Szerkesztette:
LÁSZLÓ KRISZTINA
Írta:
LÁSZLÓ KRISZTINA,GROFCSIK ANDRÁS, KÁLLAY MIHÁLY, KUBINYI MIKLÓS
Lektorálta:
TASI GYULA
FIZIKAI KÉMIA I.
KÉMIAI TERMODINAMIKA
Egyetemi tananyag
2011
LEKTORÁLTA: Dr. Tasi Gyula, Szegedi Tudományegyetem KÖZREMŰKÖDÖTT: Brátán János, Brátánné Mikics Veronika
Creative Commons NonCommercial-NoDerivs 3.0 (CC BY-NC-ND 3.0) A szerző nevének feltüntetése mellett nem kereskedelmi céllal szabadon másolható, terjeszthető, megjelentethető és előadható, de nem módosítható.
TÁMOGATÁS:
Készült a TÁMOP-4.1.2-08/2/A/KMR-2009-0028 számú, „Multidiszciplináris, modulrendszerű, digitális tananyagfejlesztés a vegyészmérnöki, biomérnöki és vegyész alapképzésben” című projekt keretében.
KÉSZÜLT: a Typotex Kiadó gondozásában FELELŐS VEZETŐ: Votisky Zsuzsa
AZ ELEKTRONIKUS KIADÁST ELŐKÉSZÍTETTE: Sosity Beáta ISBN 978-963-279-473-0
KULCSSZAVAK:
természettudomány, kémia, fizikai kémia, a termodinamika főtételei, belső energia, entalpia, entrópia, szabadenergia, szabadentalpia, fázisátalakulások, termokémia, kémiai potenciál, elegyek, fázisdiagramok, kémiai egyensúlyok, BSc képzés.
ÖSSZEFOGLALÁS:
Ez a tananyag elsősorban vegyész- és vegyészmérnök hallgatók számára készült bevezető jellegű munka.
Megértéséhez szükség van matematikai ismeretekre, beleértve a differenciál- és integrálszámítást. A fizikai kémia három nagy területe az egyensúly, a változás és a szerkezet. Ezek közül az első témát, az egyensúly kérdését járjuk körül a klasszikus termodinamika módszereivel. Ismertetjük a termodinamika három főtételét, bevezetjük a termodinamika fontos állapotfüggvényeit; a belső energiát, entalpiát, entrópiát, szabadenergiát, szabadentalpiát és a kémiai potenciált. Segítségükkel meghatározhatjuk a folyamatok irányát és az egyensúlyi állapotokat. Részletesen foglalkozunk tökéletes és reális gázok tulajdonságaival,
elegyekkel, egy- és többkomponensű fázisegyensúlyokkal, termokémiával, kémiai egyensúlyokkal és elektrolitok termodinamikai leírásával. A kidolgozott példákkal az a célunk, hogy segítsük a tananyag mélyebb megértését.
TARTALOMJEGYZÉK
1. BEVEZETÉS ... 5
1.1. A termodinamikai rendszer fogalma, típusai és jellemzése ... 5
1.2. A termodinamikai hőmérséklet és nyomás ... 11
2. A TERMODINAMIKA I. FŐTÉTELE ... 14
2.1. A belső energia, a termodinamika I. főtétele ... 14
2.2. A munka ... 15
2.3. A hő ... 18
2.4. Az entalpia ... 20
3. TÖKÉLETES GÁZOK ÁLLAPOTVÁLTOZÁSAI ... 24
3.1. Tökéletes gázok moláris hőkapacitása állandó nyomáson, ill. hőmérsékleten ... 25
3.2. Tökéletes gázok reverzibilis állapotváltozásai ... 26
4. TERMOKÉMIA ... 36
4.1. A standard reakcióhő ... 36
4.2. A reakcióhő mérése ... 38
4.3. Hess tétele ... 41
4.4. Standard entalpiák ... 43
4.5. Nyitott rendszer energiamérlege, stacionárius folyamatok ... 44
