CHAPTER THIRTEEN
ELECTROCHEMICAL CELLS
The preceding chapter dealt primarily with the physical chemistry of electrolyte solutions; we now concern ourselves with the overall chemical process that occurs when electricity is passed through a conducting solution. The emphasis will be on the work associated with this overall change, as measured by the reversible cell potential. Since reversible work at constant temperature and pressure corre
sponds to a free energy change, we will thus be able to bring the emf of cells into the general scheme of thermodynamics. The chapter concludes with a discussion of irreversible electrode processes, that is, with the physical chemistry of the approach of ions to, and their reaction àt, the surface of an electrode.
13-1 Definitions and Fundamental Relationships
A. Cell Conventions
An electrochemical cell has, as essential features, a current-carrying solution and two electrodes at which oxidation and reduction processes occur, respectively, as current flows. Figure 13-1 gives a schematic example of a fairly typical cell for this chapter; we have hydrogen and silver-silver chloride electrodes dipping into an aqueous solution of HCl. The hydrogen electrode, incidentally, typically consists of a platinized platinum metal surface arranged so that hydrogen gas bubbles past as it dips partly into the solution, the object being to provide the most intimate possible gas-solution-metal contact. Platinized platinum is merely platinum metal on which additional, very finely divided platinum has been deposited electrolytically; the result is a high area, catalytically active surface. A silver-silver chloride electrode consists of silver on which a fine-grained, adherent deposit of silver chloride has been placed, again electrolytically. The terminals of the cell might be connected to a motor, so as to provide electrochemical energy, or, in the laboratory, to a potentiometer circuit (see Section 13-2), so that the potential difference could be measured.
499
Pt
HC\(m}
Ag
AgCl
AgCl + e - ^A g + Cl -
F I G . 1 3 - 1 . Schematic diagram of an electrochemical cell. The cell reaction is given by Eq. (13-2).
It i s awkwar d t o describ e cell s i n a pictoria l manner , an d th e conventiona l representation o f th e cel l o f Fig . 13- 1 i s
Pt/H2(P atm)/HCl(m)/AgCl/Ag . (13-1)
Equation (13-1 ) i s know n a s a cell diagram. Th e rul e i s tha t on e write s i n orde r each successiv e phas e tha t make s u p th e electrica l circui t o f th e cell , usin g a diagonal ba r t o separat e phases . On e shoul d i n genera l specif y no t onl y th e tem - perature o f th e cell , bu t als o th e compositio n o f eac h condense d phas e an d th e partial pressur e o f an y gaseou s one ; w e assum e th e genera l mechanica l pressur e to b e 1 atm .
A potentiometri c measuremen t o n a cel l wil l repor t a potentia l differenc e between th e electrod e terminals , an d th e secon d conventio n neede d i s tha t t o specify th e sig n o f thi s cel l potentia l S. Ther e i s som e variatio n i n practic e an d consequent ambiguity , an d th e leas t confusin g wa y o f statin g th e conventio n use d here seem s t o b e th e following . W e first defin e th e cel l reactio n a s th e chemica l change tha t occur s pe r farada y o f electricit y passe d throug h th e cel l i n th e directio n such tha t oxidatio n occur s a t th e left-han d electrod e o f th e cel l diagram . Thi s electrode wil l b e calle d th e anode; i t i s als o th e electrod e towar d whic h anion s migrate i n th e cel l solutio n a s the y carr y current . Th e right-han d electrod e o f th e cell diagra m i s the n th e on e a t whic h reductio n occurs , an d i t i s calle d th e cathode;
it i s als o th e electrod e towar d whic h cation s migrat e a s the y carr y current . Th e cell reactio n correspondin g t o (13-1 ) i s
anode 2Hz ( l atm ) = H+(w) + e-
cathode AgC l + e ~ = A g + Cl"(m )
net JH2(1 atm ) + AgC l = A g + H+(m ) + C\-(m).
(13-2) Since th e potentia l i s a functio n o f concentratio n a s wel l a s o f th e chemica l specie s involved, statement s o f cel l reaction s shoul d includ e th e concentratio n o r partia l pressure o f eac h substance .
The sig n o f ê i s no w define d t o b e positiv e i f th e cel l reactio n occur s sponta - neously i n th e directio n writte n an d t o b e negativ e i f th e revers e directio n o f reaction i s th e spontaneou s one . I n thi s particula r exampl e ê woul d b e positive .
13-1 DEFINITION S AN D FUNDAMENTA L RELATIONSHIP S 50 1
That is , silve r chlorid e i s spontaneousl y reduce d b y hydroge n gas . Althoug h th e mechanical arrangemen t o f th e cel l i s suc h tha t th e direc t chemica l reactio n i s prevented fro m occurring , th e proces s wil l tak e plac e spontaneousl y whe n th e electrode terminal s ar e connected .
This las t i s a n importan t featur e o f electrochemica l cells . Th e cel l reactio n ha s to b e spontaneou s i n on e directio n o r th e other , an d th e cel l mus t alway s b e s o designed tha t th e direc t chemica l reactio n i s physicall y prevente d fro m occurring . In th e cas e o f th e cel l o f Eq . (13-1 ) th e reactant s H2 an d AgC l ar e isolate d a t th e separate electrodes . Anothe r wa y i n whic h direc t chemica l reactio n i s prevente d is illustrate d b y th e Daniell cell show n i n Fig . 13-2 ; thi s consist s o f a zin c anod e dipping int o Z n S 04 solutio n an d a coppe r cathod e dippin g int o C u S 04 solution . The tw o solution s ar e separate d b y a porou s diaphrag m whic h allow s electrica l contact bu t prevent s gros s mixing . Th e cel l diagra m i s the n
Zn/ZnSO^mj) / CuS04(m2)/Cu, (13-3) where th e dashe d diagona l conventionall y i s use d t o indicat e tw o miscibl e phase s that ar e physicall y prevente d fro m mixing . Thi s situatio n wil l late r b e referre d t o as on e o f a liquid junction. Th e cel l reactio n i s
anode \Zn = \2xP+(m-ù + e- cathode |Cu2 +(m2) + e ~ = C u
net \Zn + ^Cu2+(m2) = ^Zn2+(m1) + | C u .
(13-4)
Again, th e cel l ha s bee n writte n i n suc h a wa y tha t th e cel l reactio n occur s spon - taneously—if place d directl y int o th e C u S 04 solution , th e zin c electrod e woul d react a s shown—an d s o th e measure d em f o f thi s cel l woul d b e reporte d a s positive .
To retur n t o th e matte r o f sig n convention , w e not e tha t th e usua l sourc e o f confusion i s i n th e plu s an d minu s marking s o f electrodes . Th e cell s o f Figs . 13- 1 and 13- 2 ar e draw n s o tha t i n spontaneou s actio n th e left-han d electrod e i s th e anode. Thi s mean s tha t th e anod e bear s a positiv e charg e relativ e t o th e cathod e at the solution end an d a negativ e charg e relativ e t o th e cathod e at the exposed terminals. I t i s th e terminal s tha t ar e marked , henc e i t i s th e spontaneousl y operatin g
F I G . 1 3 - 2 . The Daniell cell.
Zn
Z n S 04
(/«,)
Anode
+ Cu
C u S 04 (m2)
Cathode Porous
diaphragm
+
Electrons —9
+ +
+
Cations — Solution
Anions
F I G . 1 3 - 3 . Cell conventions. The cell diagram is anode/solution/cathode.
