherrateofH2O2productionwithGMcanbe ascribedtothehigherrateofGM-inducedrespiration(Fig.1C) andtotheconsequentlyhigher m(Fig.2B)[51].Thereisa directcorrelationbetweenthemembranepotentialandH2O2 production(Figs.2A,Band3A,B)andStarkovetal.[51].Addi- tionofADPdifferentlyinhibitedH2O2formationon␣-KGand GM respiring mitochondria (Fig. 3A,B). Thehigher was the membranepotentialbeforetheADPaddition,thegreaterwas thedecreaseofH2O2production(Fig.3A,B).Takingintoconsid- erationthatthe␣-KGDHistherate-limiting(slowest)enzyme ofthecitricacidcycleinbrainmitochondria,itisobviousthat therespirationrate,aswellastherateofH2O2production,is slowerwith␣-KGthanwithGM.WhenitwasgivenafterADP, TEGDMAsignificantlyaugmentedtherateofH2O2production andthiswasvalidunderbothexperimentalconditions(with
␣-KG:18%increase,withGM:74% increase,relativetocon- trols;seeFig.3A,B).ElevatedratesofH2O2productionwere observedinthepresenceofCIinhibitors;CIpossessessev- eralsitesthatare capableofproducingROS(forreviewsee Ref.[79]).Intheelectrontransportchain,thoseelectroncarrier complexesthatareupstreamtothebindingsiteofarespira- torychaininhibitorareinhighlyreducedstates[40].Reduction ofaROS-formingsiteincreasesthelikelihoodforanelectron leakage.InhibitionofCIinCIsubstrate(glutamateplusmalate) supportedmitochondriastimulatedH2O2formationineither thepresenceorabsenceofADP.EnhancedH2O2production thereforecandepleteendogenousantioxidantssuchasglu- tathioneandinhibitglutathione-relatedantioxidantsystems [80].
Comparing the resultsdepicted inFigs. 3Band 4A,it is apparent that as a consequence of the Amplex plus HRP- freepreincubationthestimulatoryeffectofTEGDMAonH2O2
productionwasfurtherenhancedineitherthepresenceor absence ofADP.Weinterprettheseresultsthatcontinuous monitoringofmitochondrial H2O2 productionwithAmplex plusHRPnotonlydetects,butalsodetoxifiesthegenerated H2O2[44]mitigatingtheoxidativedamage.
ConsideringthatmitochondriaplayrolesinbothROSfor- mation and ROSelimination,theeffect ofTEGDMA on the elimination ofexogenous H2O2 was also investigated. Our resultsindicate(Fig.4B)thatinthepresenceofTEGDMAthe elimination was indeedslower, therefore theseresults fur- ther confirmthe hypothesis drawnfrom the preincubation experimentthatTEGDMAcanimpairtheantioxidantsystem.
This phenomenon can also beattributed to the inhibition ofComplexI,whichresultsinadecrease inthe mitochon- drial membrane potential. m is a driving force for the energy-dependenttranshydrogenaseenzymethatcantrans- ferelectronsfromNADHtoNADP+[81],thuscanmaintainthe NADPHlevel,whichisnecessaryfortheregenerationofGSH.
However,itisstillobscurewhetherTEGDMAcandirectly interferewiththeantioxidantsystem.Intheexcellentpaper ofLefeuvreet al. [52]the directeffectofTEGDMA on GSH has been investigatedand it was foundthat incubationof
TEGDMA(3mM)withGSHdecreasedtheGSHconcentration intheabsenceofcells.After1and3hofincubationsignifi- cantdecreaseof[GSH]wasdetected.Wereinvestigatedthis questionandusing5mMTEGDMAsignificantchangeswere foundafter15minofincubation,3.7%decreaseof[GSH]was detected(Fig.5).AccordingtoLefeuvreetal.incubationgin- givalfibroblastswithlowconcentrationofTEGDMA(0.3mM) generatedahighrateofGSHdisappearance,at30minGSH leveldecreasedbymorethan50%.Comparisonofinvitroand cellularexperimentssuggestthatthedecreaseofGSHlevel isnotaconsequenceofdirectinteractionwithTEGDMA.In Lefeuvre’spaperthepossibleroleofglutathione-S-transferase wasput forward. Theresults ofthe present papersuggest anadditionaltarget– mitochondria– forTEGDMA’saction.