5. A TERMODINAMIKA II. ÉS III. FŐTÉTELE ... 47
5.1. Az entrópia termodinamikai definíciója ... 47
5.2. Az entrópiaváltozás számítása zárt rendszerekben... 48
5.3. A II. főtétel megfogalmazása az entrópiával ... 51
5.4. Az entrópia statisztikus értelmezése ... 54
5.5. A termodinamika III. főtétele ... 58
6. TERMODINAMIKAI EGYENSÚLYOK ÉS A FOLYAMATOK IRÁNYA ... 61
6.1. A szabadenergia ... 61
6.2. A szabadentalpia ... 63
6.3. A termodinamikai állapotfüggvények deriváltjai ... 65
7. EGYKOMPONENSŰ RENDSZEREK ... 69
7.1. A p-T fázisdiagram ... 69
7.2. A p-T fázisdiagram termodinamikai értelmezése, a Clapeyron-egyenlet ... 71
7.3. Egykomponensű gőz-folyadék egyensúlyok, a Clausius–Clapeyron-egyenlet ... 74
7.4. A T-S diagram ... 76
7.5. Standard szabadentalpiák ... 80
7.6. A tökéletes gáz szabadentalpiája ... 81
8. ELEGYEK ÉS OLDATOK ... 84
8.1. A kémiai potenciál ... 85
8.2. A fázisegyensúlyok feltétele ... 88
8.3. A Gibbs-féle fázisszabály ... 90
8.4. Az elegyképződésre jellemző mennyiségek ... 92
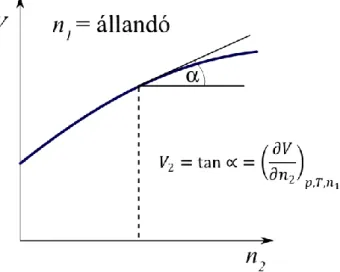
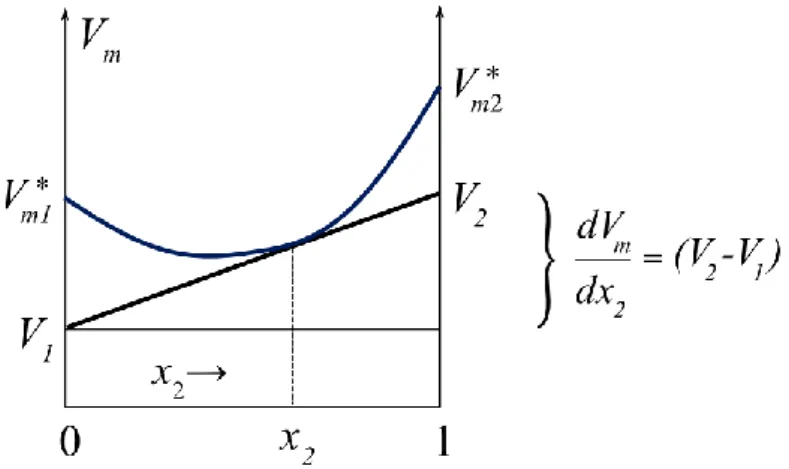
8.5. Parciális moláris mennyiségek ... 94
8.6. A parciális moláris mennyiségek meghatározása ... 97
8.7. Raoult törvénye ... 99
8.8. Eltérések az ideális viselkedéstől ... 103
8.9. Kémiai potenciál folyadékelegyekben ... 104
8.10. Elegyedési entrópia és elegyedési szabadentalpia ... 107
8.11. Korlátlanul elegyedő folyadékok tenzió- és forrpontdiagramja ... 112
8.12. Konovalov II. és III. törvényének levezetése ... 116
8.13. Korlátozottan elegyedő és nemelegyedő folyadékok forrpontdiagramja ... 119
8.14. Egyszerű eutektikumot alkotó szilárd-folyadék egyensúlyok ... 121
8.15. Szilárd-folyadék fázisdiagramok ... 123
8.16. Híg oldatok tenziócsökkenése, forrpontemelkedése és fagyáspontcsökkenése ... 126
8.17. Ozmózisnyomás ... 130
8.18. Az elegyképződés hőeffektusai ... 132
8.19. Henry törvénye, gázok oldhatósága ... 136
8.20. Az elegyek termodinamikai stabilitása ... 137
8.21. Folyadék-folyadék fázisegyensúlyok ... 138
8.22. Megoszlási egyensúlyok ... 140
8.23. Háromszög fázisdiagramok ... 142
9. REÁLIS GÁZOK ... 146
9.1. A reális gázok állapotegyenlete (van der Waals- és viriál állapotegyenlet) ... 146
9.2. A megfelelő állapotok tétele ... 149
9.3. Gázok entalpiája ... 151
9.4. A Joule–Thomson-hatás ... 153
9.5. Gázok fugacitása ... 156
10. KÉMIAI EGYENSÚLYOK ... 159
10.1. Aktivitások és standard állapotok... 159
10.2. A termodinamikai egyensúlyi állandó ... 160
10.3. Kémiai egyensúlyok gázfázisban ... 164
10.4. A nyomás hatása a kémiai egyensúlyra ... 166
10.5. Gáz-szilárd heterogén kémiai egyensúlyok ... 167
10.6. Kémiai egyensúlyok folyadékfázisban ... 169
10.7. Az egyensúlyi állandó hőmérsékletfüggése ... 171
10.8. Egyensúlyok elektrolitokban ... 173
10.9. Aktivitások és kémiai potenciálok elektrolitokban ... 176
