D . L . K E P E R T
University of Western Australia, Nedlands, Western Australia
a n d K . V R I E Z E
Koninklijke Shell-Lahoratorium, Amsterdam (Shell Research N.V.), The Netherlands 1. Introduction . . . . . . . . . . . . . . . . . . . . 1 2. F a c t o r s Influencing Metal-Metal B o n d F o r m a t i o n . . . . . . . . 4
3. Compounds B a s e d o n t h e (MeXia)^^ Core 10 A . Introduction . . . . . . . . . 10
B . Monomeric Complexes . . . . . . . . . . . . . . . . 12 C. B i n a r y H a l i d e s . . . . . . . . . . . . . . . . . . 15 D . Magnetism, Spectra a n d B o n d i n g . , . . . . . . . . . . 16 4. Compounds B a s e d o n t h e (MeX8)*+ Core . . . . . . . . . . . . 26 A . Introduction . . . . . . . 26 B . Monomeric Complexes . . . . . . . . . . . . . 28
C. B i n a r y H a l i d e s 31 D . Spectra, Magnetism a n d B o n d i n g . . . . . . . . . . . . 32
5. Compounds B a s e d o n t h e (Re3X3)e+ Core . . . . . . . . . . 3 4 A. I n t r o d u c t i o n . . . . . . . . . . . . . . . . . . 34 B . Monomeric Complexes . . . . . . . . . . . . . . . . 36
C. B i n a r y HaHdes 39 D . Spectra, Magnetism a n d B o n d i n g . . . . . . . . . . . . 40
6. Other Compounds Containing Multi-centred M e t a l - M e t a l B o n d i n g . . . . 4 4 A . Introduction . . . . . . . . . . . . . . . . . . 44 B . H a l o g e n Bridges . . . . . . . . . . . . . . . . 45
C. O x y g e n Bridges 47 D . N o Bridging A t o m s . . . . . . . . . . . . . . . . 51
References . . . . . . . . . . . . . . . . . . . . 51
1 . Introduction
T h e lower halides of n i o b i u m , t a n t a l u m , m o l y b d e n u m , t u n g s t e n a n d r h e n i u m are b a s e d on cores containing clusters of m e t a l a t o m s t i g h t l y b o n d e d together, a n d t h e y exhibit a n u m b e r of p r o n o u n c e d chemical similarities. T h e c h e m i s t r y of t h e s e a n d closely r e l a t e d c o m p o u n d s will be discussed in t h i s review.
These lower halides are fairly readily p r o d u c e d b y r e d u c t i o n or 1
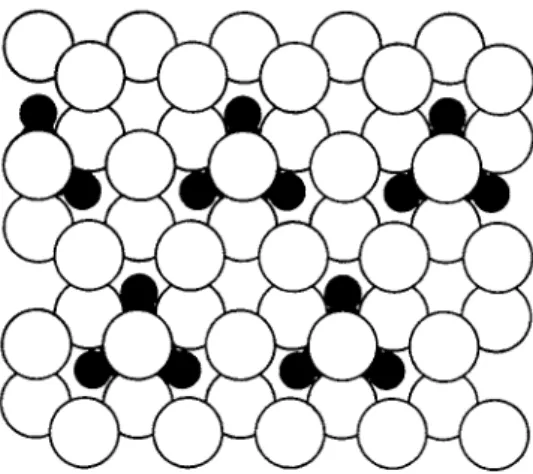
F I G . 1. T h e (MeXia)^"*" core. T h e m e t a l a t o m s in t h e octahedral cluster are s h o w n a s s h a d e d circles,while t h e bridging halogen a t o m s a b o v e t h e octahedral edges are u n s h a d e d .
F I G . 2. T h e (MeXg)*^ core. T h e m e t a l a t o m s in t h e octahedral cluster are s h o w n a s shaded circles, while t h e bridging halogen a t o m s a b o v e t h e octahedral faces are u n s h a d e d .
t h e r m a l disproportionation of t h e higher halides, a n d it h a s been k n o w n for a long t i m e t h a t t h e y are soluble in a q u e o u s acids from which well defined crystalline acids, a n d o t h e r derivatives, can be o b t a i n e d . H o w e v e r , t h e y are oxidized in alkaline solutions w i t h t h e evolution of h y d r o g e n . T h e n a t u r e of t h e s e c o m p o u n d s a w a i t e d t h e d e t e r m i n a t i o n of t h e s t r u c t u r e s of some of t h e derivatives of these acids, a n d t h e e v e n m o r e recent s t r u c t u r a l d e t e r m i n a t i o n s carried o u t on t h e halides t h e m selves. All c o m p o u n d s were found t o be b a s e d on central cores com
prising a central cluster of m e t a l a t o m s , w i t h bridging halogen a t o m s . T h e n i o b i u m a n d t a n t a l u m c o m p o u n d s are based on cores of t h e t y p e (McXig)^"^, consisting of a n o c t a h e d r a l cluster of m e t a l a t o m s w i t h a halogen a t o m a b o v e each o c t a h e d r a l edge (Fig. 1). T h e m o l y b d e n u m
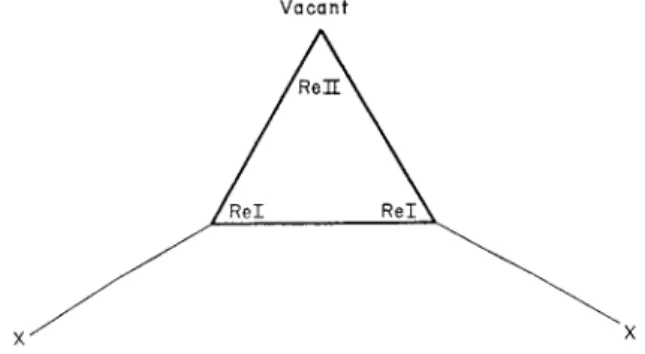
F I G . 3. T h e (Re3X3)6+ core. T h e m e t a l a t o m s in t h e triangular cluster are s h o w n as s h a d e d circles, while t h e bridging halogen a t o m s in t h e plane are u n s h a d e d .
suggesting s t r o n g m e t a l - m e t a l b o n d i n g . T h e bridging halogen a t o m s of t h e core are relatively non-labile. Useful trivial n a m e s for t h e s e cores are, for e x a m p l e , c h l o r o m o l y b d e n u m i n s t e a d of t h e m o r e s y s t e m a t i c octa-/x3-chlorohexamolybdenum(II) for t h e (MoeCls)^^ core, a n d chloro- n i o b i u m - i n s t e a d of dodeca-/x2-chlorohexaniobium (2-33) for t h e
(NbeCli2)^+ core.
T h e individual m e t a l a t o m s of t h e cluster can readily a d d a d d i t i o n a l d o n o r g r o u p s , including halide ions, forming, for e x a m p l e , t h e anionic complexes [(Nb6Cli2)Cl6]4-, [(Mo6Cl8)Cl6]2- a n d [(Re3Cl3)C]9]3-. T h e s y s t e m a t i c n a m e s for t h e salts would b e , for e x a m p l e , p o t a s s i u m h e x a - chloro-octa-jLt3-chlorohexamolybdate(II). A wide v a r i e t y of o t h e r ligands can o c c u p y t h e s e positions resulting in a fairly complex c h e m i s t r y . F o r e x a m p l e , reaction w i t h n e u t r a l u n i d e n t a t e ligands in suitable solvents p r e c i p i t a t e t h e non-electrolytes [(Nb6Cli2)Cl2(ligand)4], [(Mo6Cl8)Cl4(ligand)2] a n d [(Re3Cl3)Cl6(ligand)3] respectively. I n t h i s review, t h e central core containing t h e m e t a l a t o m s a n d t h e non-labile bridging halogen a t o m s will be s u r r o u n d e d b y p a r e n t h e s e s , a n d t h e o u t e r ligands b y s q u a r e b r a c k e t s as h a s been used a b o v e . I n t h e case of t h e o c t a h e d r a l m e t a l clusters, t h e o u t e r ligands are directed a w a y from t h e c e n t r e of t h e o c t a h e d r o n , a n d m a y for convenience b e called ''centrifugal" ligands. I n t h e case of (Re3Cl3)^+, each r h e n i u m a t o m can further coordinate t h r e e ligands, a n d only t h e one in t h e p l a n e of t h e r h e n i u m a t o m s will b e t e r m e d t h e centrifugal ligand. This distinction b e t w e e n halogen a t o m s inside a n d outside t h e core is preferred t o
" b r i d g i n g " a n d ' ' n o n b r i d g i n g " , as t h e b i n a r y halides a r e polymeric w i t h bridging centrifugal halogen a t o m s . T h e s y s t e m a t i c n a m i n g of t h e b i n a r y halides is m o r e difficult, a n d will n o t b e a t t e m p t e d .
a n d t u n g s t e n c o m p o u n d s a r e also b a s e d on o c t a h e d r a l clusters of m e t a l a t o m s , b u t in t h i s case t h e bridging halogen a t o m s lie a b o v e e a c h octa
h e d r a l face, a n d t h e formula m a y b e w r i t t e n ( M 6 X 8 ) ^ + (Fig. 2). T h e r h e n i u m c o m p o u n d s h a v e (RegXg)^^ cores, consisting of a t r i a n g u l a r cluster of r h e n i u m a t o m s w i t h a halogen a t o m outside e a c h edge (Fig. 3). T h e m e t a l - m e t a l distances are shorter t h a n in t h e m e t a l s .