Anode
oxidation Cathode reduction
anode o f a cel l tha t bear s th e negativ e sign . T o repeat , whe n w e writ e a cel l reactio n the variou s sign s an d direction s o f flo w ar e take n t o b e a s show n i n Fig . 13-3 . I f this i s th e spontaneou s directio n o f flow, th e S i s reporte d a s a positiv e number.+
A furthe r featur e o f electrochemica l cell s tha t ar e use d i n precis e measurement s is tha t the y ar e reversible. Tha t is , th e cel l reactio n mus t tak e plac e readil y i n either direction . I f th e cel l i s short-circuited , th e reactio n shoul d procee d i n it s spontaneous direction ; an d i f a n externa l potentia l i s applie d whic h override s the natura l cel l potential , the n th e reactio n shoul d jus t a s readil y procee d i n th e opposite direction . Th e reversibl e electrochemica l cel l i s thu s on e whic h ma y b e held i n a stat e o f dynami c balanc e b y applicatio n o f a n externa l counterpotentia l just equa l t o $ . Fo r example , i n th e cas e o f th e Daniel l cel l ê i s abou t 1. 1 V i f m1 = m2= 1 . Externa l applicatio n o f a n opposin g 1. 1 V wil l jus t preven t reactio n from occurring ; a slightl y smalle r opposin g potentia l wil l allo w th e cel l reactio n to occu r a s written , an d a slightl y large r opposin g potentia l wil l mak e th e reactio n go i n th e opposit e direction .
The customar y wa y o f determinin g S fo r a cel l is , i n fact , t o find tha t opposin g emf whic h put s th e cel l i n balance . Th e procedur e thu s define s S a s th e reversibl e emf o f th e cell . I f curren t i s allowe d t o flow throug h th e cel l unde r condition s such tha t applie d potentia l remain s essentiall y equa l t o <f , the n th e wor k don e i s a reversibl e work . Fro m th e definitio n o f potential , wor k i n joule s i s give n b y qV, where q i s th e amoun t o f charg e carrie d throug h potentia l differenc e V. Th e reversible wor k fo r a n electrochemica l cel l i s the n n^ê, wher e η is the number of faradays passed through the cell.
For the Daniell cell as written in Eq. (13-4), the reversible work is (96,487)(1.1) = 1.06 χ 105 J {jnx = m2 = 1, and 25°C). Had the reaction been written for 2
the reversible work would be 2.12 χ 105 J, and had the reaction been written in the opposite direction, ê woul d b e reporte d a s —1. 1 V an d th e correspondin g reversible wor k woul d b e —1.0 6 χ 105 J. Thus the sign of ê an d bot h th e sig n and magnitud e o f th e reversibl e wor k depen d o n ho w th e cel l reactio n i s written .
Zn + Cu2+(m2) - Zn2+(m!) + Cu ,
+ Thi s i s th e traditiona l America n convention— a ver y logica l on e fo r physica l chemists . Th e SI conventio n i s discusse d i n Sectio n 13-CN-l .
13-1 DEFINITIONS AND FUNDAMENTAL RELATIONSHIPS 503 A useful conversion factor is that for η = 1 an emf of 1 V = 23.06 kcal. This unit is sometimes called the electron-volt: 1 eV = 23.06 kcal m o l e- 1.
β . Thermodynamics of Cells
The reversible emf that is measured for a cell gives the reversible work associated with the cell reaction. Since this is reversible work at constant temperature and pressure, it is therefore the free energy change and, by Eq. (6-34), the sign conven
tion is such that we must write
AG = -n^£; (13-5) that is, a positive cell emf corresponds to a spontaneous cell reaction and hence
to a negative free energy change.
We may now rewrite several important equations of Section 7-4 in terms of emf's. Thus Eqs. (7-23) and (7-24) become
J s
=
( 1 3-
6)J 5 0 = Bjr
(-er),'
( 1 3-
7 )and, since G = Η — TS by definition, for a constant-temperature process we have
AH = AG+ TAS, (13-8)
AH=-n^[^-T(-±-)p], (13-9)
J j y ° = -n& [<f° - Τ(^τ) \ (13-10) Equation (13-9) or (13-10) is known as the Gibbs-Helmholtz equation.
Example. The emf of the cell Cd/solution saturated with CdCl2-2.5H20/AgCl/Ag is 0.6753 V at 25°C, and dë\dT = -0.00065 V Κ"1. If we write the cell reaction as Cd + 2AgCl = CdCl2(sat. soin.) + 2Ag, then AS = (2)(96,487)(-0.00065) = - 1 2 5 J or - 2 9 . 9 cal K "1 mole"l, AH = -(2)(96,487)[0.6753 - (298.1)(-0.00065)] = -1.677 χ 105 J or -40.08 kcal (as com
pared with —39.5 kcal from thermochemical measurements), and AG = — (2)(96,487)(0.6753) = -1.303 χ 105 J or - 3 1 . 1 4 kcal.
An interesting point is that the q for a reversibly operating cell is given by Τ AS; q would be —(29.9)(298.1) or - 8 . 9 1 kcal in the example. In terms of Eq. (13-8) the measured q is given by AH when the reaction occurs directly, as in a thermochemical experiment; in the reversible cell the energy AG goes to do useful work, and the observed q is then determined by the entropy change. Thus the statement AH = AG + Τ AS amounts to saying:
. , x /energy availablex , /energy not availablex
(total energy change) = (to o ^ ) + (d tQ do wk ) o r
C. The Nernst Equation
A ver y importan t relationshi p i s obtaine d a s follows . Fo r a genera l cel l reactio n
a A + b B + ·· · = m M + n N + ··· ,
we hav e fro m Eq . (12-91 ) tha t
AG = AG» + RTln Qih [Eq . (12-91)] ,
where, i t wil l b e remembered , 2t h ha s th e sam e for m a s a n equilibriu m constan t but contain s th e activitie s o f th e product s an d reactant s a s arbitraril y specifie d by th e state d reaction . I f th e syste m i s a t equilibrium , however , AG = 0 , an d w e then obtai n
AG0 = -RTln Km [Eq . (12-92)] , where Kth i s th e thermodynami c equilibriu m constant .
Combination o f Eqs . (13-5 ) an d (12-91 ) give s th e Nerns t equation :
ê= .£o_JV_lnQtht ( 1 3_ 1 1 )
Insertion o f th e numerica l constant s fo r 25° C yield s
β = - 0.02569 In Qth = <f0 - log Qth . (13-12) Equation (13-11) is the central equation of electrochemistry. By means of it we can determine how the emf of a cell should vary with composition, and we can also determine SQ for a cell reaction, which in turn enables us to obtain activity coefficients for electrolytes.
Example. The Nernst equation for the cell reaction of Eq. (13-2) is
g = go _ 0.02569 In °H+aci~ m (13-13)
A g and AgCl are in their standard states and hence have unit activity. The hydrogen pressure will be 1 atm, and, for the moment, we neglect activity coefficient effects, so Eq. (13-13) reduces to
g = go _ 0.02569 ln[(H+)(Cl-)] = <?° - 0.05139 In m. (13-14) The observed emf of this cell is 0.49844 at 25°C and m = 0.005 and we may use the Nernst equation
to calculate £ for s o m e other concentration, say 0.01 m. Since is a constant, it follows from Eq. (13-14) that
^ o . o i m = ^o.oosm - 0.05139 In ^ 5 = 0.46282.