ThesuggestedmechanismsforTEGMA’scytotoxicitydonot excludeeachother.
4.3.2. H2O2productionwithFADH2-linkedsubstrates (Succand˛-GP)andtheeffectofinhibitingthereverse electrontransport
FromFADH2 linkedrespiratory substratesmostoftheelec- tronsaretransportedtooxygenasthefinalelectronacceptor viatheComplexesIIIandIV.Itiswelldocumentedhowever, thatwithsuccinateintheabsenceofADPahigh misgen- eratedthatdrivesacertainfluxofelectronsfromcoenzyme Q(CoQ)toflowtowardstheCI,inadirectionthatisinverse tonormal. This phenomenon is referredto as the reverse electrontransport(RET)mechanism[82,83]thatresultsinthe conversionofNAD+toNADHattheCI.RETisthepredominant ROS-generatorwhennoADPispresentandthemitochondria are supportedbyeithersuccinateor ␣-GP [40,53]. TEGDMA loweredtherateofthemitochondrialH2O2productionby42%
(forSucc)or47%(for␣-GP)(Fig.3C,D).Thisseeminglyparadox- icalphenomenoncanbeattributedtoacontributionbythe so-calledreverseelectrontransport(RET)mechanism,aback- wardflowofelectronsfromthereducedCoQH2towardsthe CI(andNAD+)[50,84–88].CAThyperpolarizedmandthere- forerestoredtheRETintheDMSO-treatedcontrolgroup.In theTEGDMA-treatedmitochondria,however,H2O2production waslessefficientduetoinhibitionoftheCI(Fig.3C,D).
4.4. RelationshipbetweeninhibitionofComplexIand theNAD(P)HlevelinTEGDMA-treatedmitochondria
Complex I substrates (␣-KG, GM) led to NADH formation (Fig.6A,B),becausethesesubstratesare metabolizedbythe dehydrogenasestoyieldNADH.InthepresenceofADPmost oftheNADHbecameoxidized,however,additionofTEGDMA increased the steady state level of NADH/NAD (Fig. 6A,B).
ThisobservationiscrucialsinceitimpliesthatTEGDMAdoes inhibitthe CI, but does notcompromise the related dehy- drogenases.Withnon-ComplexIsubstrates(e.g.Succ,␣-GP) the formation of NADH can unambiguously be attributed to RET as the respective dehydrogenases do not produce NADH.AdditionofADPtoenergizedmitochondriadecreased the steady state level of NADH for all the tested respi- ratory substrates (Fig. 6A–D), because (i.) ADP stimulates the respiration, (ii.) a higherrate of the electronflow pro- motes NADHoxidation,and (iii.) anADP-induced decrease in the membrane potential abolishes the reverse electron
flow[89].TEGDMAinduceddifferentresponsesinmitochon- driadependingonthesubstrateusedforenergization.With CI substrates TEGDMA-mediatedinhibitionofthe Complex I withheldelectronsfrom reaching thedistal sequencesof respiratorychain;consequently,NAD+mostlygotreducedto NADH(Fig. 6A,B).In the presenceofsuccinate or ␣-GP the additionofTEGDMAprecededtheadministrationofADPto mitochondria(Fig. 6C,D).In succinate-supportedmitochon- driaadditionofADPwasunabletodropNADHconcentration tothecontrollevel,becauseCIwasinhibited,thustheaccu- mulatedNADHwasunabletobeoxidized(Fig.6C).Formore detailedexplanationseealsoRef.[44].