The preparation of compounds of these metals in the same oxidation states but from sources other than the halides (which in turn are pre
pared at elevated temperatures) do not contain clusters of metal atoms, although they m a y contain metal-metal bonds. F o r example, reduction of p e r r h e n a t e in t h e presence of neutral ligands leads to ReClg.S E t g P h P , ReCl3(Et2PhP) (PhgP CH2 CH2 PPhg), [ReCl2(Ph2P CHg CH2 PPh2)2]Cl (Chatt and Rowe, 1962) and [ReX2(o-CeH4(AsMe2)2)2]X where X is CI, Br or I (Curtis et al,, 1958). However, under other conditions the anions Re2Cl8^~ and Re2Br8^" can be obtained as salts with a variety of inorganic and organic cations (Cotton et al., 1965).
The structure shows extremely short rhenium-rhenium distances of 2-24 Â (Cotton and Harris, 1965), which are even shorter than in the (RcgCla)^"^ complexes discussed in this review, and is thought to involve the first quadruple bond between a n y two atoms (Cotton, 1965).
Related compounds containing the Tc2Cl8^~ anion have also been iso
lated (Eakins et al, 1963) (Cotton and B r a t t o n , 1965), Similarly the formally (i^-Mo(0Ac)2 prepared from molybdenum hexacarbonyl con
tains two molybdenum atoms only 2-11 Â apart, with four bridging b i d e n t a t e acetate groups (Stephenson et al., 1964; L a w t o n and Mason, 1965). However, a large number of compounds of Mo(II) w i t h o u t m e t a l - metal bonds can be obtained from t h e carbonyl or by reduction of aque
ous solutions of M o ( I I I ) , for example [MoBr2(CO)3(o-C6H4(AsMe2)2)]
and [MoBr2(o-C6H4(AsMe2)2)2] respectively (Nigam et al., 1960; Lewis et al, 1962, 1963).
2. Factors Influencing Metal-Metal Bond Formation
A number of compounds will be quoted in this section as having metal-metal bonds, but without quoting the original literature refer
ences. F o r full details of those metal-metal bonded compounds without clusters of metal atoms, the reader is referred to a more general review of metal-metal bonds (Kepert et al, 1967).
T h e distribution of metal-metal bonding throughout the periodic system can be discussed from two points of view: from thermodynamic considerations, or from the size of the bonding orbitals and the extent of metal orbital-metal orbital overlap.
T h e thermodynamics of the systems will be considered first. A n illustrative approach is to calculate the thermodynamic stability of the hypothetical ''ionic dihalides" and "ionic trihalides" of the groups I V , V, V I and V I I transition metals through the simple Born-Haber cycle ( K e p e r t et al, 1967; Vrieze, 1964). F o r example the heat of f o r m a t i o n of the hypothetical "ionic WClg" is equal to the sum of the atomization energy and the first and second ionization potentials of tungsten, the
dissociation energy a n d twice t h e n e g a t i v e of t h e electron affinity of chlorine, a n d t h e n e g a t i v e v a l u e of t h e ionic lattice energy, assuming t h a t it would be isomorphous w i t h CrClg. T h e result shows t h a t t h e h e a t of formation would be +dS kcal mole-^ a t 298°K, t h e positive value indicating t h a t it would be u n s t a b l e w i t h respect t o t h e elements.
I n t h e case of t h e h y p o t h e t i c a l "ionic MoClg", a l t h o u g h t h e h e a t of f o r m a t i o n w o u l d b e n e g a t i v e (—7 kcal mole-^) a n d i t w o u l d b e stable w i t h respect t o t h e elements, calculations show t h a t it would be u n stable t o disproportionation, each mole of "ionic MoClg" liberating 30 kcal on formation of t h e p e n t a h a l i d e a n d t h e m e t a l . T h e experi
m e n t a l h e a t of formation of M0CI2 is —44 kcal mole-^ a n d it is stable w i t h respect t o d i s p r o p o r t i o n a t i o n .
Similar calculations show t h a t t h e h y p o t h e t i c a l " i o n i c " dihalides
"ZrCla", "HfClg", "NbCla", "TaCl^" a n d "ReClg", as well as t h e h y p o thetical " i o n i c " trihalides "HfCl3", "NbCla", "TaClg", "WCI3" a n d
"ReCla" would b e u n s t a b l e w i t h respect t o t h e elements a n d / o r t o disproportionation. T h e h y p o t h e t i c a l " i o n i c " trihalides "ZrCl3",
"M0CI3" a n d "TcClg" would be e x p e c t e d t o b e t h e r m o d y n a m i c a l l y stable, b u t of course t h i s does n o t rule o u t t h e possibility of gaining e x t r a stability t h r o u g h t h e formation of m e t a l - m e t a l b o n d s .
A l t h o u g h t o o m u c h reliance should n o t be placed on t h e s e detailed figures, these B o r n - H a b e r cycle calculations do p o i n t t o t h e m o r e i m p o r t a n t energy factors, for e x a m p l e , a l t h o u g h t h e ionization p o tentials a r e large, t h e y a r e u n i m p o r t a n t , as t h e y will be t h e s a m e w h e t h e r or n o t t h e halide formed h a s m e t a l - m e t a l b o n d s . T h e m a i n factor which causes these h y p o t h e t i c a l halides t o be u n s t a b l e is t h e high a t o m i z a t i o n energy of t h e second a n d t h i r d row t r a n s i t i o n m e t a l s . T h e lattice energy is of lesser i m p o r t a n c e , a l t h o u g h t h e high lattice energy of t h e fluorides suggests t h a t for c o m p o u n d s which are b a s e d on one of t h e n o r m a l fluoride s t r u c t u r e s , m e t a l - m e t a l b o n d s will n o t form because of t h e rigidity of t h e lattice, a l t h o u g h m e t a l - m e t a l b o n d s c o m m o n l y cause distortion of lattices containing m o r e polarizable anions. This a r g u m e n t does n o t of course a p p l y t o t h e m u l t i c e n t r e d m e t a l - m e t a l b o n d e d cluster n o t b a s e d on close p a c k e d anion s t r u c t u r e s which is t h e m a i n t h e m e of this review, as shown b y NbFg.s which h a s o c t a h e d r a l clusters of niobium a t o m s .
Since these calculations show t h a t t h e a t o m i z a t i o n energy is t h e m o s t i m p o r t a n t factor in t h e destabilization of t h e " i o n i c " halides m e n t i o n e d a b o v e , a n d since t h e s e halides h a v e all b e e n found t o c o n t a i n m e t a l - m e t a l b o n d s , it is worthwhile t o look i n t o t h i s q u a n t i t y , t h e a t o m i z a t i o n energy, in m o r e detail.
A n interesting theoretical a p p r o a c h t o t h e cohesive energies of
t r a n s i t i o n m e t a l s h a s been given b y Griffith (1956). T h e a s s u m p t i o n s in his t h e o r y are t h a t t h e valence electrons in t h e gaseous a t o m s a r e coupled as far as possible w i t h parallel ones, while in t h e m e t a l t h e electrons are coupled as far as possible antiparallel t o electrons from o t h e r m e t a l a t o m s . T h e essential features of his t h e o r y are t h a t in order t o form a m e t a l in which all t h e electrons are coupled antiparallel, it is necessary t o b r e a k d o w n t h e parallel i n t r a - a t o m i c spin couplings.
Neglecting a small crystal field contribution, Griffith derives a formula s t a t i n g t h a t t h e a t o m i z a t i o n energy ΔΗ is t h e s u m of t w o c o m p o n e n t s :
-ΔΗ = p + Ρ
I n t h i s formula Ρ is positive a n d r e p r e s e n t s t h e energy required t o bring t h e a t o m s i n t o a n o n - s t a t i o n a r y s t a t e M, in w h i c h t h e i n t r a - a t o m i c spin couplings are uncoupled. This q u a n t i t y could be calculated for t h e first t r a n s i t i o n m e t a l series. T h e q u a n t i t y p, which is n e g a t i v e , is t h e h e a t of formation of t h e m e t a l from t h e m e t a l a t o m s in s t a t e M.
ρ w a s calculated as t h e s u m oi ΔΗ a n d Ρ a n d varies s m o o t h l y along t h e first t r a n s i t i o n m e t a l series w i t h m a x i m a a t v a n a d i u m a n d m a n g a n e s e a n d w i t h a small d i p a t c h r o m i u m . T h e q u a n t i t y ρ per u n p a i r e d valence electron varies b e t w e e n 30 a n d 40 kcal a n d is a m o r e funda
m e n t a l q u a n t i t y t h a n t h e a t o m i z a t i o n energy per valence electron.