10.10. A Debye–Hückel-elmélet alapjai ... 179
FÜGGELÉK ... 183
F1. Táblázatok ... 183
F2. Feladatok ... 192
ÁBRÁK, ANIMÁCIÓK, TÁBLÁZATOK JEGYZÉKE ... 201
Ábrák ... 201
Animációk ... 201
Táblázatok ... 204
1. BEVEZETÉS
A termodinamika a világ egy jól körülhatárolható részének, a rendszernek és a rendszer környezetének a kölcsönhatásaival, valamint a rendszer makroszkopikus tulajdonságai közötti összefüggésekkel foglalkozó tudományág. A termodinamika a XIX. század folyamán indult fejlődésnek, és a XX.
századra gyakorlatilag elérte végleges formáját. A tudományág kialakulásának a gyors iparosodás volt a hajtóereje, amely szükségessé tette a hőerőgépek működési elvének pontos megismerését.
Általánosságban azt mondhatjuk, hogy a termodinamika mai formájában minden olyan jelenség leírására alkalmas, ahol a hő vagy a hőmérséklet központi szerepet játszik. Így a termodinamika elvei alapján magyarázhatóak az alapvető termikus jelenségek (pl. melegítés, hűtés, fázisátalakulások stb.), de a termodinamika szolgáltatja az alapot a kémiai folyamatok megértéséhez is.
A jegyzet keretein belül elsősorban a fenomenologikus termodinamikával fogunk foglalkozni, amely kísérleti tapasztalatokon alapuló deduktív diszciplína. A fenomenologikus termodinamika a tapasztalatokból leszűrt néhány egyszerű alapelvet axiómaként mond ki, ezek a termodinamika főtételei. A főtételekből és további empirikus megfigyelésekből kiindulva matematikai módszerekkel jut el a különböző jelenségek magyarázatához. A fenomenologikus termodinamika nem tesz semmi feltételezést az anyag szerkezetére vonatkozóan. Az anyagot makroszkopikusan vizsgálja, függetlenül attól, hogy milyen részecskék alkotják, és a részecskék között milyen kölcsönhatások hatnak. Az anyag makroszkopikus sajátságai és mikroszkopikus szerkezete között a termodinamika egy másik területe, a statisztikus termodinamika teremt kapcsolatot, amelynek az alapjaival az 5. fejezetben foglalkozunk érintőlegesen.
A termodinamikában központi szerepet játszik az egyensúly fogalma, ami azt jelenti, hogy a rendszerben nem észlelhető makroszkopikus változás, azaz a rendszer tulajdonságai időben állandóak.
A különböző jelenségek tárgyalása során sokszor azzal a feltevéssel fogunk élni, hogy a rendszer minden időpillanatban egyensúlyban van. A termodinamikának azt az ágát, amely ezen a feltéte- lezésen alapszik, egyensúlyi vagy reverzibilis termodinamikának nevezik. Az egyensúly kikötése durva közelítésnek tűnik, de látni fogjuk, hogy az egyensúlyi termodinamika a természetben lejátszódó nem túl gyors folyamatokat jól modellezi. Az egyensúlyi termodinamika hiányosságait egy másik tudományterület, a nemegyensúlyi vagy irreverzibilis termodinamika hivatott kiküszöbölni, amellyel a jegyzet keretein belül nem foglalkozunk.