The observed value is 0.46419, a difference we will shortly be attributing to the nonideality of aqueous HCl.
A s a different kind of example, for the reaction of the Daniell cell, Eq. (13-4), we have 1/2
g = go 0.02569 In = go - 0.01285 In .
a^+ aCu>+
If Z n2+ and C u2+ are at unit activity (or, very roughly, 1 m), S = 1.10 V at 25°C. This means that S30 is also 1.10 V, since the log term is zero. If an excess of zinc metal is placed in a solution of copper sulfate which is initially at unit activity, the direct spontaneous reaction will occur to
13-2 EXPERIMENTA L PROCEDURE S 50 5 form coppe r meta l an d zin c ion . Eventuall y th e solutio n wil l consis t o f roughl y 1 m o r uni t activity Z n2+ an d som e smal l equilibriu m concentratio n o f Cu2 +. W e ca n us e th e Nerns t equatio n to calculat e thi s last . Sinc e th e fina l stat e i s a t equilibrium , AG an d henc e ê mus t b e zero , an d s o
<T> = 0 =L 0.0128 5 I n 1
\ « C u2+ / equi l
= 0.0128 5 I n ( —1— )
\ tfCu2+ / e q u i l
The equilibriu m (Cu2 + ) i s the n e x p ( - l . 10/0.01285) , o r abou t 1 0 "37 m(\).
13-2 Experimenta l Procedure s
A. The Potentiometer
The reade r i s referre d t o experimenta l text s fo r details , bu t th e principl e employe d in th e measuremen t o f emf' s o f cell s shoul d b e describe d a t leas t briefly . A n elementary potentiomete r arrangemen t i s show n i n Fig . 13-4 . On e set s u p a close d circuit involvin g a workin g battery , usuall y a we t cel l capabl e o f deliverin g a reasonable amoun t o f curren t withou t changin g it s voltage . Th e circui t contain s a moderatel y hig h resistance , perhap s 100 0 ohms , whic h i s eithe r i n th e for m o f a slid e wire , o r whic h ma y b e tappe d a t clos e intervals . Thi s resistanc e R the n has a n ohmi c dro p i n potentia l iR acros s it . Th e electrochemica l cel l i s connecte d as shown , wit h th e electrode s i n th e sam e directio n a s fo r th e workin g cell . On e now move s th e poin t o f contac t A unti l th e galvanomete r G show s n o curren t flow. A t thi s poin t th e potentia l dro p AB i s th e sam e a s S fo r th e cell .
The circui t i s usuall y calibrate d b y mean s o f a standar d cell , o r on e o f accuratel y known em f <^r ef . B y determinin g poin t A' whe n th e standar d cel l i s i n balance , one therefor e know s th e voltag e dro p A'B. Th e desire d em f i s the n ^Γ Θ ^ Α Β / ^ Α ' Β ) ·
In actual practice one adjusts subsidiary resistances in the circuit (not shown) in calibrating with the standard cell so that the tapped or slide wire resistance posi
tion A will read directly in volts.
F I G . 1 3 - 4 . A potentiometer circuit.
A' A
<!ΛΑΑΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛΛ^ C
X I
Hllr
HI Standard cell
\
β . Standard Cells
The most widely accepted reference cell is the Weston cell, illustrated in Fig. 13-5.
The anode consists of a layer of solid cadmium amalgam containing 12.5% cad
mium, and the cathode consists of a pool of mercury layered with a thick paste of H g2S 04. The solution is saturated with C d S 04 · f H20 , with some excess crystals present on both sides, to maintain saturation. The cell diagram is
Pt/Cd(Hg)/saturated CdS04 · f HaO/Hg/Pt and the corresponding cell reaction is
f H20 + Cd(amalgam) + Hg2S04(s) = CdS04 · f H20 ( J ) + 2 Hg.
The emf at 25°C is 1.0183 V; the temperature dependence is small, —4.06 X 1 0- 5 V K -1.
F I G . 1 3 - 5 . The Weston cell.
Potentiometric measurements are among the most accurate of physical chemistry.
Cell potentials may be measured to about 10~5 V, the limiting accuracy usually being that of the voltage of the reference cell. If the unknown cell is one of very high resistance, then the sensitivity of the galvanometer may become the limiting factor. In the case of a pH meter, for example, the glass electrode may have 105 ohms resistance, and one must use a vacuum-tube null-meter to detect the balance point (early users of pH meters struggled with quadrant electrometers).
The potentiometric method is a null method—when the circuit is balanced the galvanometer shows no current flow, and a slight shift in the position of the contact A to one side or the other causes a galvanometer deflection in one direction or the other. A barely detectable galvanometer deflection need correspond to no more than perhaps 10~12 A, so the change in condition needed to reverse the current flow is very small and the measured potential is essentially the reversible one. If a relatively large current, say 10~3 A, is drawn through a cell, then the potential drops as various irreversible processes occur, such as polarization, discussed in Section 13-9. The magnitude of the measured potential is therefore at a maximum at zero current.
13-2 EXPERIMENTAL PROCEDURES 507
Hg2Cl;
KC1
(m) -KC1 in a g a r - a g a r
HCl
(a) (b)
F I G . 1 3 - 6 . Reference electrodes, (a) Calomel half cell, (b) Complete cell that includes a calomel half cell.
C. Reference Electrodes
An electrochemical cell consists essentially of the two parts defined by the two electrodes, and one often constructs a cell in which one electrode is that under investigation and the other is a conventional electrode of known properties. This last is called a reference electrode. A very common and easily constructed one is the calomel reference electrode, illustrated in Fig. 13-6(a). Platinum wire dips into a pool of mercury which is layered with a paste of H g2C l2, followed by a solution which is usually 0.1 TV KCI, 1 TV KCI, or saturated KCI. Electrolytic connection must be made to the rest of the cell, and this is done through a side arm in which the KCI solution has usually been stiffened with agar-agar or gelatine.
This type of electrolytic connection is known as a salt bridge (see Special Topics section).
A complete cell might then appear as in Fig. 13-6(b). The cell diagram is Pt/Hg/Hg2Cl2(s)/l WKCl or saturated KCl///HCl(m)/H2(l atm)/Pt.
If the HCl is at unit activity, then
^298 = - 0 . 2 8 0 V (1JVKC1) and <T2 98 = - 0 . 2 4 2 Y (saturated KCI).
The boundary between the KCI solution and that of the electrolyte of the second part of the cell is known as a liquid junction. Since ions are carrying the current in solution, passage of electricity means that ions move across the junction, just as in a transference experiment. In terms of this cell some K+ ions must move from the KCI solution into the HCl one, and some C I- ions must move from a concentration m in the HCl to that in the KCI. Some net changes thus occur at the liquid junction, whose free energy requirement contributes to the emf of the cell as a whole. This contribution is known as the junction potential, and fortunately it is small if the bulk of the current is carried by oppositely charged ions of the same mobility. This is essentially the situation in the case of a KCI salt bridge;
K+ and Cl~ do have nearly the same mobility. The more detailed treatment of junction potentials is given in the Special Topics sections, and it is sufficient
here to note that for most purposes the junction potential for a saturated KCI (or a concentrated N H4N 03) salt bridge can be neglected. However, this eifect does impair the accuracy of a cell involving a calomel reference electrode.