T h e q u a n t i t y Ρ was n o t calculated for t h e second a n d t h i r d t r a n s i t i o n m e t a l series, b u t it a p p e a r s t o decrease on m o v i n g d o w n t h e columns of t h e periodic s y s t e m , t h a t is, t h e i n t r a - a t o m i c spin couplings are b r o k e n m o r e easily. This decrease is i n d i c a t e d b y t h e absence of residual u n p a i r e d spins in t h e second a n d t h i r d row t r a n s i t i o n m e t a l s , a n d in t h e m u c h g r e a t e r t e n d e n c y of their c o m p o u n d s t o form m e t a l - m e t a l b o n d s , b o t h in t h e lower a n d in t h e higher oxidation s t a t e s .
C h r o m i u m a n d m a n g a n e s e are therefore r a t h e r averse t o forming m e t a l - m e t a l b o n d s , n o t because of t h e r a t h e r low a t o m i z a t i o n energy, b u t because t h e factor Ρ is v e r y large for t h e s e elements.
A l t h o u g h t h e a b o v e a r g u m e n t is a v e r y q u a l i t a t i v e one a n d neglects factors such as spin-orbit coupling which becomes m o r e i m p o r t a n t in t h e heavier elements, a n d also t h e influence of ligands a n d anion packing, it affords some insight i n t o t h e f u n d a m e n t a l difference in b e h a v i o u r b e t w e e n t h e c o m p o u n d s of t h e first r o w t r a n s i t i o n m e t a l series on t h e one h a n d a n d t h e c o m p o u n d s of t h e second a n d t h i r d row t r a n s i t i o n m e t a l series o n t h e o t h e r h a n d .
Some a t t e m p t s h a v e been m a d e t o e s t i m a t e t h e m e t a l - m e t a l b o n d energies in c o m p o u n d s . Schafer a n d Schnering (1964), for e x a m p l e , h a v e t a k e n t h e a t o m i z a t i o n energy, which is a less f u n d a m e n t a l q u a n t i t y t h a n t h e q u a n t i t y ^ , as a m e a s u r e of t h e b o n d energy in t h e
2 4 0
2 0 0
160
<3 120
Τ Ί
• Nb-O
2 3 Formal v a l e n c e
F I G . 4. H e a t s of formation ΔΗ of v a n a d i u m , n i o b i u m a n d t a n t a l u m chlorides a n d oxides as a function of stoichiometry.
metallic s t a t e . T h e y considered t h a t t o form chlorides containing m u l t i - centred clusters of m e t a l a t o m s , t h e m e t a l - m e t a l b o n d energy m u s t b e g r e a t e r t h a n t h e m e t a l - c h l o r i n e b o n d energy (which is a b o u t 80-90 kcal mole-^ for a n y t r a n s i t i o n m e t a l chloride), t h a t is, t h e y m u s t be restricted t o t h e second a n d t h i r d rows of t h e t r a n s i t i o n m e t a l s , from zirconium t o a b o u t p a l l a d i u m a n d from hafnium t o a b o u t gold respectively.
H o w e v e r , A r i y a a n d K h e r n b u r g (1964) h a v e found t h a t t h e b o n d i n g energy per electron in t h e metallic s t a t e is c o n s t a n t for a large n u m b e r of m e t a l s . T h e y observed t h a t for a n u m b e r of m e t a l s t h e h e a t s of formation of t h e oxides are p r o p o r t i o n a l t o t h e degree of oxidation, as shown b y t h e s t r a i g h t line o b t a i n e d for t h e n i o b i u m oxides in F i g . 4. I t was concluded t h a t t h e g r a d u a l o x i d a t i o n of t h e m e t a l results in t h e g r a d u a l r e p l a c e m e n t of m e t a l - m e t a l b o n d s b y m e t a l - o x y g e n b o n d s . Similar s t r a i g h t line plots were o b t a i n e d , for e x a m p l e , w i t h t i t a n i u m a n d t u n g s t e n oxides. H o w e v e r , o t h e r m e t a l s such as v a n a d i u m show a d i s c o n t i n u i t y (Fig. 4) indicating t h a t a t a certain degree of o x i d a t i o n all t h e m e t a l - m e t a l b o n d s are b r o k e n a n d n o further energy is available
from t h i s source. T h e position of t h i s b r e a k w a s considered t o be t h e valency of t h e a t o m in t h e metallic s t a t e which w a s found t o agree w i t h e s t i m a t e s derived from t h e m a g n e t i c p r o p e r t i e s of alloys. If t h e n u m b e r of valency electrons w a s divided i n t o t h e a t o m i z a t i o n e n e r g y a c o n s t a n t figure of 30-40 kcal p e r b o n d i n g electron w a s o b t a i n e d . F o r e x a m p l e , c h r o m i u m w a s found t o h a v e t h r e e b o n d i n g electrons p e r a t o m , m o l y b d e n u m t o h a v e four or five b o n d i n g electrons, a n d t u n g s t e n t o h a v e six b o n d i n g electrons.
T h e s a m e t r e n d s are shown if t h e h e a t s of f o r m a t i o n of t h e chlorides (Schafer a n d Schnering, 1964) are used i n s t e a d of t h e oxides. T h e p l o t for t h e v a n a d i u m chlorides shows a b r e a k in a b o u t t h e s a m e position a s for t h e v a n a d i u m oxides (Fig. 4). A linear p l o t is again o b t a i n e d for niobium (and t a n t a l u m ) despite t h e profound s t r u c t u r a l changes which a c c o m p a n y chlorination:
Stoichiometry Compound Structure N b - N b D i s t a n c e (A)
N b Metal B o d y centred cubic 2-86
NbCl2.33 NbeCli4 Nbg Octahedra 2-89 a n d 2-95
NbCla.67 Nb3Cl8 Nbg Triangles 9.7»
NbCl3.i3 Nb^.ssCle D e f e c t NbgClg ύ ί Ό
NbCl^ (NbCl4)oo N b - N b Pairs 2-94
NbClg Nb^Clio N o N b - N b B o n d s —
I t is concluded t h a t t h e m e t a l - m e t a l b o n d energy is insensitive t o small changes in i n t e r n u c l e a r s e p a r a t i o n .
A l t h o u g h t h e conclusions of A r i y a a n d K h e r n b u r g a b o u t t h e v a l e n c y m a y b e a b i t t o o optimistic, it can be observed from t h e s e t h e r m o d y n a m i c plots t h a t t h e first row t r a n s i t i o n m e t a l s are r e l u c t a n t t o u n c o u p l e t h e i n t r a - a t o m i c spin couplings. These w o r k e r s d r e w t h e interesting conclusion t h a t t h e m e t a l - m e t a l b o n d energy p e r electron seems t o b e a b o u t t h e s a m e for t h e m e t a l a t o m s of t h e s y s t e m s i n v e s t i g a t e d , t h a t is, t h e s a m e for all t h r e e t r a n s i t i o n m e t a l rows. I t would therefore seem worthwhile t o calculate t h e ρ v a l u e s for t h e second a n d t h i r d r o w t r a n sition m e t a l s , where possible, in order t o see if t h e y would b e t h e s a m e as t h e respective jp values of t h e first t r a n s i t i o n m e t a l s in t h e s a m e column.
H a v i n g n o w discussed t h e t h e r m o d y n a m i c s which h a v e given us a r o u g h idea of t h e essential energy factors which are favourable for m e t a l - m e t a l b o n d formation, i t is enlightening t o discuss t h i s subject from t h e a t o m i c p o i n t of view.
Several a u t h o r s h a v e tried t o use semi-empirical e q u a t i o n s r e g a r d i n g t h e size of orbitals involved in t h e m e t a l - m e t a l b o n d i n g . F o r e x a m p l e ,
Sheldon (1964b) calculates t h e r a d i u s of t h e b o n d i n g electron b y using Slater's rules, a n d finds t h a t m e t a l - m e t a l b o n d i n g occurs if t h e m e t a l - m e t a l d i s t a n c e is less t h a n 0-4 Â g r e a t e r t h a n twice t h e o r b i t a l r a d i u s . G o o d e n o u g h (1963) also uses a semi-empirical a p p r o a c h a n d found t h a t t h e critical d i s t a n c e b e y o n d which m e t a l - m e t a l b o n d i n g c a n n o t occur for a first r o w t r a n s i t i o n m e t a l is given b y :
R,{U) - [3-05 - 0·03(Ζ - Ζτι)] Â
w h e r e a n d Ζ a r e t h e a t o m i c n u m b e r s of t i t a n i u m a n d t h e o t h e r 3d-element in question, respectively. ( I t is a s s u m e d in t h i s expression t h a t t h e electrons d o n o t b e c o m e completely delocalized on m e t a l - m e t a l b o n d formation.)
Similarly,
i? e( 4 d ) - R,{U) + 0-88 Â Rci^d) - Roi^d) + 1-36 Â
T h e s e schemes h a v e a limited p r e d i c t i v e ability for c o m p o u n d s b a s e d o n close p a c k i n g of t h e non-polarizable oxide or chloride ions w h e r e t h e d i s t a n c e b e t w e e n t h e m e t a l a t o m s is p a r t l y g o v e r n e d b y a n i o n p a c k i n g , for e x a m p l e for t h e clusters t h a t occur in NbgClg a n d ZnaMogOg. H o w ever, t h e a b o v e criteria h a v e n o p r e d i c t i v e ability for t h e f o r m a t i o n of clusters of m e t a l a t o m s w h e r e t h e r e is n o r e s t r a i n t o n t h e m e t a l a t o m s a p p r o a c h i n g e a c h o t h e r .