4.5. ATPproductioninTEGDMA-challenged mitochondria
Inordertominimizethecontributionoftheadenylatekinases (AKs)tothemitochondrialATPgenerationtheAKinhibitor2- amino-phoshponophentanoate(AP5)[90]wasappliedinthe experimentsthatassessedATPproduction.Inthepresenceof ADPandtheabsenceofanyrespiratorysubstratesonlytheAK- relatedATPsynthesiscouldbedetected;theAKactivitywas onlypartiallyinhibited.Additionoftherespiratorysubstrate (GMorSucc)ledtotheinitiationofOXPHOS.InGM-supported mitochondriaTEGDMAloweredtherateofATPsynthesisby 86%(Fig.7).ThisobservationsuggeststhatTEGDMA,which wasinhibitingtheCI,causeddepolarizationandtheremain- ingprotongradientwasnotsufficienttosupportahighrateof OXPHOS;compromisedATPsynthesisleadstocellularbioen- ergeticinsufficiencyandeventuallynecroticcelldeath.
4.6. ComplexIactivityinTEGDMA-treated mitochondria
ThedirectmeasurementofCIactivityprovedthatTEGDMA canindeedinhibittheCI.TheimmediateeffectofTEGDMA suggeststhatCIcaninfactbeaprimarytargetofthisresin monomer. In rotenone-treated(CI inhibitor) cells oxidative stress,glutathionedepletion,andDNAdamageweredetected withasubsequentlyincreasedprevalenceofapoptosis [80].
It hasbeen described that various inhibitors ofthe CI are abletostimulatethemitochondrialROSproductionandcause MAPK/NF-Bactivation [91]. Furthermore, the extent of CI inhibitioncandeterminethetypeofcelldeathinduced.Low dosesofCIinhibitorsgenerallyinduceapoptosis,whilehigher dosesofthesamecompoundmayevokenecroticcelldeath [37].
Our studyfocused onthe acuteeffects ofTEGDMA and theresultsdonotcontradicttheobservationsmadebyYeh et al. [31]. The development of TEGDMA-induced changes whichwereattributedtoNOX4activationrequiredhoursand changesinthegeneexpressionofNOX4wereobserved[31].
Recently,itwasreportedthatinhibitionoftheCIbyrotenone canstimulateNOX2expression[92].Thus,weproposethatthe CIisanimmediatetargetofTEGDMAandthattheactivation ofvarioussignaltransductionmechanismsmayberequired toactivatetheNOXisoenzymes.
The effects and consequences of inhibitory action of TEGDMAonrespiratoryComplexIaresummarizedonFig.8.
Fig.8–SchematicsummaryofTEGDMA-inducedmitochondrialdamage.TheprimarytargetofTEGDMAisidentifiedasthe CI.TheeffectsofCIinhibitionaredecreasedO2consumption,stimulationofH2O2production,impairmentofmitochondrial H2O2eliminationwithaconsequentoxidativestress,andapoptosis.ThedecreaseofATPsynthesisathighTEGDMA concentrationscancreatenecrotictypeofcelldeath.Forsimplicitythereverseelectronflow(seeSection4.3.2)isnot indicatedonthefigure.Abbreviations:IM:innermitochondrialmembrane;IMS:intermembranespace;OM:outer membrane;CI,II,III,IV:respiratorycomplexes, m:mitochondrialmembranepotential.
5. Conclusions
WeidentifiedtheCIofthemitochondrialelectrontransport chain as a target of TEGDMA resin monomer. Addition of TEGDMAresultsininhibitionoftheCI-dependentoxygencon- sumption, depolarizationof mitochondrial membrane, and consequentlydecreaseinATPproductionviaOXPHOS,there- foreTEGDMAcausesbioenergeticinsufficiency.Inhibitionof CIbyTEGDMA enhancesthemitochondrial H2O2(ROS) for- mationandimpairsROSelimination.Thedirectinteraction betweenTEGDMAandreducedglutathioneisunlikelytobea majorcomponentinthetoxicity.
Acknowledgments
Thisworkwas supportedbytheHungarian BrainResearch Program (KTIA13NAP-A-III/6 and 2017-1.2.1-NKP-2017- 00002), OTKA (NK 112230), and the Hungarian Academy of Sciences (MTA TKI 02001), all to Vera Adam-Vizi. We thankKatalinTakacsandAndreaVarnagyfortheirexcellent technicalassistance.
references
[1] FerracaneJL.Resincomposite—stateoftheart.DentMater 2011;27:29–38.
[2] SchmalzG,GallerKM.Biocompatibilityofbiomaterials– lessonslearnedandconsiderationsforthedesignofnovel materials.DentMater2017;33:382–93.