Other referenc e electrode s includ e th e hydroge n electrod e itself , whic h i s ver y accurate bu t somewha t inconvenien t t o use , an d well-know n reversibl e electrode s such a s th e silver-silve r io n o r th e silver-silve r chlorid e ones . Fo r accurat e wor k a referenc e electrod e shoul d b e a direc t par t o f th e cell , bu t i t ma y b e mor e con - venient t o connec t th e electrod e bein g studie d t o th e referenc e electrod e vi a a sal t bridg e an d t o eithe r neglec t o r tr y t o estimat e th e valu e o f th e junctio n potential.
13-3 Determinatio n o f
&° Value s an d Activit y Coefficient s
Equation (13-11 ) ma y b e writte n i n th e for m
RT RT
# = £o __HJ^]IlQ-Jl±rinQ (13-15 )
where
flM"W - ( M ) m( N )w - y MmyN" - n mn , ~ Q th = = ( A ) W - y AW - = Q Q" • ° 3"1 6) We no w writ e
S> = S + ~ l n Q = <f ° - ^ l n Qv . (13-17 ) The quantit y ê' i s determine d fo r a serie s o f concentration s an d plotte d agains t
concentration. A t infinit e dilutio n th e activit y coefficient s an d henc e Qy approac h unity, an d I n Qy approache s zero ; th e extrapolate d valu e o f S" i s thu s equa l t o
The procedur e ma y b e illustrate d fo r th e cel l correspondin g t o reactio n (13-2 )
13-4 ADDITIVITY RULES FOR EMF'S. STANDARD OXIDATION POTENTIALS 509
at 25°C. We write Eq. (13-14) in the form
β = 0 - 0.05139 In m - 0.05139 In y± or
β' = β + 0.05139 In m = <f0 - 0.05139 In y± . (13-18) According to the Debye-Huckel limiting law, Eq. (12-89), In γ± should be propor
tional to y/m, so ê' i s plotte d agains t y/m a s show n i n Fig . 13-7 . Extrapolatio n to zer o concentratio n give s = —0.2223 9 V . Havin g determine d th e <f ° fo r th e cell reaction , on e ma y the n inser t it s valu e bac k int o Eq . (13-18 ) an d thu s obtai n γ± for HCl at each concentration.
This procedure illustrates how a number of very accurate values of <f0 and of activity coefficients have been obtained. One may also, of course, estimate Qv either theoretically or from other activity coefficient data and thus calculate <o°
from the measured S.
13-4 Additivity Rules for
Emf s. Standard Oxidation Potentials
Since the S or «f0 for a cell reaction is just the free energy change per equivalent, emf's obey essentially the same additivity rules as do free energies. For example,
(a) Zn + 2H+ = Zn2+ + H2, (b) Cu + 2H+ = Cu2+ + H2, (c) = (a) - (b) Zn + Cu2+ = Z n2+ + Cu.
We know that AG°{C) = AG°{&) - AG°ih), hence
or ^?c) — ^!a) ~ ^?b) · In general, if two cell reactions are added (subtracted), the resultant emf is the sum (difference) of those for the two reactions.
We make use of this attribute in much the same way as is done in formulating enthalpies and free energies of formation. First, all emf data are expressed as
<f0 relative to the hydrogen electrode as cathode. Second, a cell reaction is expressed
as a combination of two half-cells. For example, reaction (a) is broken down as
Zn = Zn2+ + 2e- <^n/zn»+
2H+ + 2e- = H2 - < ^ .2/ H +
Zn + 2H+ = Zn2+ + H2 = ^n / Z n,+ - ^ / + H
Thus any cell reaction may be written as the difference between two half-cell oxidation reactions and any *f0 as the difference between two standard half-cell oxidation potentials. This combination of equations may therefore be written as
*?c> = ^ z „ / Z „ - ~ ^ ° H2/ H+) ~ ^ ° C u / C u - ~ ^ ° H2/ H+)
= ^ Z n / Z n2+ ^ C u / C u2+ *
Some indirect estimates suggest that the absolute standard half-cell potential for H2/H+ is small, but it is apparent that its actual value is immaterial in com
bining equations, since it cancels out. The third step is, accordingly, to make the convenient, arbitrary assignment that <^H2/H+ = 0 and to report the measured <f°
values for reactions such as (a) and (b) as the actual values for the half-cells.
Thus ^298 = 0.763 and —0.337 for reactions (a) and (b), respectively, and we report that
^ n / z n - = °-763 and < f «u / C u 2+ = - 0 . 3 3 7 .
Then <^298 for reaction (c) is 0.763 — (—0.337) = 1.10 V; this result is independent of the assumption regarding <^Η2/Η+·
The convention is that the emf of a cell is given by
* ° = * l ° e f t - * ? ig M. d3'1? )
where <f°e ft and ^i g ht are the standard oxidation potentials of the half-cells corre
sponding to the anode and cathode of the full cell, respectively.
The general mass of emf data has been reduced by means of this formalism and a number of standard half-cell potentials are given in Table 13-1. Their use follows the example just given. Each value is actually the for the cell whose anode is the stated half-cell and whose cathode is the standard hydrogen electrode.
The right-hand column of the table gives the corresponding standard electrode potentials, i^0. This alternative definition is described in Section 13-CN-l.
As one other further example, the reaction (c) 2Hg + 2AgCl = Hg2Cl2 + 2Ag
can be written as the difference between
(a) 2Hg + 2C1- = Hg2Cl2 + 2e~, S\w = - 0 . 2 6 7 6 and
(b) Ag + CI- = AgCl + e-, <f2°98 = - 0 . 2 2 2 4 .
Then <s?°c) = -(0.2676) - (-0.2224) = - 0 . 0 4 5 2 . Note that although we must multiply reaction (b) by 2 before subtracting it from reaction (a) in order to obtain reaction (c), we subtract the emf's directly. This is because an emf corresponds to the free energy change per faraday, so that all emf's are on the same basis.
There is one situation where the additivity procedure must be handled with care, namely in the combining of two half-cell reactions to give a third half-cell reaction. Each emf should be weighted by the number of faradays for which the half-cell reaction is written. As an example, consider the case
(a) Fe = Fe2+ + 2e~, AG°(3i) =
-2Fe%$ ,
(b) F e2+ - Fe3+ + e", AG φ) = (b) (c) Fe = Fe3+ + 3e-, JG(°C) = - 3 ^(°c) .