T h e factors influencing t h e size of t h e m e t a l orbitals a n d c o n s e q u e n t l y t h e m e t a l o r b i t a l - m e t a l o r b i t a l o v e r l a p are t h e effective nuclear charge on t h e m e t a l a t o m , w h i c h is d e p e n d e n t u p o n t h e o x i d a t i o n s t a t e , a n d t h e ligands b o n d e d t o t h e m e t a l a t o m . F o r e x a m p l e , a single σ electron pair m e t a l - m e t a l b o n d is f o u n d in t h e isoelectronic [Fe^^(C0)8]^~, Co§(CO)8 a n d [Ni2(CN)6]^-, w h e r e a s t h e isoelectronic Cu^^ forms n o m e t a l - m e t a l b o n d s of t h i s t y p e d u e t o its higher o x i d a t i o n s t a t e a n d smaller orbital size.
I f a n isovalent series is n o w considered, for e x a m p l e TigOg, VgOg, CrgOg, t h e progressively poorer shielding of t h e increasing n u c l e a r charge b y t h e a d d i t i o n a l cZ-electrons will r e s u l t in a c o n t r a c t i o n of t h e iZ-electron cloud. T h u s t h e first t w o m e m b e r s of t h i s series form m e t a l - m e t a l b o n d s , b u t n o t CrgOg. Similarly, TiOg c o n t a i n s infinite strings of t i t a n i u m a t o m s close t o g e t h e r a n d shows metallic b o n d i n g w h e n d o p e d w i t h T i ( I I I ) . VO2 h a s V - V p a i r s which a r e easily b r o k e n b y h e a t i n g t o a b o u t 70'', while n o m e t a l - m e t a l b o n d i n g occurs in CrOg (Morin, 1959).
T h e d i s t a n c e over which m e t a l - m e t a l b o n d s can s p a n is also s t r o n g l y d e p e n d e n t u p o n t h e anion. A n increase in polarizability of t h e a n i o n s
increases t h e screening of t h e ίί-electrons from t h e nucleus, allowing t h e d-orbitals t o e x p a n d . On t h e o t h e r h a n d in close p a c k e d anion lattices, t h e larger m o r e polarizable anions force t h e cations further a p a r t . W h i c h of t h e t w o factors is m o r e i m p o r t a n t is difficult t o d e t e r m i n e . H o w e v e r , t h e y seem t o cancel each o t h e r fairly well, as is shown b y t h e fairly large r a n g e of i n t e r n u c l e a r distances over which m e t a l - m e t a l b o n d i n g can occur. F o r e x a m p l e , T i ( I I I ) - T i ( I I I ) b o n d i n g isfoundinTigOgiTi—Ti
= 2 - 5 9 Â ) , jS-TiClg (2-96 A), TiBrg (3-05 Â), Tilg (3-23 A), c o m p a r e d w i t h 2-90 A in t h e m e t a l . This feature is also found w i t h o t h e r polariz
able anions, for e x a m p l e t h e t r i a n g u l a r clusters of iron a t o m s in F e S are 3-08 A from each other, c o m p a r e d w i t h 2-48 A in t h e m e t a l .
T h e combined effect of increasing size of orbitals, a n d m o r e impor
t a n t l y t h e increasing n u m b e r of electrons available for m e t a l - m e t a l bonding, is shown in t h e series NbCI^, a n d MoCl^; as χ is decreased. T h u s m e t a l a t o m s occur as pairs in NbCl4, t r i a n g u l a r triplets in NbClg-e? a n d o c t a h e d r a l clusters in NbClggg. Similarly no m e t a l - m e t a l b o n d s are found in M0CI5, pairs of m o l y b d e n u m a t o m s occur in M0CI4 a n d MoClg, while o c t a h e d r a l clusters of m e t a l a t o m s occur in MoClg. T h e ability t o form m e t a l - m e t a l b o n d s , however, will reach a m a x i m u m a n d t h e n decrease as t h e n u m b e r of electrons in t h e valence shell increases. This can be seen from t h e series TaClg, ReClg, OsClg, IrClg, which h a v e 2, 4, 5 a n d 6 iZ-electrons p e r m e t a l a t o m respectively. T h e first t w o m e m b e r s contain m e t a l - m e t a l b o n d s while IrClg h a s a filled dc shell which c a n n o t be used for m e t a l - m e t a l b o n d i n g .
Finally, Cotton a n d H a a s (1964) h a v e p o i n t e d o u t t h a t t h e t y p e of m u l t i c e n t r e - b o n d e d m e t a l - m e t a l cluster will be limited if t h e species a r e n o t t o a t t a i n w h a t t h e y consider t o b e a n i m p r o b a b l y large charge.
F o r e x a m p l e , if t h e formally d^ Mo(II) a n d R e ( I I I ) in t h e cores (MogClg)^"^ a n d (RegClg)^+ were t o a d o p t each o t h e r ' s s t r u c t u r e , we would h a v e t h e h y p o t h e t i c a l cores (MogClg)'^^ a n d (RegCig)^^"^. T h e former would be unlikely t o form [(Mo3Cl3)Cl9]^~ w i t h t h e negatively charged chloride ion, while t h e charge on t h e l a t t e r was considered t o b e i m p r o b a b l y high.
3. Compounds Based on the {M^X-^^)^^ Core A. Introduction
T h e lowest halides formed b y t h e r e d u c t i o n of n i o b i u m a n d t a n t a l u m p e n t a h a l i d e s u n d e r fairly vigorous conditions a r e :
NbFa-s NbCl2.33 NbBrg Nbia?
— TaClg.s TaBr^.s Tal2.33
T h e formulation a n d s t r u c t u r e of t h e s e halides h a v e only r e c e n t l y been d e t e r m i n e d (Schafer a n d Schnering, 1964), a l t h o u g h t h e i r reactions h a v e b e e n s t u d i e d for some t i m e . F o r a n u n d e r s t a n d i n g of t h e n a t u r e of these c o m p o u n d s , a n d for historical reasons, it is c o n v e n i e n t t o discuss t h e n a t u r e of t h e d e r i v a t i v e s before dealing w i t h t h e halides t h e m s e l v e s . F o r t h e p r e p a r a t i o n of t h e d e r i v a t i v e s it is n o t necessary t o isolate t h e p u r e halides t h e m s e l v e s , a n d n o r m a l l y t h e r e d u c e d m a s s of t h e chlorides or b r o m i d e s is e x t r a c t e d a n d recrystallized from a q u e o u s hydrochloric or h y d r o b r o m i c acids respectively, yielding t h e c o m p o u n d s NbgCli4.
7H2O, NbgBri^.VHaO, Ta6Cli4.7H20 a n d Ta6Bri4.7H20 respectively.
Metallic c a d m i u m a p p e a r s t o be a p a r t i c u l a r l y suitable reducing a g e n t for t h e p e n t a c h l o r i d e s a n d p e n t a b r o m i d e s , a n d w i t h t h e exception of t h e b r o m o n i o b i u m c o m p o u n d , a b o u t 3 0 % yields h a v e been o b t a i n e d ( H a r n e d et al,, 1960). T h e only r e a c t i o n w i t h o t h e r halides a p p e a r s t o be t h e o b s e r v a t i o n t h a t Talg-aa is stable in acid solution w i t h a green colour b u t decomposes in alkaline solution w i t h t h e evolution of h y d r o gen, in a n e x a c t l y analogous m a n n e r t o t h e chloro a n d b r o m o com
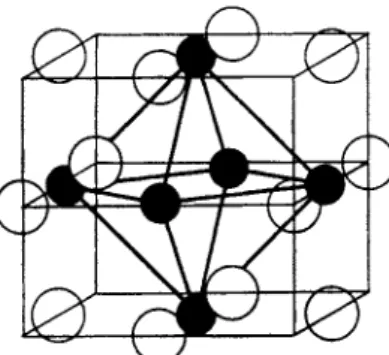
p o u n d s i K o r o s y , 1939). A l t h o u g h t h e s t r u c t u r e of t h e s e p a r t i c u l a r species h a v e n o t b e e n d e t e r m i n e d , t h e r e is n o d o u b t t h a t t h e s t r u c t u r e is b a s e d on t h e (MeXi2)^+ core s h o w n in Fig. 1, where t h e six m e t a l a t o m s are s i t u a t e d a t t h e corners of a n o c t a h e d r o n , w i t h a halogen a t o m a b o v e each o c t a h e d r a l edge, each halogen bridging t w o m e t a l a t o m s .
A v a r i e t y of d o n o r molecules can a p p a r e n t l y a d d o n t o t h e (NL^^^'^+
cage, one t o each m e t a l a t o m , in a similar m a n n e r as h a s b e e n m o r e conclusively d e m o n s t r a t e d w i t h t h e c o m p o u n d s containing t h e analo
gous (MoeClg)^^ a n d (Re3Cl3)^+ cages, indicating t h a t t h e c o m p o u n d s should be considered t o b e composed of [(M6Xi2)L6]'^- ions, where L h a s been found t o be a n e u t r a l or negatively charged ligand (Fig. 5).