[3] GeurtsenW.Substancesreleasedfromdentalresin compositesandglassionomercements.EurJOralSci 1998;106:687–95.
[4] SpahlW,BudzikiewiczH,GeurtsenW.Determinationof leachablecomponentsfromfourcommercialdental compositesbygasandliquidchromatography/mass spectrometry.JDent1998;26:137–45.
[5] VanLanduytKL,SnauwaertJ,DeMunckJ,PeumansM, YoshidaY,PoitevinA,etal.Systematicreviewofthe chemicalcompositionofcontemporarydentaladhesives.
Biomaterials2007;28:3757–85.
[6] HumeWR,GerzinaTM.Bioavailabilityofcomponentsof resin-basedmaterialswhichareappliedtoteeth.CritRev OralBiolMed1996;7:172–9.
[7] SchmalzG.Thebiocompatibilityofnon-amalgamdental fillingmaterials.EurJOralSci1998;106:696–706.
[8] GeurtsenW,LeyhausenG.Chemical–biologicalinteractions oftheresinmonomertriethyleneglycol-dimethacrylate (TEGDMA).JDentRes2001;80:2046–50.
[9] SeissM,LangerC,HickelR,ReichlFX.Quantitative determinationofTEGDMA,BHT,andDMABEEineluates frompolymerizedresin-baseddentalrestorativematerials byuseofGC/MS.ArchToxicol2009;83:1109–15.
[10] VanLanduytKL,NawrotT,GeebelenB,DeMJ,SnauwaertJ, YoshiharaK,etal.Howmuchdoresin-baseddental materialsrelease?Ameta-analyticalapproach.DentMater 2011;27:723–47.
[11] BouillaguetS,WatahaJC,HanksCT,CiucchiB,HolzJ.Invitro cytotoxicityanddentinpermeabilityofHEMA.JEndod 1996;22:244–8.
[12] SchweiklH,SpagnuoloG,SchmalzG.Geneticandcellular toxicologyofdentalresinmonomers.JDentRes
2006;85:870–7.
[13] SanterreJP,ShajiiL,TsangH.Biodegradationofcommercial dentalcompositesbycholesterolesterase.JDentRes 1999;78:1459–68.
[14] HanksCT,StrawnSE,WatahaJC,CraigRG.Cytotoxiceffects ofresincomponentsonculturedmammalianfibroblasts.J DentRes1991;70:1450–5.
[15] NodaM,WatahaJC,KagaM,LockwoodPE,VolkmannKR, SanoH.Componentsofdentinaladhesivesmodulateheat shockprotein72expressioninheat-stressedTHP-1human monocytesatsublethalconcentrations.JDentRes 2002;81:265–9.
[16] AboutI,CampsJ,MitsiadisTA,BotteroMJ,ButlerW, FranquinJC.Influenceofresinousmonomersonthe differentiationinvitroofhumanpulpcellsinto odontoblasts.JBiomedMaterRes2002;63:418–23.
[17] GallerKM,SchweiklH,HillerKA,CavenderAC,BolayC, D’SouzaRN,etal.TEGDMAreducesmineralizationindental pulpcells.JDentRes2011;90:257–62.
[18] KrifkaS,SpagnuoloG,SchmalzG,SchweiklH.Areviewof adaptivemechanismsincellresponsestowardsoxidative stresscausedbydentalresinmonomers.Biomaterials 2013;34:4555–63.
[19] EngelmannJ,VolkJ,LeyhausenG,GeurtsenW.ROS formationandglutathionelevelsinhumanoralfibroblasts exposedtoTEGDMAandcamphorquinone.JBiomedMater ResBApplBiomater2005;75:272–6.
[20] StanislawskiL,LefeuvreM,BourdK,Soheili-MajdE, GoldbergM,PerianinA.TEGDMA-inducedtoxicityinhuman fibroblastsisassociatedwithearlyanddrasticglutathione depletionwithsubsequentproductionofoxygenreactive species.JBiomedMaterResA2003;66:476–82.
[21] EckhardtA,GerstmayrN,HillerKA,BolayC,WahaC, SpagnuoloG,etal.TEGDMA-inducedoxidativeDNAdamage andactivationofATMandMAPkinases.Biomaterials 2009;30:2006–14.