1.1. A termodinamikai rendszer fogalma, típusai és jellemzése
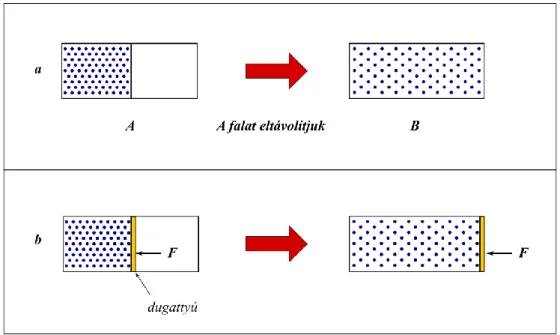
A termodinamikában a világ egy képzelt vagy valós határfelülettel elkülönített részének a tulaj- donságait vizsgáljuk. A világnak ezt a részét rendszernek vagy más néven termodinamikai rendszer- nek nevezzük. Ebben az értelemben a rendszer lehet egy lombik, maga a laboratórium vagy akár a Föld légköre. A világnak a rendszeren kívüli részét környezetnek nevezzük. Nyilvánvaló, hogy végte- len sok rendszert lehet definiálni, de a rendszer és a környezet közötti kölcsönhatás alapján három alapvető típust különíthetünk el: elszigetelt, zárt és nyílt rendszert (1.1. ábra).
Elszigetelt rendszer esetében sem anyag, sem energia nem léphet át a rendszer határfelületén. Ez azt jelenti, hogy egy elszigetelt rendszerből nem vonhatunk ki anyagot, és nem is adhatunk hozzá, de nem vonhatunk ki energiát a rendszerből, és nem közölhetünk vele, tehát pl. nem melegíthetjük, vagy nem változtathatjuk a térfogatát. Elszigetelt rendszerre egy példa a környezetétől hőszigetelő falakkal elválasztott, állandó térfogatú zárt edény. Ilyen például a bombakaloriméter, amivel a 4. fejezetben ismerkedünk majd meg.
Zárt rendszer határfelületén energia átléphet, de anyag nem. A zárt rendszereket tovább csoportosítjuk aszerint, hogy a rendszer térfogata állandó vagy változó, és megkülönböztetünk állandó térfogatú és változó térfogatú zárt rendszereket. Előbbire példa egy lezárt lombik, amelyből nem távozhat anyag, és a térfogata is állandó, de energiát cserélhet a környezetével, pl. melegíthetjük.
Változó térfogatú zárt rendszer pl. egy dugattyús henger, amelyben az anyagmennyiség szintén állandó, de energiája változhat, hiszen pl. hőt közölhetünk a rendszerrel, vagy változtathatjuk a térfogatát, és így munkát végzünk a rendszeren.
Legáltalánosabb esetben mind anyag-, mind energiatranszport lehetséges a rendszer határfelületén. Az ilyen rendszereket nyílt rendszereknek nevezzük. Nyílt rendszerre példa egy nyitott főzőpohár, amiben változtathatjuk az anyag mennyiségét, és energiát is közölhetünk vagy vonhatunk el belőle pl. melegítéssel vagy hűtéssel.
1.1. ábra. A termodinamikai rendszer típusai. Elszigetelt (a), állandó térfogatú zárt (b), változó térfogatú zárt (c) és nyílt (d) rendszerek
A termodinamikai rendszereket a rendszer makroszkopikus tulajdonságainak térbeli eloszlása szerint is csoportosíthatjuk. Homogén rendszer makroszkopikus tulajdonságai minden pontban azonosak. Ilyen pl. egy jól elkevert konyhasóoldat (1.2. ábra). Ennek minden pontjában azonos a hőmérséklet, a koncentráció és az összes többi fizikai mennyiség. Inhomogén rendszerben egyes makroszkopikus tulajdonságok helyről helyre változnak, de eloszlásukat folytonos függvény írja le. Például, ha egy rézrudat az egyik végén melegítünk, a hőmérséklet változik a rúd hossza mentén, de a változás folytonos, a hőmérséklet egyik pontban sem változik ugrásszerűen. Heterogén rendszerben egy vagy több makroszkopikus tulajdonság ugrásszerűen változik a tér bizonyos pontjaiban. Ilyen pl. egy olvadó víz-jég rendszer, ahol általában a hőmérséklet állandó a rendszer minden pontjában, de a legtöbb fizikai mennyiség, pl. hőkapacitás, törésmutató, sűrűség ugrásszerűen változik a jég-víz határfelületen.