T h e c o m p o u n d s of t h e t y p e Nb6Cli4.7H20 r e t a i n t h e i r w a t e r t e n a ciously, a n d do n o t completely d e h y d r a t e even a t 300°, suggesting t h a t some of t h e w a t e r is strongly b o n d e d t o t h e m e t a l a t o m s . H o w e v e r , care m u s t be t a k e n a b o u t t h e formulation of t h e s e c o m p o u n d s a n d t h e i r derivatives, as m a n y of t h e essential properties h a v e n o t been m e a s u r e d . F o r e x a m p l e , a l t h o u g h t h e a b o v e c o m p o u n d is conveniently f o r m u l a t e d as [(Nb6Cli2)Cl2(H20)4]3H20, t h e necessary chemical a n d physical properties h a v e n o t b e e n d e t e r m i n e d .
I t should b e n o t e d t h a t t h e d e r i v a t i v e s p r e p a r e d h a v e t h e a v e r a g e m e t a l a t o m in t h e formal o x i d a t i o n s t a t e of 2-33. H o w e v e r , t h e y a p p e a r t o b e p r e p a r e d from b i n a r y halides where t h e m e t a l a t o m s h a v e formal o x i d a t i o n s t a t e s of 2-00, 2-33 a n d 2-50. A s t u d y of t h e o x i d a t i o n - r e d u c t i o n processes is therefore clearly i n d i c a t e d , a n d it should also b e possible t o p r e p a r e d e r i v a t i v e s w i t h o t h e r formal o x i d a t i o n s t a t e s . I t
F I G . 5. T h e structure of [(M6Xi2)(Hgand)6]^". One Hgand a t o m (hatched ch-cle) is a t t a c h e d t o each m e t a l a t o m of Fig. 1 in a centrifugal position.
T h e correct formulation of t h e s e c o m p o u n d s h a s always been a n interesting problem, d u e t o t h e non-integral n u m b e r of t h e valency of t h e average m e t a l a t o m . T h e m a g n e t i s m a n d m o d e of b o n d i n g in these c o m p o u n d s will be discussed separately after t h e chemistry h a s been summarized.
B. Monomeric complexes
T h e ' ' h y d r a t e d t a n t a l u m dichloride" was first p r e p a r e d b y Chabrie (1907) who r e d u c e d t a n t a l u m pentachloride w i t h 3 % sodium a m a l g a m a t red h e a t , a n d e x t r a c t e d t h e m i x t u r e w i t h hydrochloric acid. T h e c o m p o u n d is green, b u t t u r n s b r o w n on prolonged exposure t o air, a n d was formulated as TaCl2.2H20.
Chapin (1910) similarly p r e p a r e d this c o m p o u n d a n d no less t h a n 20 g of t h e corresponding b r o m i d e b y reduction w i t h h y d r o g e n , b u t formu
lated t h e p r o d u c t s as Ta6Cli4.7H20 a n d Ta6Bri4.7H20 respectively. T h e h a s recently been shown (McCarley et al, 1964) t h a t Τ8ϋοΒτΐ2^+ can b e oxidized w i t h h y d r o g e n peroxide t o form Ta>QBr^2^+,
compounds were correctly described as h e x a m e r i c species on the basis of a molecular weight determination on the bromo compound in boiling propanol (found 1750, TagBri4.7H20 requires 2332). A molecular weight determination in freezing water (found 720) indicated that this com
pound dissociated into three ions. This was confirmed b y showing t h a t only t w o of the bromide ions in Ta6Bri4.7H20 could be precipitated with silver nitrate from cold aqueous solutions, and also that only t w o bromide ions were replaceable after repeated evaporation with hydro
chloric acid. T h e compounds Ta6Bri2(OH)2.10H2O and Ta6Bri2l2.71120 were similarly obtained. More recently (Allen and Sheldon, 1965) it has been shown that these compounds are 2:1 electrolytes in water and ethanol.
H a r n e d ( 1 9 1 3 ) similarly prepared the green chloroniobium compound, which is also obtained as the green heptahydrate Nb6Cli4.7H20. I t was again found that two of these chlorine atoms could be replaced, and Nb6Cli2(OH)2.8H20 and Nb6Cli2Br2.7H20 were prepared. H a r n e d noted that the green N b 6 C l i 4. 7 H 2 0 dissolved in alkali forming a green-brown solution, which on addition of concentrated hydrochloric acid produced a brown crystalline material thought to be N b 6 C l i 4. 9 H 2 0 . H o w e v e r , the relationship between the t w o species was not understood; the hepta
hydrate could not be formed b y dehydrating the enneahydrate, but was formed if aqueous solutions of the enneahydrate were boiled. T h e enneahydrate was less soluble than the heptahydrate. I t has been suggested (Allen and Sheldon, 1 9 6 5 ) that the brown form is similar to t h e green form, e x c e p t t h a t t h e r e is a slight r e p l a c e m e n t of t h e bridging chloride groups w i t h hydroxide groups.
L i n d n e r and F e i t (1924) again prepared the chlorotantalum com
pound, b y reduction of t h e p e n t a c h l o r i d e w i t h lead at 600°. R a t h e r t h a n formulate t h e compound in terms of t h e metal atom having a non- integral valence number, t h e y preferred t h e formula H(TaÇCl7.H20).
3H2O. T h e y found that only 1/14 of the chloride was precipitated from ethanolic solution, but 4/14 was precipitated from aqueous solution under vigorous conditions. These authors produced a number of com
plexes which appear largely analogous to the better known molybdenum compounds, but which were formulated as derivatives of the acid H(Ta3Cl7.H20).3H20, a few examples of which are shown on the left hand side of the following reaction scheme. I n the absence of critical measurements such as conductivity and molecular weight, these com
pounds can be reformulated as shown on the right hand side of t h e reaction scheme. Six centrifugal ligands have tentatively been used with compounds obtained from solution, but the compounds obtained on heating must be polymeric with bridging centrifugal ligands.
L i n d n e r a n d F e i t Possible reformulation H(Ta3Cl7.H20).3H20
1 A q u e o u s H B r
H(Ta3Cle.Br.H20).3H20
I H e a t
HiTasCle-Br.H^O)
p y in E t O H l sat. w i t h HCl
pyH(Ta3Cl,) Η(Τα3θΐ7.Η2θ).3Η2θ
ivy
(pyH)[Ta3Cl,py]
H(Ta3Cl7.H20).3H20
j E t O H
H(Ta3Cl,.EtOH).EtOH
[(TaeCl,,)Cl,(H,0)4].3H,0
I A q u e o u s H B r
[(TaeCl,2)Br,(H,0)J.3H,0
j H e a t
(TaeCli,)Br,(H,0), [(TaeCli,)Cl2(H20)J.3H,0
p y in E t O H j sat. w i t h HCl
H2(pyH)2[(TaeCli2)Cle]
j 2 9 0 °
(TaeCli2)Cl2py2
[(TaeCl,2)Cl2py4]
[(TaeCli2)Cl2(H20)4].3H20
j E t O H
[(TaeCli2)Cl2(EtOH)J
Similar c o m p o u n d s were o b t a i n e d b y r e p l a c e m e n t of t h e o u t e r centri
fugal chlorine a t o m s w i t h b r o m i n e , w h i c h can b e r e f o r m u l a t e d H2(pyH)2 [(TaeCli2)Bre], (TaeCli2)Br2py2 a n d [(TaeCli2)Br2pyJ.
Ruff a n d T h o m a s (1925) also preferred t h a t t h e m e t a l a t o m s in t h e s e c o m p o u n d s h a v e a n integral valence, b u t preferred t r i v a l e n t t a n t a l u m . F o r e x a m p l e , t h e y r e w r o t e t h e H(Ta^3Cl7.H20)3H20 of L i n d n e r a n d F e i t as ( T a | " C l 7 0) 3 H 2 0 .
T h e formulae a n d s t r u c t u r e of t h e s e c o m p o u n d s r e m a i n e d in d o u b t u n t i l V a u g h a n et al. (1950) m e a s u r e d t h e X - r a y diffraction of alcoholic solutions of NbgCli^.VHgO, TaeCli^.THgO a n d Ta6Bri4.7H20, a n d d e d u c e d t h a t t h e s t r u c t u r e s were b a s e d on a ( M 6 X i 2 ) ^ + core, w i t h t h e m e t a l a t o m s in t h e form of a n o c t a h e d r o n a n d w i t h a halogen a t o m a b o v e each edge (Fig. 1). T h e i n t e r a t o m i c distances are given below.