[22] BlasiakJ,SynowiecE,TarnawskaJ,CzarnyP,PoplawskiT, ReiterRJ.Dentalmethacrylatesmayexertgenotoxiceffects viatheoxidativeinductionofDNAdoublestrandbreaksand theinhibitionoftheirrepair.MolBiolRep2012;39:7487–96.
[23] ChangHH,GuoMK,KastenFH,ChangMC,HuangGF,Wang YL,etal.Stimulationofglutathionedepletion,ROS productionandcellcyclearrestofdentalpulpcellsand gingivalepithelialcellsbyHEMA.Biomaterials 2005;26:745–53.
[24] EckhardtA,MullerP,HillerKA,KrifkaS,BolayC,Spagnuolo G,etal.InfluenceofTEGDMAonthemammaliancellcycle incomparisonwithchemotherapeuticagents.DentMater 2010;26:232–41.
[25] SchweiklH,HartmannA,HillerKA,SpagnuoloG,BolayC, BrockhoffG,etal.InhibitionofTEGDMAandHEMA-induced genotoxicityandcellcyclearrestbyN-acetylcysteine.Dent Mater2007;23:688–95.
[26] ReichlFX,EstersM,SimonS,SeissM,KeheK,KleinsasserN, etal.Celldeatheffectsofresin-baseddentalmaterial compoundsandmercurialsinhumangingivalfibroblasts.
ArchToxicol2006;80:370–7.
[27] SpagnuoloG,GallerK,SchmalzG,CosentinoC,RengoS, SchweiklH.Inhibitionofphosphatidylinositol3-kinase amplifiesTEGDMA-inducedapoptosisinprimaryhuman pulpcells.JDentRes2004;83:703–7.
[28] ChangHH,ChangMC,HuangGF,WangYL,ChanCP,Wang TM,etal.Effectoftriethyleneglycoldimethacrylateonthe cytotoxicity,cyclooxygenase-2expressionandprostanoids productioninhumandentalpulpcells.IntEndodJ 2012;45:848–58.
[29] HarorliOT,BayindirYZ,AltunkaynakZ,TatarA.Cytotoxic effectsofTEGDMAonTHP-1cellsinvitro.MedOralPatol OralCirBucal2009;14:e489–93.
[30] HuangFM,KuanYH,LeeSS,ChangYC.Cytotoxicityand genotoxicityoftriethyleneglycol-dimethacrylatein
macrophagesinvolvedinDNAdamageandcaspases activation.EnvironToxicol2015;30:581–8.
[31] YehCC,ChangJZ,YangWH,ChangHH,LaiEH,KuoMY.
NADPHoxidase4isinvolvedinthetriethyleneglycol dimethacrylate-inducedreactiveoxygenspeciesand apoptosisinhumanembryonicpalatalmesenchymaland dentalpulpcells.ClinOralInvestig2015;19:1463–71.
[32] LefeuvreM,AmjaadW,GoldbergM,StanislawskiL.TEGDMA inducesmitochondrialdamageandoxidativestressin humangingivalfibroblasts.Biomaterials2005;26:5130–7.
[33] OttM,GogvadzeV,OrreniusS,ZhivotovskyB.Mitochondria, oxidativestressandcelldeath.Apoptosis2007;12:913–22.
[34] KimJS,HeL,LemastersJJ.Mitochondrialpermeability transition:acommonpathwaytonecrosisandapoptosis.
BiochemBiophysResCommun2003;304:463–70.
[35] RasolaA,BernardiP.Mitochondrialpermeabilitytransition inCa(2+)-dependentapoptosisandnecrosis.CellCalcium 2011;50:222–33.
[36] ParsonsMJ,GreenDR.Mitochondriaincelldeath.Essays Biochem2010;47:99–114.
[37] HartleyA,StoneJM,HeronC,CooperJM,SchapiraAH.
ComplexIinhibitorsinducedose-dependentapoptosisin PC12cells:relevancetoParkinson’sdisease.JNeurochem 1994;63:1987–90.