Since AG°i0) = AG°(a) + AG°ih) , it follows that <f(°c) = (2<sf(°a) + ^(% ) / 3 . At 25°C, we get <f(°c) - [(2)(0.440) + (—0.771)]/3 = 0.036. The general equation is
= W(a)^?a) + M ( b ) ^ ? b ) (13-20)
13-4 ADDITIVITY RULES FOR EMF'S. STANDARD OXIDATION POTENTIALS
T A B L E 1 3 - 1 . Standard Oxidation Potentials at 25°Ca
Half-cell reaction ^ 0 b
' 2 9 8
Li = Li+ + e" 3.045 -3.045
K - K+ + e- 2.925 -2.925
Ca = C a2+ + 2e- 2.87 - 2 . 8 7
Na = Na+ + e~ 2.714 -2.714
Mg = Mg2+ + 2e- 2.37 - 2 . 3 7
Al = A l3+ + 3e~ 1.66 - 1 . 6 6
Zn = Z n2+ + 2e- 0.763 -0.763
Fe = F e2+ + 2e~ 0.440 - 0 . 4 4 0 Cd = C d2+ + 2e~ 0.403 - 0 . 4 0 3 Pb + SO2" - PbS04 + 2e- 0.356 -0.356
Tl - T1+ + e~ 0.3363 -0.3363
Pb = P b2+ + 2e- 0.126 -0.126
Ag + I- = Agi + e- 0.156 - 0 . 1 5 6
Fe = F e3+ + 3e~ 0.036 - 0 . 0 3 6
H2 - 2H+ + 2e- 0.0000 0.0000
Ag + Br- = AgBr + e~ -0.0713 0.0713
Cu+ - C u2+ + e~ -0.153 0.153
Ag + CI" = AgCl + e- -0.22239 0.22239
Saturated calomel -0.242 0.242
2Hg + 2C1- = Hg2Cl2 + 2e~ -0.2676 0.2676
Normal calomel -0.280 0.280
0.1 Ν calomel -0.3358 0.3358
Cu = C u2+ + 2e- -0.337 0.337
Cu = Cu+ + e- -0.521 0.521
21- = IA + 2e- -0.5355 0.5355
F e2+ = F e3+ + e~ -0.771 0.771
2Hg = H g2+ + 2e- -0.789 0.789
Ag = Ag+ + e~ -0.7991 0.7991
H g2+ = 2Hg2+ + 2e- -0.920 0.920
2Br~ = Br2 + 2e~ -1.0652 1.0652
i HaO = J 02 + H+ + e- - 1 . 2 3 1.23
T1+ = T l3+ + 2e- - 1 . 2 5 1.25
2C1- = Cla + 2e- - 1 . 3 6 1.36
PbS04 + 2 H20 =
P b 02 + SOI" + 4 H+ + 2e" -1.685 1.685
2F- = F2 + 2e~ - 2 . 8 7 2.87
Basic solutions
SO2" + 2 0 H - = SO*- + H20 + 2e~ 0.93 - 0 . 9 3 H2 + 2 0 H - - 2 H20 + 2e- 0.8281 -0.8281 Ni + 2 0 H - - Ni(OH)2 + 2e~ 0.72 - 0 . 7 2 3 0 H - = H20 + H 02- H- 2e~ - 0 . 8 8 0.88 a Largely adapted from G. N. Lewis and M. Randall, "Thermo
dynamics," 2nd ed. (revised by K. S. Pitzer and L. Brewer). McGraw- Hill, New York, 1961.
b See Commentary and Notes section.
where η denotes number of faradays. Whenever one has any question about combining cell emf's, he should always do so first in terms of AG's since these are always additive.
13-5 Emf and Chemical Equilibria
A. Thermodynamic Relationships
Combination of Eqs. (13-5) and (12-92), AG0 = —RTln Kth , gives an impor
tant relationship between the <f0 for a cell and the equilibrium constant for the cell reaction:
RT
ê« = ^lnKth (13-21 )
or, fo r 25° C
Thus fo r η = 1, 0.059 V corresponds to one power of ten in K.
β . Direct Applications
A very direct and useful application of <f0 values is to the treatment of oxidation- reduction equilibria in solution. The following examples illustrate typical situa
tions.
Example. To what extent will Zn reduce 0.01 m F e2+ at 25°C? The reaction is Zn + Fe2+ - Zn2+ + Fe
and, from Table 13-1, <?°298 = 0.763 - 0.440 = 0.323. Then Kth = exp[(2)(0.323)/(0.02569)] = 8.3 x 101 0, or
* Z n2 +
8.3 x 1010
aFe2+
The reaction will go virtually to completion, or, if activity coefficients are neglected, until (Zn2 +) is essentially 0.01 m\ the equilibrium (Fe2+) is then 0.01/8.3 χ 1010 = 1.2 χ IO"13.
Example. To what extent should 0.01 m H g |+ disproportionate into Hg and H g2+ at 25°C?
We combine the following half-cell reactions:
(a) -|-Hg2+ = Hg2+ + e-, <?(°a> = - 0 . 9 2 0 , (b) |Hg2+ + e- = Hg(/), <f ?b> = 0.789,
(c) Hg2+ = Hg2+ + Hg(/), <f(°c) = - 0 . 1 3 1 .
= exp(-0.131/0.02569) = 6.10' x 10"3, so we have
from which ( H g2 +) present.
13-5 EMF AND CHEMICAL EQUILIBRIA 513
= 6.06 x 10"5. The calculation supposes that some Hg(/) is formed or is
C. Determination of Solubility Products
Several of the half-cell reactions of Table 13-1 are written as the reaction of a metal with an anion to give a slightly soluble salt. The potential for such a half-cell reaction may be combined with the one for the simple oxidation of the metal, to give the solubility product of the salt. If we have
(a) Μ + Χ" = MXO) + e-, <^a) ,
(b) M = M+ + e~, ^°
(c) = (b) - (a) M X ( J ) = M + + X - , «f(°c) = <?ob) - df(°a) , then, neglecting activity coefficients, Ksv = exp(#(°c )/0.02569) at 25°C.
An alternative and sometimes very useful approach is the following. We write the Nernst equation for reaction (b):
- <f(°b) - 0.02569 In αΜ+ . (13-23)
The potential at a metal-metal ion electrode must always reflect the chemical potential of that ion; in the presence of X-, MX(s) forms, which decreases aM+ according to the solubility constant, aM+ = Kth/ax~ . The standard potential of reaction (a) must therefore be the same as the potential of the metal-metal ion electrode for that value of aM+ which is present when ax- is unity. Thus when ax- is unity *f( b) in Eq. (13-23) may be replaced by and #M+ by Km :
* & ) = * 8 » ~ 0.02569 In Kth , Kth = exp , which is the same result as before. This alternative treatment, although longer as presented here, becomes very advantageous when one is dealing with more com
plicated situations.
Example. Calculate the solubility product for A g2S 04 if <^598 = —0.627 V for the cell Ag/Ag2S04/H2S04(m)/H2/Pt.
The «f° is that for the half-cell reaction
2Ag -1- S 04~ = A g2S 04 + 2e~, = -0.627.
Subtraction of this from
2Ag - 2Ag+ + 2e~, ^M = -0.799 gives
A g2S 04 = 2Ag+ + SOÎ", ^2 98 = - 0 . 1 7 2 .
The solubility product is then exp[(2)(-0.172)/0.02569] = 1.53 x 10"6. Note that η = 2 in this case.
Application of the alternative procedure is as follows. We write é> = - 0 . 7 9 9 - «0.02569) In a2Ag+ .
13-6 Concentration Cells
The term concentration cell is used to designate a cell whose net reaction involves only changes in composition of species (or of gas pressures) and no net oxidation or reduction. The for such a cell must be zero since all species in the cell reaction are then to be at unit activity, in which case no change at all accompanies the pas
sage of electricity.
When aSol~ is unity, £ = —0.627 V, and aAg+ = KlaSol~ = K, so - 0 . 6 2 7 = - 0 . 7 9 9 - £(0.02569) In K, which gives the same result as before.
D. Determination of Dissociation Constants
The alternative procedure described in the treatment of solubility equilibria may be applied to a homogeneous equilibrium in solution. If a metal ion is com- plexed in solution, then the half-cell potential M/Mz+ gives the activity of that ion, and this often allows the calculation of the equilibrium constant for complexa- tion.