M — M (A) M — X (A)
Nb6Cli4.7H20 2-85 2-41
TaeCl^.TH^O 2-88 2-44
TaeBrj^.VHaO 2-92 2-62
NbeFis NbeCli4 NbeBr^^ mj.^^
(NbF^.so) (NbCl2.33) (NbBr^.oo) (Nbl^.oo)
— TaeCli5 TaeBr^s Tagl^^
— (TaCla.so) (TaBr^.^o) (Tal^.ga)
T h e s t r u c t u r e of NbgFis shows t h e presence of (NbgFia) cores s h o w n in Fig. 1 (Schafer a n d Schnering, 1964). T h e centrifugal fluorine a t o m o n e a c h n i o b i u m a t o m is s h a r e d w i t h a centrifugal position of t h e neighbouring (NbgFig) core, so t h a t a three-dimensional n e t w o r k is formed. T h e formula c a n b e conveniently w r i t t e n [(Nb6Fi2)F6/2]- N o distortion of t h e cage from regular g e o m e t r y w a s n o t e d , t h e i n t e r a t o m i c distances being: N b — N b = 2 - 8 0 Â , N b — b r i d g i n g F 2-05 Â, N b — centrifugal F = 2-11 Â. T h e lowest t a n t a l u m chloride a n d b r o m i d e also h a v e t h i s stoichiometry, a n d a r e i s o m o r p h o u s w i t h each o t h e r (Schafer et al, 1964, 1965). T h e existence of TaCl2 which h a s b e e n claimed b y earlier workers could n o t be confirmed (e.g. see Y o u n g a n d B r u b a k e r (1952); F r e r e a n d Michel (1961, 1962) ).
T h e s t r u c t u r e of NbeCli4 also shows t h e presence of Nb^ o c t a h e d r a , b u t it is interesting t o n o t e t h a t in t h i s case t h e o c t a h e d r o n is flattened along one of its fourfold axes, such t h a t t h e r e a r e four n i o b i u m - n i o b i u m distances of 2-95 Â, a n d eight n i o b i u m - n i o b i u m distances of 2-89 Â (Schafer a n d Schnering, 1964). I n a d d i t i o n t o t h e six centrifugal chlorine a t o m s a c t i n g as bridges b e t w e e n different (Nb6Cli2)^+ cores, t w o of t h e inner chlorine a t o m s of t h e core itself a r e s h a r e d w i t h t h e centrifugal positions of a d j a c e n t (NbeClig)^"*" clusters. E a c h Nbg cluster is therefore linked t o eight n e i g h b o u r s in a three-dimensional n e t w o r k , a n d Nb6Cli4 can b e conveniently f o r m u l a t e d [(Nb6ClioCl2/2)Cl6/2]- E o r t h e t w o n i o b i u m a t o m s a t t h e flattened corners of t h e o c t a h e d r o n , N b — b r i d g i n g CI = 2 · 3 6 - 2 · 3 9 A, a n d N b — c e n t r i f u g a l CI = 3-04 Â, while for t h e o t h e r four n i o b i u m a t o m s , Nb—^bridging CI = 2 - 4 0 - 2-47 Â, a n d Nb—centrifugal CI = 2-58. Earlier r e p o r t s of a lower C. Binary halides
T h e r e h a v e been n o crystal s t r u c t u r e d e t e r m i n a t i o n s of t h e n u m e r o u s solid crystalline d e r i v a t i v e s referred t o a b o v e . Confirmation of t h e existence of t h e (MgXja)^"^ cage a w a i t e d t h e p r e p a r a t i o n of single crystals of t h e b i n a r y halides b y r e d u c t i o n of t h e higher halides w i t h t h e m e t a l in a t e m p e r a t u r e g r a d i e n t . W h e r e a s all t h e d e r i v a t i v e s referred t o a b o v e h a v e t h e a v e r a g e m e t a l a t o m in t h e formal o x i d a t i o n s t a t e of t h e lowest halides which h a v e been p r e p a r e d h a v e stoichiometry d e p e n d e n t b o t h on t h e m e t a l a n d t h e halogen:
90°K 195°K 2 9 5 ° K 2 9 7 ° K
10«x(TaCl2.5) 295 178 129
10«x(TaBr2.5) 113 89 70
These values for t h e susceptibility are consistent w i t h a single u n p a i r e d electron for t h e six t a n t a l u m a t o m s (/Xeff(Ta6Xi5) ' ^ 1 - 5 B), w h i c h is in a g r e e m e n t w i t h t h e o d d n u m b e r of electrons which m u s t b e p r e s e n t in t h e s e c o m p o u n d s .
C o m p o u n d s of t h e G r o u p V m e t a l s of t h e general formulae (MeXig)^^
h a v e 30 electrons on t h e six m e t a l a t o m s . Of t h e s e , 12 will b e used for binding t h e 12 halogen a t o m s , a n d t w o t o p r o v i d e t h e n e t charge, leaving 16 electrons p o t e n t i a l l y available for m e t a l - m e t a l b o n d i n g . H o w e v e r , if electron p a i r m e t a l - m e t a l b o n d s are considered t o lie along chloride NbClg (Schafer a n d D o h m a n n , 1959; F r e r e a n d Michel, 1961, 1962) h a v e n o t been confirmed.
Ta6li4 h a s a similar s t r u c t u r e t o Nb6Cli4, t h e Tag o c t a h e d r o n again being d i s t o r t e d b y a flattening along one of t h e principal axes. T h e r e a r e four t a n t a l u m - t a n t a l u m distances of 3-10 Â, a n d eight of 2-82 Â. T h e centrifugal iodine a t o m s are again significantly further a w a y from t h e t w o fiattened corners of t h e o c t a h e d r o n t h a n from t h e o t h e r four corners, t h e distances being 4-33 a n d 3-12 Â respectively. T h e t a n t a l u m - i o d i n e distances w i t h i n t h e (Ta^Iia)^^ core r a n g e from 2-72 t o 2-82 Â (Schafer a n d Schnering, 1964).
T h e s t r u c t u r e s of NbBrg ( G u t m a n n a n d T a n n e n b e r g e r , 1956) a n d N b i g (Chaigneau, 1957b) are n o t k n o w n .
D. Magnetism, spectra and bonding
T h e formally 2-33-valent Nb6Cli4.7H20 e x h i b i t s a t e m p e r a t u r e - i n d e p e n d e n t p a r a m a g n e t i s m of 10^x(Nb6Cli4.7H2O) = 240 c.g.s.u. from 42 t o 290°K (Robin a n d K u e b l e r , 1965). A lower r o o m t e m p e r a t u r e susceptibility of 102 c.g.s.u. h a s been r e p o r t e d for t h e c h l o r o t a n t a l u m analogue (Schafer a n d Schnering, 1964). T h e higher t e m p e r a t u r e d e p e n d e n t values found earlier are p r e s u m a b l y d u e t o p a r a m a g n e t i c impurities (Krylov, 1958). A slight p a r a m a g n e t i s m h a s also been found for Nb6Cli4, w h e r e a s Ta6li4 is d i a m a g n e t i c (Schafer a n d Schnering, 1964).
T h e formally 2-5-valent TagClis (Schafer et al,, 1964) a n d TagBrig (Schafer et al., 1965), show a slight p a r a m a g n e t i s m ; t h e v a l u e s e x t r a p o l a t e d t o infinite field s t r e n g t h b u t n o t corrected for d i a m a g n e t i s m a r e :
F I G . 6. Square antiprismatic hybridization s h o w n o n one of t h e m e t a l a t o m s in (M6Xi2)2+. T h e hybrid orbitals are at angles β a n d γ t o t h e eight fold inversion a x i s of t h e square antiprism.
each of t h e o c t a h e d r a l edges, 24 b o n d i n g electrons are required. These c o m p o u n d s are therefore a p p a r e n t l y electron deficient, a n d a n y descrip
t i o n of t h e b o n d i n g m u s t t a k e t h i s i n t o a c c o u n t , m u s t also a c c o u n t for t h e observed d i a m a g n e t i s m (or t e m p e r a t u r e i n d e p e n d e n t p a r a m a g n e t i s m ) , a n d should also b e of some assistance in t h e i n t e r p r e t a t i o n of t h e visible a n d u l t r a v i o l e t spectra.
T h e valence b o n d a p p r o a c h will b e briefly discussed first. H o w e v e r , for t h i s p a r t i c u l a r class of inorganic c o m p o u n d s , t h e r e are considerable a d v a n t a g e s in using a molecular orbital a p p r o a c h , a n d t h i s will b e discussed in m o r e detail.
I n t h e simplest valence b o n d a p p r o a c h each m e t a l a t o m would b e considered t o be b o n d e d t o four m e t a l a t o m s a n d four halogen a t o m s . T h e p r o b l e m is therefore raised of t h e existence of a n electron deficient system as i n d i c a t e d a b o v e , b u t in a d d i t i o n t h e m e t a l a t o m would require a stereochemistry which would b e related t o cubic stereo
chemistry, a n d suitable h y b r i d orbitals could only be formed b y t h e utilization of/-orbitals, which would n o t be e x p e c t e d t o be energetically favourable for n i o b i u m or t a n t a l u m .