[38] AndreyevAY,KushnarevaYE,MurphyAN,StarkovAA.
MitochondrialROSmetabolism:10yearslater.Biochemistry (Mosc)2015;80:517–31.
[39] Adam-ViziV,TretterL.Theroleofmitochondrial dehydrogenasesinthegenerationofoxidativestress.
NeurochemInt2013;62:757–63.
[40] AndreyevAY,KushnarevaYE,StarkovAA.Mitochondrial metabolismofreactiveoxygenspecies.Biochemistry(Mosc) 2005;70:200–14.
[41] TretterL,Adam-ViziV.ModeratedependenceofROS formationonDeltaPsiminisolatedbrainmitochondria supportedbyNADH-linkedsubstrates.NeurochemRes 2007;32:569–75.
[42] PestaD,GnaigerE.High-resolutionrespirometry:OXPHOS protocolsforhumancellsandpermeabilizedfibersfrom smallbiopsiesofhumanmuscle.MethodsMolBiol 2012;810:25–58.
[43] KomaryZ,TretterL,Adam-ViziV.H2O2generationis decreasedbycalciuminisolatedbrainmitochondria.
BiochimBiophysActa2008;1777:800–7.
[44] TretterL,AmbrusA.MeasurementofROShomeostasisin isolatedmitochondria.MethodsEnzymol2014;547:199–223.
[45] TretterL,BiagioniAE,ArdestaniMR,GoracciG,Adam-ViziV.
Reversibleinhibitionofhydrogenperoxideeliminationby calciuminbrainmitochondria.JNeurosciRes
2011;89:1965–72.
[46] RahmanI,KodeA,BiswasSK.Assayforquantitative determinationofglutathioneandglutathionedisulfide levelsusingenzymaticrecyclingmethod.NatProtoc 2006;1:3159–65.
[47] TretterL,HorvathG,HolgyesiA,EssekF,Adam-ViziV.
Enhancedhydrogenperoxidegenerationaccompaniesthe beneficialbioenergeticeffectsofmethyleneblueinisolated brainmitochondria.FreeRadicBiolMed2014;77:317–30.
[48] RaganCI,HatefiY.Isolationoftheiron-sulfur-containing polypeptidesofNADH:oxidoreductaseubiquinone.Methods Enzymol1986;126:360–9.
[49] KlingenbergM,FalknerG,ErdeltH,GrebeK.Ontherelation betweenadeninenucleotidecarriersitesandatractyloside bindinginmitochondria.FEBSLett1971;16:296–300.
[50] Adam-ViziV.Productionofreactiveoxygenspeciesinbrain mitochondria:contributionbyelectrontransportchainand non-electrontransportchainsources.AntioxidRedoxSignal 2005;7:1140–9.
[51] StarkovAA,PolsterBM,FiskumG.Regulationofhydrogen peroxideproductionbybrainmitochondriabycalciumand Bax.JNeurochem2002;83:220–8.
[52] LefeuvreM,BourdK,LoriotMA,GoldbergM,BeauneP, PerianinA,etal.TEGDMAmodulatesglutathione transferaseP1activityingingivalfibroblasts.JDentRes 2004;83:914–9.
[53] TretterL,Adam-ViziV.HighCa2+loadpromoteshydrogen peroxidegenerationviaactivationof
alpha-glycerophosphatedehydrogenaseinbrain mitochondria.FreeRadicBiolMed2012;53:2119–30.
[54] ChanceB,HollungerG.Theinteractionofenergyand electrontransferreactionsinmitochondria.I.General propertiesandnatureoftheproductsofsuccinate-linked reductionofpyridinenucleotide.JBiolChem
1961;236:1534–43.
[55] GyulkhandanyanAV,PennefatherPS.Shiftinthe localizationofsitesofhydrogenperoxideproductionin brainmitochondriabymitochondrialstress.JNeurochem 2004;90:405–21.
[56] TretterL,TakacsK,KoverK,Adam-ViziV.Stimulationof H(2)O(2)generationbycalciuminbrainmitochondria respiringonalpha-glycerophosphate.JNeurosciRes 2007;85:3471–9.