Example. The potential for the cell
Cu/0.02/Cu(II) in 0.5/NH3/normal calomel electrode
is 0.26 V at 25°C. [We use formalities since the copper is largely present as the complex Cu(NH3)2+
and we wish merely to describe the overall makeup of the solution.] We treat the left-hand electrode as a Cu/Cu2+ electrode whose emf is determined by #C U2 + in the solution: The emf of the cell is therefore written
* = * c u / c u * + - ^ref - «0.02569) In *0 α. + or
0.26 = -0.337 - (-0.280) - 0 , 0 2 5 69 m aC U2+ ,
Virtually all of the C u2+ is in the form of Cu(NH3)2 +; therefore (NH3) = 0.5 - (4)(0.02) = 0.42, and we evaluate the equilibrium constant as
( Ο ι ( Ν Η3) Γ ) = 0.02 = 3 37 χ 1 Q 1. (Cu2+)(NH3)4 (1.91 X l O -1 1) ^ )4
(neglecting activity coefficients).
A number of equilibrium constants for the dissociation of complex ions have been determined in this way. We see in Section 13-8 that an analogous procedure may be applied to the determination of aH+ in a solution.
A. Electrode Concentration Cells
13-6 CONCENTRATION CELLS 515
A very straightforward type of concentration cell is the following:
Pt/H2(A)/HCl(m)/H2(P2)/Pt, for which the cell reaction is
anode Η2( Λ ) = 2H+(m) + 2e~, cathode 2H+(m) + 2e~ = H2CP2), net reaction Η2( / \ ) = H2(P2), with
- 0.02569, Λ 0 298 — 2 P[ '
In the case of a metal electrode the metal may be present as an amalgam:
C d ( X i , in Hg)/CdS04(m)/Cd(;c2, in Hg), with
0.02569 ax%
? 298 = 9 n — A
B. Simple Electrolyte Concentration Cells
In a simple electrolyte concentration cell liquid junctions are avoided by setting up two opposing cells which differ only in their electrolyte concentration. The following is an example:
Ag/AgCl/HCl(m1)/H2(l atm)/Pt—Pt/H2(l atm)/HCl(m2)/AgCl/Ag, first anode Ag + Cl~(/«i) = AgCl -f e-, first cathode H+(mi) + e~ = JH2(1 atm), second anode atm) = H+(m2) + e_, second cathode AgCl + e- = Ag -+- Cl_(m2), net reaction H+imO + Cl-CmO = H+(ra2) + Q - ( m2) .
Note that it is important to write each ionic species separately if the electrolyte is in fact treated as fully dissociated. The emf for this cell is
<f2 98 = - 0 . 0 2 5 6 9 In ^ ' ^ " ^ ,
αΈΙ +,τηχβα',πΐ!
<f2 98 = - 0 . 0 5 1 3 9 In ^ - 0.05139 In .
Cells of this type may be used to obtain the ratio of activity coefficients of an electrolyte at two different concentrations.
Electrolyte concentratio n cell s havin g a liqui d junctio n ar e discusse d i n th e Special Topic s section .
13-7 Oxidation-Reductio n Reaction s
We conside r her e th e situatio n i n whic h bot h th e oxidize d an d reduce d form s o f th e half-cel l couple ar e solutio n species . Example s ar e th e couple s F e2 +/ F e3+ an d C u+/ C u2 +. Bot h partner s of th e redo x coupl e ar e i n solution , an d sinc e the y ca n b e expose d onl y t o o n e o f th e electrode s (or els e th e cel l woul d b e short-circuited) , i t i s mandator y tha t th e secon d electrod e b e connecte d by mean s o f a sal t bridge . Thu s w e hav e
P t / C u+( mx) , C u2 +( m2) / s t a n d a r d calome l electrode . The potentia l o f suc h a cel l i s give n b y th e genera l for m
^ ^ n o d e - ^ r - ^ ^ l n ^ , (13-24 ) where th e anod e reactio n i s
M(red) = M(oxid ) + m~
and tfoxid an d aTea refe r t o th e activitie s o f th e oxidize d an d th e reduce d form s o f th e specie s o f th e redox couple , respectively . A s mentione d i n Sectio n 13-2C , th e presenc e o f a liqui d junctio n introduces s o m e unavoidabl e inaccurac y i n th e measure d emf . (Se e als o th e Specia l Topic s section.)
Cells o f thi s typ e hav e bee n use d i n th e determinatio n o f th e redo x em f no t onl y fo r meta l io n couples bu t als o fo r a variet y o f inorgani c coordinatio n c o m p o u n d s an d fo r man y organi c systems . A n exampl e o f th e secon d typ e i s th e F e ( C N ) J ~ / F e ( C N ) 6 ~ couple , fo r whic h <^g98 — —0.3 6 V , and a n exampl e o f th e latte r i s th e coupl e
[ H O - C6H4- O H ] = [ 0 = C 6H4= 0 ] + 2H + + 2e~ , <?2 93 = - 0 . 6 9 9 4 . (13-25 ) It wil l b e see n i n th e nex t sectio n tha t th e quinone-hydroquinon e coupl e i s usefu l i n pH deter - minations.
Returning t o inorgani c examples , w e not e tha t mos t element s exis t i n severa l oxidatio n state s and tha t th e potential s betwee n thes e state s ar e generall y determine d b y cell s o f th e typ e repre - sented b y Eq . (13-24) . Tabl e 13- 2 summarize s a fe w o f thes e em f relationships . Th e numbe r between oxidatio n state s i s th e standar d oxidatio n potentia l a t 25°C . W e assum e aci d solution s and us e H + an d HaO a s neede d i n writin g th e balance d half-cel l reactions .
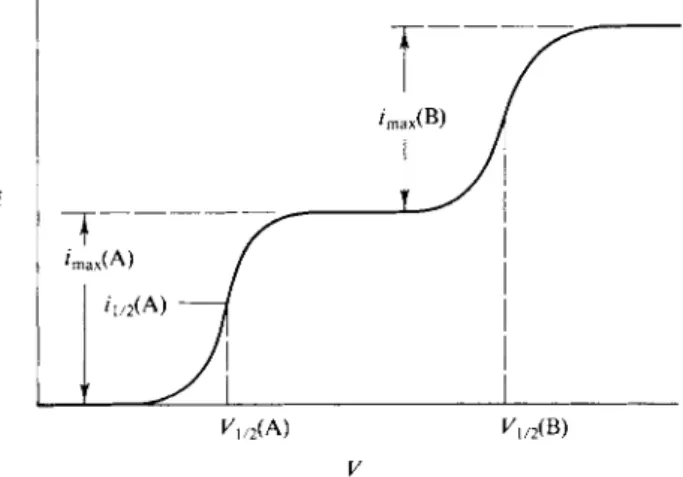
A cel l correspondin g t o Eq . (13-24 ) m a y als o b e use d t o follo w a redo x titration . Le t u s sa y that io n M i i s initiall y presen t i n reduce d for m an d tha t a n oxidizin g agent , io n M 2, i s adde d progressively. Th e reactio n
MjOred) + M2(oxid) = M ^ o x i d ) + M2(red)
occurs. W e assum e tha t th e reactio n i s rapi d s o tha t a t eac h stag e o f th e titratio n th e syste m i s i n equilibrium. Sinc e ther e ca n b e onl y on e potentia l a t th e platinu m anode , thi s mean s tha t « M1( o x i d ) / û M ^ r e d ) i s alway s equa l t o aM 2( o x i d ) / « M2( r e d ) .