Dufffey (1951), however, suggested t h a t t h e h y b r i d orbitals projected t o w a r d s t h e centres of t h e o c t a h e d r a l faces r a t h e r t h a n along t h e octa
hedral edges (Fig. 6). T h e h y b r i d i z a t i o n required is n o w r e l a t e d t o t h e square a n t i p r i s m r a t h e r t h a n t o t h e cube, a n d such h y b r i d i z a t i o n can b e o b t a i n e d using only s-, ρ- a n d cZ-orbitals. T h a t is, t h e r e is overlap of orbitals from t h r e e different m e t a l a t o m s a t t h e c e n t r e of each octa
hedral face, a n d t h e 16 available electrons are j u s t a c c o m m o d a t e d if t w o b o n d i n g electrons a r e placed in each set of t h r e e overlapping orbitals. Dufffey also p o i n t e d o u t t h a t t h e s q u a r e a n t i p r i s m considered
here is considerably d i s t o r t e d from t h a t e x p e c t e d for n o r m a l s q u a r e a n t i p r i s m a t i c coordination. F o r (NbgClia)^"'', t h e angles t o t h e principal axis which are m a d e b y t h e lines joining t h e m e t a l t o t h e chlorine a t o m a n d t o t h e centre of t h e o c t a h e d r a l face, ή a n d γ respectively in Fig. 6 are 81° a n d 145° respectively, c o m p a r e d w i t h 33° a n d 147° e x p e c t e d for n o r m a l s q u a r e a n t i p r i s m a t i c coordination (Kepert, 1965). I t h a s therefore been suggested t h a t t h e m e t a l - h a l o g e n σ-bonds are consider
a b l y b e n t (Gillespie, 1961), b u t t h e degree of overlap still a p p e a r s favourable (Duffey, loc. cit.).
T h e first molecular orbital t r e a t m e n t w a s d u e t o Grossman et al.
(1963), w h o defined t h e axes a b o u t t h e m e t a l a t o m as s h o w n in F i g . 7,
F I G . 7. T h e a x e s chosen for t h e molecular orbital description of (M6Xi2)^''"are s h o w n o n one of t h e m e t a l a t o m s only. E a c h individual 2-axis p o i n t s radially o u t w a r d from t h e centre of t h e octahedron, a n d t h e individual x- a n d y-axes point over t h e edges of t h e octahedron a n d in t h e direction of t h e bridging halogen a t o m s .
a n d t h i s s y s t e m h a s been r e t a i n e d for t h e following discussion. These a u t h o r s considered p r e l i m i n a r y h y b r i d i z a t i o n of t h e a t o m i c orbitals in suitable directions, before c o m b i n a t i o n t o form t h e molecular orbitals.
T h e y found t h e m o s t stable molecular orbital w a s of A^g s y m m e t r y formed from t h o s e orbitals w i t h c o m p o n e n t s along t h e individual 2;-axes {s, p ^ , ά^ή. T h e n e x t m o s t stable w a s formed from t h e six d^y- orbitals w i t h lobes over t h e o c t a h e d r a l faces (A^u)- F i n a l l y {T^g a n d T^u) were formed from t h o s e orbitals w i t h c o m p o n e n t s directed t o w a r d s t h e o c t a h e d r a l edges (p^, py, dr^^ a n d dy^). These molecular orbitals t h e n a c c o m m o d a t e t h e 16 electrons accounting for t h e observed d i a m a g n e t i s m .
T h e general t r e a t m e n t b y Grossman, Olsen a n d Duffey leads t o difficulties in t h e q u a n t i t a t i v e calculations, a n d a simplified a p p r o a c h w a s later used b y C o t t o n a n d H a a s (1964). These a u t h o r s first of all
'Eu /hybrids Ligand
X 2u
F i g . 8. Molecular orbital e n e r g y levels for (MeXia)^^, after Cotton a n d H a a s .
i t w a s a s s u m e d t h a t t h e ^^-orbital w a s t o t a l l y involved w i t h t h e b o n d i n g of t h e centrifugally directed ligand, a n d did n o t i n t e r a c t w i t h t h e iZ22-orbital. F i n a l l y l i g a n d - m e t a l 7r-bonding w a s neglected. These last t w o a s s u m p t i o n s could h a r d l y b e e x p e c t e d t o b e justified a n d a r e of critical i m p o r t a n c e ; t h e y will b e discussed in m o r e detail later. T h e r e m a i n i n g a t o m i c orbitals, d^^^ da.z, dy^ a n d d^y, were c o m b i n e d t o form t h e molecular orbitals used for t h e m e t a l - m e t a l b o n d i n g , a n d t h e over
l a p integrals a n d relative energies calculated. T h e r e s u l t a n t molecular orbital scheme is i l l u s t r a t e d q u a l i t a t i v e l y in F i g . 8. One r a t h e r r e m a r k able feature of t h e r e s u l t a n t molecular orbital d i a g r a m is t h a t t h e r e a r e only eight b o n d i n g orbitals c o m p a r e d w i t h 16 a n t i b o n d i n g orbitals (the
T2g{dxy) is e x p e c t e d t o b e m o r e u n s t a b l e t h a n shown, d u e t o i n t e r a c t i o n w i t h t h e b o n d i n g T^gid^^z, dy^) ). T h e 16 available electrons were t h e n a c c o m m o d a t e d as indicated, w h i c h is in a g r e e m e n t w i t h t h e observed d i a m a g n e t i s m . These filled orbitals are t h e s a m e as t h o s e derived earlier b y Grossman, Olsen a n d Duffey, a l t h o u g h t h e ordering of t h e T^g{daiz, dyzY a n d Τ-^^(^^ζ9 -orbitals w a s reversed.
Allen a n d Sheldon (1965) s t u d i e d t h e s p e c t r a of t h e s e c o m p o u n d s in t h e visible region (8000-45000 cm-^), a n d eight or m o r e b a n d s were observed. T h e y considered t h a t all observed b a n d s were d u e t o m e t a l - a s s u m e d t h a t h y b r i d s from t h e s-, p^-, py- a n d d^j^-i/a-orbitals b o n d e d t h e halogen a t o m s in a s q u a r e p l a n a r a r r a n g e m e n t . A t t h e t i m e t h i s a s s u m p t i o n a p p e a r e d justified, as t h e only s t r u c t u r e w a s b a s e d on t h e analysis of t h e s c a t t e r i n g of X - r a y s b y ethanolic solutions of t h e com
p o u n d s , b u t since t h i s t i m e a n u m b e r of c o m p o u n d s h a v e h a d t h e i r s t r u c t u r e s a c c u r a t e l y d e t e r m i n e d in t h e solid s t a t e . T h e results show t h a t t h e angle ή (Fig. 6) t h e m e t a l - h a l o g e n b o n d m a k e s w i t h t h e 2;-axis is 88° for Nb6Fi5, 81° for Nb6Cli4 a n d alcohoHc solutions of Nb6Cli2^+
a n d Ta6Cli22+, 79° for alcohohc solutions of T a 6 B r i 2 ^ + , a n d 73° for Ta6li4; t h i s t r e n d w o u l d b e e x p e c t e d on simple steric g r o u n d s . Secondly
m e t a l b o n d i n g t o m e t a l - m e t a l a n t i b o n d i n g transitions, a n d fitted t h e spectra t o t h e molecular orbital d i a g r a m of C o t t o n a n d H a a s b y assum
ing t h a t t h e v a l u e of a, t h e Slater orbital e x p o n e n t , h a s a value of 1-7 for t h e (Z^2-orbitals, a n d 1-2 for t h e o t h e r orbitals. All possible transitions were considered t o be allowed. F o r e x a m p l e t h e lowest energy b a n d , which is strong (extinction coefficient '^3000), was assigned t o t h e t r a n s i t i o n ΤΊ„(ίία;ζ? ^yz) — T^g{d^y), while t h e t h i r d lowest energy b a n d which a p p e a r s only as a shoulder (extinction coefficient ^ 7 0 0 ) was assigned t o t h e t r a n s i t i o n A^uidxyyT^gidr^y), a l t h o u g h t h e a b s o r p t i o n d u e t o such a t r a n s i t i o n m a y be e x p e c t e d t o b e m o r e intense. I t is im
p o r t a n t t o n o t e t h a t t h e s e molecular orbitals derived from different a t o m i c cZ-orbitals do n o t m i x even a l t h o u g h t h e y m a y be of t h e s a m e molecular orbital s y m m e t r y , since t h e y are forbidden b y t h e local a t o m i c orbital s y m m e t r y . Transitions b e t w e e n s u b s y s t e m s are therefore generally forbidden, a n d each set of molecular orbitals can be considered separately (Robin a n d K u e b l e r , 1965). T h e only exceptions are t h e mixing of t h e T^^{dç^'i_y^ w i t h t h e T^uidxz, dy^), or b y mixing t h r o u g h t h e halogen bridges.
R o b i n a n d K u e b l e r (1965) modified t h e C o t t o n a n d H a a s p i c t u r e b y i n t r o d u c i n g those ligand-metal interactions d u e t o t h e 12 halogen a t o m s of t h e (MgXig)^"^ core, a n d derived t h e molecular orbital d i a g r a m shown in F i g . 9. T h e allowed m e t a l - m e t a l t r a n s i t i o n s are shown. F o r t h e
F I G . 9. Molecular orbital energy levels for (Μ^Χ^ί)^^, after R o b i n a n d Kuebler.
molecular orbitals derived from t h e m e t a l d^^y orbitals, if only m e t a l - m e t a l interactions are considered t h e ordering will b e A^u, T^g, E^, b u t if ligand ρ 7r-metal d^^y i n t e r a c t i o n s a r e considered, t h e i n v e r t e d order E^y T^g, A^u is o b t a i n e d . T h e correct order w a s decided b y considering t h e e x p e r i m e n t a l l y d e t e r m i n e d a b s o r p t i o n spectra. (In addition t h e s e a u t h o r s i n t r o d u c e d t h e iZa.2_y2-orbitals i n t o t h e i r calculations, a n d also
reversed t h e order of Εg[dz'^ a n d ^1^(^-52), b u t t h e s e differences a r e u n i m p o r t a n t for t h e p r e s e n t discussion.)