[57] KrifkaS,PetzelC,HillerKA,FrankEM,BoslC,SpagnuoloG, etal.Resinmonomer-induceddifferentialactivationofMAP kinasesandapoptosisinmousemacrophagesandhuman pulpcells.Biomaterials2010;31:2964–75.
[58] NodaM,WatahaJC,LockwoodPE,VolkmannKR,KagaM, SanoH.Sublethal,2-weekexposuresofdentalmaterial componentsalterTNF-alphasecretionofTHP-1monocytes.
DentMater2003;19:101–5.
[59] SchweiklH,SchmalzG,RackebrandtK.Themutagenic activityofunpolymerizedresinmonomersinSalmonella typhimuriumandV79cells.MutatRes1998;415:119–30.
[60] InamitsuH,OkamotoK,SakaiE,NishishitaK,MurataH, TsukubaT.ThedentalresinmonomersHEMAandTEGDMA haveinhibitoryeffectsonosteoclastdifferentiationwithlow cytotoxicity.JApplToxicol2017;37:817–24.
[61] BakopoulouA,LeyhausenG,VolkJ,TsiftsoglouA,GarefisP, KoidisP,etal.EffectsofHEMAandTEDGMAontheinvitro odontogenicdifferentiationpotentialofhumanpulp stem/progenitorcellsderivedfromdeciduousteeth.Dent Mater2011;27:608–17.
[62] KrifkaS,PetzelC,BolayC,HillerKA,SpagnuoloG,Schmalz G,etal.Activationofstress-regulatedtranscriptionfactors bytriethyleneglycoldimethacrylatemonomer.Biomaterials 2011;32:1787–95.
[63] SchweiklH,HillerKA,EckhardtA,BolayC,SpagnuoloG, StempflT,etal.Differentialgeneexpressioninvolvedin oxidativestressresponsecausedbytriethyleneglycol dimethacrylate.Biomaterials2008;29:1377–87.
[64] SpagnuoloG,DesiderioC,RivieccioV,AmatoM,RossettiDV, D’AntoV,etal.Invitrocellulardetoxificationoftriethylene glycoldimethacrylatebyadductformationwith
N-acetylcysteine.DentMater2013;29:e153–60.
[65] WaltherUI,SiagianII,WaltherSC,ReichlFX,HickelR.
AntioxidativevitaminsdecreasecytotoxicityofHEMAand TEGDMAinculturedcelllines.ArchOralBiol2004;49:125–31.
[66] BrandMD,NichollsDG.Assessingmitochondrial dysfunctionincells.BiochemJ2011;435:297–312.
[67] NoccaG,DePF,MinucciA,DeSP,MartoranaGE,CallaC, etal.Alterationsofenergymetabolismandglutathione levelsofHL-60cellsinducedbymethacrylatespresentin compositeresins.JDent2007;35:187–94.
[68] ChappellJB,GrevilleGD.Effectsofoligomycinonrespiration andswellingofisolatedlivermitochondria.Nature
1961;190:502–4.
[69] EstabrookRW.EffectofoligomycinonthearsenateandDNP stimulationofmitochondrialoxidations.BiochemBiophys ResCommun1961;4:89–91.
[70] KomaryZ,TretterL,Adam-ViziV.Membrane potential-relatedeffectofcalciumonreactiveoxygen speciesgenerationinisolatedbrainmitochondria.Biochim BiophysActa1797;2010:922–8.
[71] VignaisPV,DouceR,LauquinGJ,VignaisPM.Bindingof radioactivelylabeledcarboxyatractyloside,atractylosideand bongkrekicacidtotheADPtranslocatorofpotato
mitochondria.BiochimBiophysActa1976;440:688–96.
[72] JuaristiI,Garcia-MartinML,RodriguesTB,SatrusteguiJ, Llorente-FolchI,PardoB.ARALAR/AGC1deficiency,a neurodevelopmentaldisorderwithsevereimpairmentof neuronalmitochondrialrespiration,doesnotproducea primaryincreaseinbrainlactate.JNeurochem 2017;142:132–9.
[73] ObergKE.Theinhibitionoftherespirationofbrain mitochondriaofrotenone-poisonedfish.ExpCellRes 1964;36:407–10.