The situatio n i s mos t easil y delineate d b y mean s o f a concret e example . Conside r th e titratio n of Cu + wit h F e3 +:
C u+ + F e3+ = Cu2+ + F e2 +. (13-26 )
13-7 OXIDATION-REDUCTION REACTIONS 517
T A B L E 1 3 - 2 . Selected Standard Oxidation Potentials for the Elements in Their Various Valence States"
Cr - î i i - Cr2+ -1ÎÎ- Cr3+ - ^ i - Crtf-
Mn - î i M n « Mn3+ M n 02 MnO*- MnCV
Co Co2+ - ^ i . Co3+ ^ ± 1 CoO,"
N ^ Li N u ^ Li N ! i 0 C u ^ i - C u + ^ î - C u "
Cl' α , -rLÎL HCIO HCI02 CIO," -=i^- C104"
B r- JUL Br2 - i u - HBrO ^ Br03"
I- ι, _ ^ i L HIO - ^ i - IO," - t ± 2 - H5I 06
α Adapted from W. M. Latimer, "The Oxidation States of the Elements and Their Potentials in Aqueous Solution," 2nd ed.
Prentice-Hall, Englewood Cliffs, New Jersey, 1952.
The cell would be
Pt/Cu+(plus C u2 +, F e3 +, F e2+ as the titration proceeds)/calomel reference electrode.
The cell potential is #c e li = < ^Pt — < ^re f > where < fPt is the half-cell potential at the platinum electrode. The statement is that at all stages of the titration
<fpt = ( ^F e2 + / F e 3 + = ^ C u + / C u 2 + , (13-27)
fCn2 + ï (Fe3 + >)
^ P t = <*& i + / c u « + - 0.0256 9 I n ^ j + y = *ί·« + / ρβ» + - 0.02569 In ^ ^ + ) , (13-28) where, for simplicity, activity coefficients are neglected. Equation (13-28) rearranges to
(Cus+VFeïi + }
*8«/c»« + - ^ΐβ» + /ρβ»+ = 0.02569 In (cu + ) ( F e3 +) = 0 , 6 1 8* ( 1 3"2 9) Equation (13-29) defines the equilibrium constant for reaction (13-26) and may be solved for each
stage of the titration. One then inserts the equilibrium ratio of (Cu2 +)/(Cu+) or that of (Fe3 +)/
(Fe2 +) in Eq. (13-28) to give <?Pt and thence «?cen .
At the end-point, F et ot = C ut o t, and since ( F e2 +) = ( C u2 +) by the stoichiometry of Eq.
(13-26), it follows that ( F e3 +) = ( C u+) . Equation (13-29) then reduces to
? g u / c u * + - <*Ρβ· + / ρ β · + = 0.05139
Insertion of this relationship into the right-hand side of Eq. (13-28) to eliminate the log term leads to
^ P t . e n d p o i n t = i ( ^ F e2/ F e3 ++ + ^ C u + / C u2 +) - (13-30) Equation (13-30) gives the value of and hence of &o e ll at the endpoint. The plot of this par
ticular titration is shown in Fig. 13-8, where F i s the degree of progress of the titration.
0
- 0 . 2
- 0 . 6
- 0 . 8 I 1 1 1 1 1 1 —
0 0.2 0.4 0.6 0.8 1.0 1.2 F
FIG. 13-8. Potentiometric titration curve for Cu+ + F e3+ = C u2+ + F e2 +.
13-8 Determination of pH
The term pYL was coined by S. Sorensen in 1909 to mean — log(H+), and we have come to use the symbol p{X) as an operator meaning —log X, where X may be a concentration, an activity, or an equilibrium constant; ρΚΆ means —log Κ& , for example. A potential measurement always reflects the activity of the species present, and only at infinité dilution can activity be equated with concentration.
Potentiometric methods for pH determination therefore measure some type of hydrogen ion activity, although it turns out that the exact nature of what is measured depends on the cell that is used. The modern procedure is to define pH as essentially
pH = - l o g t fH+ (13-31)
but with the exact meaning of aH+ determined by the cell used.
Consider first a cell such as (13-1):
Pt/H2(l atm)/HCl(m)/AgCl/Ag.
The corresponding Nernst equation is
β = g« - 0.05139 In a± t H C 1. (13-32)
The procedure described in Section 13-3 allows the determination of <o°, and so the cell measurements provide values for the mean activity of HCl in any solution, which might be thought to correspond to aH+ . Suppose that now we replace the electrolyte by [HAc(mx) + KCl(w2)]. The S0 remains the same, as does Eq. (13-32).
However, the calculated mean activity for HCl will now depend on both m1 and m2. Thus such a cell is not suited for pYL determination.
Next, consider the cell
Pt/H2(l atm)/solution /calomel electrode.
13-8 DETERMINATION OF pH 519
This emf is
298 — <fr ef - 0.02569 In αΗ+ = + 0.05916 pH (13-33) and it appears that our goal is achieved. The problem is that < fr ef contains the junction potential at the solution/KCl interface (see Section 13-2C and Special Topics section) and is not rigorously measurable. What is actually done is to determine Sref such that aH+ corresponds to ( H+) γ± as observed, by independent means, to apply to various solutions of weak acids. In effect, it is assumed, not quite correctly, that yH+ = y± ; it is also assumed, not quite correctly, that the junction potential incorporated in êref wil l no t var y wit h th e natur e o f th e solutio n
studied.
The resul t o f al l thi s i s tha t th e operationa l definitio n o f pU i s give n b y Eq . (13-33 ) with th e <fr ef value s o f Tabl e 13- 1 fo r th e variou s calome l referenc e electrodes . The definitio n allow s ver y precis e pH measurements , bu t one s whos e accurac y is subjec t t o some , bu t probabl y no t muc h uncertaint y s o lon g a s fairl y dilut e solutions ar e involved . Th e interpretatio n o f pH i n concentrate d solution s o r i n nonaqueous solvent s ca n b e quit e a problem .
The hydroge n electrod e i s a demandin g on e t o us e experimentally ; th e platinize d platinum ca n b e poisone d (los e it s catalyti c abilit y du e t o adsorptio n o f solutio n components), s o tha t th e electrod e fail s t o functio n well . A n alternativ e pH- determining cel l i s tha t correspondin g t o Eq . (13-25) :
Pt/solution wit h quinon e (Q ) plu s hydroquinon e ( H a Q ^ / c a l o m e l electrode .
One convenienc e i s tha t quinon e form s a 1: 1 compoun d wit h hydroquinone , quinhydrone, s o tha t b y dissolvin g th e compoun d i n th e solutio n t o b e tested , on e establishes equa l concentration s o f bot h species . Wit h th e adde d assumptio n that , being nonelectrolytes , thei r activit y coefficient s wil l b e unity , th e Nerns t term , (RT/n^) l n( 0Q/ 0 H2Q )5 drop s ou t an d on e ha s fo r 25° C
(subject t o th e sam e reservation s abou t liqui d junction s a s state d earlier) .