Egy heterogén rendszerben a rendszer homogén kémiai összetételű és homogén vagy inhomogén fizikai szerkezetű részét fázisnak nevezzük. Például, ha egy jégkocka olvad egy pohár vízben, az egyik fázis a jég, a másik a víz, még akkor is, ha a jég vagy a víz belsejében nem egyenletes, de legalább folytonos a hőmérséklet vagy más fizikai mennyiség eloszlása. A fázis lehet diszpergált (széttöredezett), ilyenkor egy fázisba soroljuk az azonos összetételű részeket. Pl. ha több jégkocka olvad a vízben, akkor ezeket nem soroljuk külön fázisba, hanem egy fázisnak tekintjük. A rendszernek a kémiai tulajdonság alapján megkülönböztethető részét komponensnek nevezzük. A fenti olvadó jég- víz rendszerben egy komponens van jelen, hiszen mind a két fázist azonos kémiai összetételű anyag, a víz alkotja. Azt mondjuk, hogy az ilyen rendszer egy egykomponensű, kétfázisú rendszer. Tekintsünk egy kicsit bonyolultabb példát, egy kétkomponensű, háromfázisú rendszert (1.3. ábra). Öntsünk össze vizet és étert egy zárt edényben! A víz és az éter korlátozottan elegyedik egymással. Három fázis alakul ki, két folyadék- és a gőzfázis, mindkét komponens jelen van mind a három fázisban. Az egyik folyadékfázisban a víz van többségben és az éter móltörtje kicsi, a második folyadékfázisban fordított a helyzet.
1.2. ábra. A termodinamikai rendszer típusai a makroszkopikus tulajdonságok eloszlása szerint.
Homogén (a), inhomogén (b) és heterogén (c) rendszerek
1.3. ábra. Példa egy kétkomponensű, háromfázisú rendszerre: az éter-víz rendszer
A termodinamikai rendszer állapotának a mérhető fizikai tulajdonságok összességét nevezzük. A rendszer állapotától függő makroszkopikus jellemzők az állapotjelzők (állapothatározók). A legfontosabb állapotjelzők az anyagmennyiség, a térfogat, a nyomás, a hőmérséklet és a koncentráció (1.1. táblázat).
1.1. táblázat. Az alap-állapotjelzők
Elnevezés Jelölés Mértékegység
anyagmennyiség n mol
térfogat V m3
nyomás p Pa
hőmérséklet T K
koncentráció c mol m-3
Megjegyezzük, hogy az anyagmennyiség helyett gyakran a tömeget (jele: m, mértékegysége: kg) használják a rendszerben lévő anyag mennyiségének jellemzésére. A koncentráció megadása is többféleképpen történhet. A laboratóriumban leggyakrabban az anyagmennyiség-koncentrációt, más
néven molaritást (jele: c, mértékegysége: mol m-3 vagy mol dm-3), iparban inkább a Raoult-féle koncentrációt, vagy más néven molalitást (jele: m, mértékegysége: mol kg-1) alkalmazzák. Ez utóbbi az egy kilogramm oldószerben oldott anyag anyagmennyisége. Gyakran használjuk még a móltörtet is (dimenzió nélküli), ami az adott komponens anyagmennyiségének és a teljes anyagmennyiségnek a hányadosa. A móltört jelölésére x-et vagy y-t használunk, az előbbit általában folyadékokra, az utóbbit pedig gázokra szokás alkalmazni.
A termodinamikában központi szerepet játszik az egyensúly fogalma. Egy rendszer termo- dinamikai egyensúlyban van, ha az állapothatározók egyike sem változik. Másképp megfogalmazva:
az egyensúlyban nem játszódnak le makroszkopikus folyamatok. Fontos azonban megjegyezni, hogy ez a definíció nem zárja ki a mikroszkopikus folyamatokat, ilyenek lejátszódhatnak egyensúlyban is, azt mondjuk, hogy az egyensúly dinamikus. Azonban a mikroszkopikus folyamatok nem változ- tathatják meg az állapothatározók értékét. Pl. egy olvadó jég-víz rendszerben a jég és a víz anyag- mennyisége állandó, ha a rendszer egyensúlyban van. A gyakorlatban azonban adott idő alatt valamennyi vízmolekula a szilárd fázisból átkerül a folyadékfázisba, de ugyanannyi molekula folyadékfázisból átvándorol a szilárd fázisba. Az olyan rendszereket, amelyek állapothatározói változnak, nemegyensúlyi rendszereknek nevezzük.