[74] ParkerJrWD,HaasR,StumpfDA,ParksJ,EgurenLA,Jackson C.Brainmitochondrialmetabolisminexperimental thiaminedeficiency.Neurology1984;34:1477–81.
[75] MitchellP.Chemiosmoticcouplinginoxidativeand photosyntheticphosphorylation.BiochimBiophysActa 2011;1807:1507–38.
[76] PalmieriF,PierriCL.Mitochondrialmetabolitetransport.
EssaysBiochem2010;47:37–52.
[77] NeupertW,HerrmannJM.Translocationofproteinsinto mitochondria.AnnuRevBiochem2007;76:723–49.
[78] KomlódiT,TretterL.Methylenebluestimulates
substrate-levelphosphorylationcatalysedbysuccinyl-CoA ligaseinthecitricacidcycle.Neuropharmacology 2017;123:287–98.
[79] BrandMD.Mitochondrialgenerationofsuperoxideand hydrogenperoxideasthesourceofmitochondrialredox signaling.FreeRadicBiolMed2016;100:14–31.
[80] ShererTB.AninvitromodelofParkinson’sdisease:linking mitochondrialimpairmenttoalteredalpha-synuclein metabolismandoxidativedamage.2002.
[81] AlbrachtSP,MeijerAJ,RydstromJ.Mammalian NADH:ubiquinoneoxidoreductase(ComplexI)and nicotinamidenucleotidetranshydrogenase(Nnt)together regulatethemitochondrialproductionof
H(2)O(2)—implicationsfortheirroleindisease,especially cancer.JBioenergBiomembr2011;43:541–64.
[82] AzzoneGF,ErnsterL,WeinbachEC.Succinate-linked acetoacetatereduction.I.Endergonicreductionof
acetoacetatebysuccinateinlivermitochondria.JBiolChem 1963;238:1825–33.
[83] HinklePC,ButowRA,RackerE,CHANCEB.Partialresolution oftheenzymescatalyzingoxidativephosphorylation.XV.
Reverseelectrontransferintheflavin-cytochromebeta regionoftherespiratorychainofbeefheart
submitochondrialparticles.JBiolChem1967;242:5169–73.
[84] CinoM,DelMaestroRF.Generationofhydrogenperoxideby brainmitochondria:theeffectofreoxygenationfollowing postdecapitativeischemia.ArchBiochemBiophys 1989;269:623–38.
[85] HansfordRG,HogueBA,MildazieneV.DependenceofH2O2
formationbyratheartmitochondriaonsubstrateavailability anddonorage.JBioenergBiomembr1997;29:89–95.
[86] KwongLK,SohalRS.Substrateandsitespecificityof hydrogenperoxidegenerationinmousemitochondria.Arch BiochemBiophys1998;350:118–26.
[87] ShabalinaIG,NedergaardJ.Mitochondrial(‘mild’)
uncouplingandROSproduction:physiologicallyrelevantor not?BiochemSocTrans2011;39:1305–9.
[88] VotyakovaTV,ReynoldsIJ.DeltaPsi(m)-dependentand -independentproductionofreactiveoxygenspeciesbyrat brainmitochondria.JNeurochem2001;79:266–77.
[89] KushnarevaY,MurphyAN,AndreyevA.ComplexI-mediated reactiveoxygenspeciesgeneration:modulationby
cytochromecandNAD(P)+oxidation–reductionstate.
BiochemJ2002;368:545–53.
[90] MelnickRL,RubensteinCP,MotzkinSM.Measurementof mitochondrialoxidativephosphorylation:selective inhibitionofadenylatekinaseactivitybyP1, P5-di-(adenosine-5)-pentaphosphate.AnalBiochem 1979;96:7–11.
[91] YeJ,JiangZ,ChenX,LiuM,LiJ,LiuN.Electrontransport chaininhibitorsinducemicrogliaactivationthrough enhancingmitochondrialreactiveoxygenspecies production.ExpCellRes2016;340:315–26.
[92] HuW,TianH,YueW,LiL,LiS,GaoC,etal.Rotenone inducesapoptosisinhumanlungcancercellsbyregulating autophagicflux.IUBMBLife2016;68:388–93.