The mos t widel y use d /?H-determinin g cel l i s tha t know n a s th e pH meter o r glass electrode. Th e cel l diagra m i s
where th e anod e ma y b e eithe r Ag/AgC l o r a calome l electrode , bu t wit h th e solution buffere d a t som e constan t pW. Th e glas s membran e ha s th e propert y o f passing essentiall y onl y hydroge n ions , and , pe r faraday , th e cel l reactio n i s
< f2 98 = - 0 . 6 9 9 4 - <fr ef + 0.0591 6 prl (13-34)
glass membran e reference electrod e / /
// u n k n o t
in solutio n A / / // unknow n solutio n / calome l electrode , (13-35)
H+( s o l u t i o n A ) = H+( u n k n o w n solution ) SO
S w = constan t + 0.0591 6 prl, (13-36) where th e constan t contain s £ ° fo r th e cell , a constan t Nerns t term , an d th e junction potential . I n practice , on e calibrate s th e pH mete r scal e b y measurin g the em f whe n a know n buffe r solutio n i s use d i n plac e o f th e unknow n solution ,
I 1
<^rev %q
V
F I G . 13-9. Variation of current i with voltage V.l-2: current determined by diffusion of electrode products away from the electrode region; 2-3 current determined by Ohm1 s law.
and one finds that it gives almost the same pH values for other solutions as a hydrogen electrode does [see Dole (1941) and Bates (1954) for details].
Recently other ion-selective membranes have been developed that pass only one or another cation or anion, and an electrode incorporating such a membrane allows the determination of a pM or a /?X, where M or X is the ion passed. The selectivity is not always rigorous, however, and one must test such electrodes carefully in the actual situation before accepting the results of their use.
13-9 Irreversible Electrode Processes
The material so far has dealt with reversible emf's, the experimental measurements being made under conditions of virtually no current flow through the cell. Practical applications of electro
chemistry, except for standard reference cells, involve appreciable current flows and we now encounter a new set of phenomena which involve irreversible processes. One is that of over- voltage, important in electrodepositions and also in the study of the kinetics of electrode reactions.
Another is polarography, discussed in the Special Topics section.
A. Electrodeposition
Electrodeposition is the production of an electrolysis product, usually a metal, on a pre
parative scale. The situation is illustrated in Fig. 13-9. When a potential is applied across the terminals of a cell very little happens until a critical voltage is reached, beyond which the current increases about linearly with potential. This is what occurs: Suppose that we have two platinum wires dipping in an HCl solution. The cell reaction and reversible potential are
anode 2Cl~(m) = C12(P) + 2e~, cathode 2H+(m) + 2e~ = H2(P), and
* = <?° - In — ' g 1η(β Η ι«α,), (13-37)
±,HC1
13-9 IRREVERSIBLE ELECTRODE PROCESSES 521 where <f° = —1.3595 V at 25°C. This would, in principle, be established if hydrogen and chlorine gas were bubbling past the respective electrodes, so that aHi and aC\2 corresponded to a Ρ of 1 atm.
In the present experiment, however, no gases are being supplied, and Ρ is initially zero, so that é i s some larg e positiv e number , infinit y i n theory . Whe n th e electrode s ar e inserte d int o th e solutio n and connected , bu t wit h n o potentia l applie d som e fluctuation wil l decid e on e t o b e th e anod e and th e othe r th e cathode , an d som e minut e amoun t o f electrolysi s wil l occur , formin g a littl e dissolved hydroge n an d chlorin e nex t t o th e electrode s an d bringin g thei r loca l activitie s u p t o some low , bu t nonzer o value . Th e activit y o f th e dissolve d gase s i s fa r belo w tha t fo r 1 at m pressure an d n o bubble s ca n form , bu t th e dissolve d gase s wil l diffus e steadil y awa y fro m th e electrodes int o th e solution . Th e resul t i s tha t a smal l curren t wil l flow, an d i f a ver y sensitiv e probe coul d b e used , eac h electrod e woul d b e observe d t o hav e som e potentia l relativ e t o th e probe. Th e electrode s are sai d t o b e polarized, an d th e smal l curren t i s calle d th e residual polariza- tion current.
If w e no w appl y a smal l potentia l V betwee n th e electrodes , additiona l electrolysi s wil l tak e place, buildin g u p aH2 an d aCi2 s o tha t a bac k em f £Q develop s i n oppositio n t o th e applie d potentia l difference. Th e increase d hydroge n an d chlorin e activitie s lead t o a n increase d diffusio n rat e away fro m th e electrode s an d a consequen t increas e i n th e ver y lo w steady-stat e current . W e thu s observe sectio n 1- 2 o f th e curv e show n i n Fig . 13-9 . Wit h continue d increas e i n applie d voltag e
#H2 a n cl aci2 wiH becom e equa l t o th e valu e o f th e gase s a t 1 atm , an d actua l bubbl e formatio n will occur . The n êq i s a t it s maximu m valu e <^(m ax) an d canno t increas e further , an d beyon d thi s point th e increas e i n applie d potentia l goe s int o a n ohmi c potentia l dro p throug h th e solution , that is , V — ^ (m a x) = iR. Th e sectio n 2- 3 o f th e plo t i s thu s linear .
This analysi s indicate s tha t extrapolatio n o f th e linea r portio n o f th e plo t o f Fig . 13- 9 give s
^e(max) ; thi s i s calle d th e decomposition potential. Ideally , <^(max ) i s equa l t o ^r ev fo r th e cell , but i n practic e i t ma y b e mor e positive , an d th e differenc e betwee n th e tw o i s the n calle d th e overvoltage. I n electrodepositio n on e i s operatin g i n thi s linea r region , an d althoug h th e reversibl e potential i s th e idea l decompositio n potential , i n practic e on e mus t kno w wha t th e overvoltag e will be .
Suppose tha t w e wis h t o deposi t cadmiu m fro m a 0. 1 m solutio n o f a cadmiu m sal t a t pH 1.
The reversibl e potentia l fo r th e reactio n C d2+ - f 2e ~ = C d wil l be , a t 25° C an d wit h activit y coefficient effect s neglected ,
^c d= -0.40 2 - 2 ϋ| 6 ? 1 ^η=_ 0 2 v 4 3
The value for hydrogen, that is, £ for evolving hydrogen gas, is - Λ 0.02569, / 1 \ i f\ A \ Α \τ
^ H2 = 0 _ i n^ _ j = - 0 . 4 1 4 V.
One thus expects the evolution of hydrogen to occur first as potential is applied to the electrolysis cell and that, in fact, it would be virtually impossible to cause cadmium metal to deposit on the electrode. The actual situation is just the reverse because the overvoltage for hydrogen evolution from a cadmium surface is about 0.5 V; as a consequence, the cadmium deposits and no hydrogen evolution occurs.
Some representative hydrogen evolution overvoltages are as follows: platinized platinum, zero or small; smooth platinum, 0.09; silver, 0.15; copper, 0.23; lead, 0.64; zinc, 0.70; and mercury, 0.78 (for zero current). An interesting observation is that the hydrogen overvoltage parallels the heat of adsorption of atomic hydrogen on the metal. The overvoltage thus seems to be related to the energy required to produce surface hydrogen atoms as the intermediate to H2 formation.
β . Theory of Overvoltage
The excess of an applied potential over the reversible decomposition potential, or the over
voltage, is usually given the symbol 17, and Fig. 13-9 could be redrawn so as to show a plot of i versus η. However, there are three general types of contribution to an observed overvoltage, only two of which are of fundamental interest. As mentioned earlier, the linear portion of Fig.
13-9 arises primarily from the Ohm's law drop in potential across the solution and is trivial from a theoretical point of view. Second, as active decomposition occurs, electrolysis products accumu-