A termodinamikai rendszerekben lejátszódó folyamatokat többféleképpen osztályozhatjuk.
Elméleti szempontból a legfontosabb felosztás a folyamatok megfordíthatósága szerinti csoportosítás.
Reverzibilisnek nevezünk egy változást, ha a rendszer minden pillanatban egyensúlyban van, és a rendszer a végállapotból ugyanazon közbülső egyensúlyi állapotokon keresztül képes visszajutni a kezdeti állapotba. Ez azt jelenti, hogy a rendszer állapotjelzői minden pillanatban csak infi- nitezimálisan (végtelenül) kis mértékben változnak, a folyamat a változók infinitezimális módo- sításával megfordítható. Az olyan folyamatot, amelyik nem elégíti ki a fenti feltételeket, irrever- zibilisnek nevezzük.
Nézzünk néhány példát reverzibilis és irreverzibilis folyamatokra! Töltsünk valamilyen gázt egy dugattyúba, és nyomjuk össze (1.4. ábra)! Ahhoz, hogy össze tudjuk nyomni a gázt, a pk külső nyomásnak nagyobbnak kell lennie a dugattyú belsejében mérhető p belső nyomásnál. Ha a folyamatot úgy vezetjük, hogy minden pillanatban a külső nyomás csak infinitezimálisan nagyobb, mint a belső, azaz a dugattyút végtelenül lassan nyomjuk össze, a változás reverzibilis. Tekintsünk egy másik példát, egy gáz kiterjesztését 5 bar nyomásról 1 bar nyomásra! Az első esetben egy szelepen keresztül engedjük kiterjedni a gázt a vákuumba úgy, hogy a végső nyomás 1 bar legyen (1.5a. ábra). A szelep kinyitása után megindul a gáz áramlása a tartály nagy nyomású részéről a másik oldalra, ahol erede- tileg vákuum volt. A folyamat egészen addig tart, míg a tartály két fele között ki nem egyenlítődik a nyomás. A folyamat végén a rendszer egyensúlyba kerül, de a folyamat lejátszódása során a rendszer nincs egyensúlyban, hiszen a tartály két fele között mindig nyomáskülönbség áll fenn, azaz a folyamat irreverzibilis. A második esetben egy dugattyú folyamatos mozgatásával terjesztjük ki a gázt (1.5b.
ábra). Az előző példához hasonlóan, hogyha végtelen lassan mozgatjuk a dugattyút, a rendszer minden pillanatban egyensúlyban van, és a folyamat reverzibilis. A dugattyú gyors mozgatása esetén viszont ez nem lesz igaz, és a folyamat ekkor irreverzibilis.
1.4. ábra. Példa egy reverzibilis folyamatra: egy gáz reverzibilis összenyomása
1.5. ábra. Példa reverzibilis és irreverzibilis folyamatokra: egy gáz kiterjesztése szelepen keresztül (a), illetve dugattyú mozgatásával (b)
A fenti példákból is nyilvánvaló, hogy a természetben lejátszódó folyamatok szigorúan véve mindig irreverzibilisek, hiszen infinitezimálisan kis változások nem lehetségesek. Például a dugattyút végtelenül lassan kéne mozgatnunk, de ez végtelenül hosszú ideig is tartana. A termodinamikában mégis nagyon sokszor feltételezni fogjuk, hogy egy folyamat reverzibilis. Ezt, mint látni fogjuk, az indokolja, hogy a reverzibilitás feltételezése nagymértékben egyszerűsíti a tárgyalást, illetve sok esetben a jelenségek leírása nem is lehetséges e nélkül a feltevés nélkül. A reverzibilitás feltételezése természetesen közelítés, de a gyakorlatban a nem túl gyorsan lejátszódó folyamatok esetében nem okoz számottevő hibát. Nagyon gyors folyamatok, mint például robbanások persze nem tekinthetők reverzibilisnek.
Gyakorlati szempontból különösen érdekesek azok a folyamatok, amikor a rendszer valamelyik állapotjelzője nem változik. Ezek jelentősége abban áll, hogy a természetben, a laboratóriumban vagy egy technológiai eljárás során gyakran előfordul, hogy valamilyen fizikai mennyiség értéke nem változik. Izotermnek nevezzük az olyan folyamatokat, amikor a rendszer hőmérséklete nem változik.