R o b i n a n d K u e b l e r t h e n s e p a r a t e d t h e observed spectral b a n d s of t h e n i o b i u m c o m p o u n d s (in e t h a n o l a t —100°) i n t o t w o g r o u p s : t h o s e w h i c h change only slightly as we go from (NbeClig)^^ t o (NbeBria)^^, a n d t h o s e which are shifted b y t h e order of 1000 cm*^ t o lower energies in t h e n o r m a l m a n n e r for halogen t o m e t a l charge transfer b a n d s (see first t h r e e columna-of T a b l e I ) . T h e first b a n d a t 10,870 cm-^ (underlined in T a b l e I) is therefore assigned t o t h e first m e t a l - m e t a l t r a n s i t i o n , n a m e l y
^u(dxy) — T^gidoiy). A d d i t i o n a l d a t a o b t a i n e d in w a t e r show a slight shift t o higher energy w h e n c o m p a r i n g t h e b r o m o complex w i t h t h e chloro complex, confirming t h a t t h i s is a m e t a l - m e t a l t r a n s i t i o n (last t h r e e columns of T a b l e I ) .
T A B L E I . V i s i b l e s p e c t r a ( c m - ^ ) o f (NbeXi2)^+ i n e t h a n o l a n d w a t e r
(Nb6Xi2)2+ in ethanol»
X = Cl Χ = B r Difference
(Nb6Xi2)2+ in water&
X = CI X = B r Difference
10870 (m) 10640 - 2 3 0 11200 11400 + 200
16670 (w) 15270 - 1 4 0 0 16500 15500 - 1 0 0 0
20410 (w) — — 21000 21000 0
24690 (m) 23360 - 1 3 3 0 25200 23700 - 1 5 0 0
31250 (w) 30300 - 9 5 0 31000 31000 0
35650 (m) 35210 - 4 4 0 36500 35300 - 1 2 0 0
46390 (s) 45300 - 1 0 9 0 43000
4 8 2 0 0 α R o b i n a n d K u e b l e r (1965).
» A l l e n a n d Sheldon (1965).
T h e underlined b a n d is t h a t assigned t o a m e t a l - m e t a l b o n d i n g t o m e t a l - m e t a l anti- b o n d i n g transition.
T h e n e x t four b a n d s , which were considered t o show considerable shifts for changing chloride t o b r o m i d e , w e r e assigned t o charge transfer t r a n s i t i o n s (although T a b l e I shows t h a t t h e spectral results o b t a i n e d in w a t e r d o n o t really confirm t h i s view). T h e w e a k b a n d s a t 16,670, 20,410 a n d 31,250 cm-^ were considered t o b e s y m m e t r y forbidden t r a n s i t i o n s , while t h e first intense charge transfer b a n d a t 24,690 cm-^
i n d i c a t e d t h a t t h i s w a s a n allowed t r a n s i t i o n , a n d w a s assigned t o a t r a n s i t i o n from a ligand ^^-orbital t o one of t h e m e t a l molecular orbitals originating from t h e cia-^-atomic orbitals. I t w a s t h e fact t h a t t h i s t r a n s i t i o n w a s observed as a singlet t h a t led R o b i n a n d K u e b l e r t o propose t h a t t h e t r a n s i t i o n w a s d u e t o t h e A^y^{dr^y) o r b i t a l ; for t h i s t o b e e m p t y t h e s e b a n d s m u s t b e i n v e r t e d relative t o t h e order of
C o t t o n a n d H a a s , so t h a t t h e d o u b l y d e g e n e r a t e EJdr^y) was of t h e lowest energy, t h a t is, m e t a l - l i g a n d i n t e r a c t i o n s a r e m o r e i m p o r t a n t t h a n m e t a l - m e t a l i n t e r a c t i o n s (see above). F o r d i a m a g n e t i s m t o b e r e t a i n e d w i t h t h e s a m e n u m b e r of valence electrons this level m u s t b e filled, a n d t h e only place t h e necessary electrons can come from is t h e lowest lying A•^g{dzή, so t h a t t h e stabilization achieved b y filling t h e EJdxy) t o form t h e closed shell configuration w a s t h o u g h t sufficient t o overcome t h e energy r e q u i r e d t o p r o m o t e t h e s e electrons from t h e A-^g{dz^ level.
Again on t h e basis of comparison of t h e chloride a n d b r o m i d e s p e c t r a in e t h a n o l , t h e b a n d a t 35,650 cm~^ w a s assigned t o a m e t a l - m e t a l transition, while t h a t a t 46,390 cm~^ t o a l i g a n d - m e t a l t r a n s i t i o n .
W i t h t h e exception of t h e lowest energy b a n d a t 10,870 cm-^, a l t h o u g h t h e assignment of t h e b a n d s on t h e basis of t h e shifts observed in ethanolic solution is feasible, it is n o t necessarily convincing, as shown, for e x a m p l e , b y a comparison of t h e s p e c t r a o b t a i n e d in w a t e r (see last t h r e e columns of T a b l e I ) .
T A B L E I I . V i s i b l e s p e c t r a (cm-^) o f (TaeXi2)^+ i n w a t e r a n d a l k a l i ( A l l e n a n d S h e l d o n , 1 9 6 5 )
(TaeXia)^+ in w a t e r (TaeXia)^^ in alkaU
X = CI X = B r Difference X = CI X = B r Difference
11600 12000 + 400 11000 11100 + 100
12800
— — — —
— -14000 13700 - 3 0 0 13900 13900 0
16000 15800 - 2 0 0 18000 ?
—
22000 21000 - 1 0 0 0 23000 21500 - 1 5 0 0
25200 24000 - 1 2 0 0 27800 25800 - 2 0 0 0
30200 28600 - 1 6 0 0 33900 31000 - 2 9 0 0
37000 34000 41000 39000
42500 37000 44500 43400
43200 47200 47500
T h e underlined b a n d s are t h o s e assigned t o m e t a l - m e t a l b o n d i n g t o m e t a l - m e t a l antibonding transitions.
T h e spectra of t h e t a n t a l u m complexes are similar t o those of t h e n i o b i u m c o m p o u n d s ; n e a r l y all b a n d s are shifted t o higher energy w i t h respect t o t h e corresponding n i o b i u m c o m p o u n d . H o w e v e r , t h e r e is one i m p o r t a n t difference. T h e lowest energy b a n d which h a s been assigned t o a m e t a l - m e t a l t r a n s i t i o n is split, b o t h in solution a n d in t h e solid.
F o r e x a m p l e , t h e b a n d a t 10,640 cm*^ for (NbgBria)^"^ in e t h a n o l a t
—100° is split i n t o c o m p o n e n t s a t 13,000 a n d 15,290 cm"^ for (TagBrig)^"^ u n d e r t h e s a m e conditions. T h a t b o t h p o r t i o n s of t h i s split b a n d are d u e t o m e t a l - m e t a l t r a n s i t i o n s can b e confirmed using t h e s a m e criteria as before, n a m e l y t h e small or even positive shift w h e n m o v i n g from t h e chloride t o t h e b r o m i d e (Table I I ) .
R o b i n a n d K u e b l e r a t t r i b u t e d t h i s splitting t o a t e t r a g o n a l distortion resulting from a localization of t h r e e positive u n i t charges on t w o of t h e m e t a l a t o m s , r a t h e r t h a n a uniform d i s t r i b u t i o n of 2-33 positive charges on all m e t a l a t o m s (Fig. 10). T h e i n t e n s i t y of t h e high energy t r a n s i t i o n w a s observed t o b e double t h e i n t e n s i t y of t h e low energy t r a n s i t i o n , which is e x p e c t e d on t h i s model.
F I G . 10. Tetragonal elongation of t h e Mg octahedron in (MgXia)'*"^.
On t h i s basis a t w o electron oxidation would be e x p e c t e d t o form a flattened o c t a h e d r o n (Fig. 11). This flattened o c t a h e d r o n would be e x p e c t e d t o h a v e t h e reverse i n t e n s i t y ratios, a n d t h i s w a s in fact n o t e d
F I G . 11. Tetragonal flattening of t h e Mg octahedron in (MeXja)*"''.
![TABLE V. Structural parameters (average values) for some molecules based on the (Re^Cls)" + core Re—Re (Â) Re—X (out of Re—X (cen- X (out of plane)—Re^ plane) (Â) trifugal) (Â) X (out of plane)* bridging X—Re bridging X^ Cs3[(Re3Cl3)Cl,]i' [(Re3Cl3](https://thumb-eu.123doks.com/thumbv2/9dokorg/1130776.80151/43.658.151.469.108.896/table-structural-parameters-average-molecules-trifugal-bridging-bridging.webp)