Jó példa erre egy szilárd anyag olvadása. Az anyaggal hőt kell közölnünk, hogy megolvadjon, de a hőmérséklete nem változik, amíg az összes anyag el nem olvad. Ha egy folyamatban nem változik a rendszer nyomása, izobár folyamatról beszélünk. Ezek a legfontosabb folyamatok, hiszen a gyakorlatban a legtöbb átalakulás állandó, leggyakrabban légköri nyomáson megy végbe. Példaként gondoljunk egy nyitott lombikban lejátszódó kémiai reakcióra. Ha egy folyamat során a rendszer térfogata nem változik, a folyamat izochor (más szóval izosztér). Erre példa egy zárt edényben lezajló átalakulás (pl. reakcióhő mérése bomba-kaloriméterben, lásd a 4. fejezetben). Fontosak még az adiabatikus folyamatok is. Ebben az esetben a rendszer és környezete között nincs hőcsere a folyamat lejátszódása alatt. Például, ha egy gázt összenyomunk egy dugattyúban, amelyet a környezettől megfelelő szigetelés választ el, a gáz általában felmelegszik, de a környezetnek nem tud hőt leadni, a folyamat adiabatikus.
A rendszer fizikai állapotának, illetve a rendszerben lejátszódó folyamatoknak a jellemzésére az állapothatározók mellett további fizikai mennyiségeket szoktak bevezetni. Az állapotfüggvény az állapothatározók olyan többváltozós függvénye, amelynek változása csak a rendszer kezdeti és végállapotától függ, és független az úttól, amelyen a rendszer a kezdeti állapotból a végállapotba jutott. Az állapotfüggvények a fizikából jól ismert potenciális energiához hasonlóak. Valamely erőtérben (pl. elektromos vagy gravitációs) mozgó test potenciális energiája független az úttól, amin azt a tér egyik pontjából a másikba juttatjuk, csak attól függ, hogy a test honnan hova jutott. A legfontosabb állapotfüggvények a belső energia (U), az entalpia (H), az entrópia (S), a szabadenergia (A) és a szabadentalpia (G), amelyeket a következő fejezetekben részletesen meg fogunk vizsgálni.
Mivel egy állapotfüggvény értéke csak a kezdeti és végállapottól függ, beszélhetünk az állapot- függvény megváltozásáról egy adott folyamatban. Az állapotfüggvény megváltozása az állapot- függvény vég- és kezdeti állapothoz tartozó értékének különbsége, jelölésére a Δ görög betűt használjuk. Például ΔU a belső- energia-változás, ΔH az entalpiaváltozás stb. Infinitezimálisan kis változásokat a d szimbólummal jelölünk, pl. dU vagy dH a belső energia, illetve az entalpia végtelen kis mértékű változását jelenti. Mivel ezek az infinitezimális mennyiségek az adott állapotfüggvény infinitezimális megváltozásai, egyenlőek az állapotfüggvény teljes differenciáljával. Látni fogjuk majd, hogy speciális folyamatok (pl. izochor, izobár stb.) esetében az állapotfüggvények megváltozása összefüggésbe hozható valamilyen jól mérhető fizikai mennyiséggel, és a folyamat jól jellemezhető az állapotfüggvény megváltozásával. Ez adja az állapotfüggvények bevezetésének értelmét.
Az állapotfüggvényekkel szemben az útfüggvények értéke függ a kezdeti és a végállapot között megtett úttól. Útfüggvény pl. a munka (W) és a hő (Q). Tekintsünk például egy vízszintes súrlódó felületen mozgó testet (1.6. ábra). Nyilvánvaló, hogy ha a testet az 1-es számú úton juttatjuk el A pontból B-be, akkor kevesebb munkát kell végeznünk, mint ha a másik utat választatnánk. A két folyamat kezdeti és végpontja ugyanaz, de az 1-es és a 2-es úton végzett W1 és W2 munka különbözik.
Útfüggvények változását nem értelmezzük, az útfüggvények értéke egy adott folyamathoz kapcso- lódik, és már magában is változást fejez ki. Nem helyes tehát a „munkaváltozás” vagy a „hőváltozás”
szóhasználat, hanem az adott folyamatban végzett munkáról, a rendszerrel közölt vagy a rendszer által leadott, ill. a rendszer és a környezet között kicserélt hőről szoktunk beszélni, és ennek megfelelően nem írunk „ΔW”-t vagy a „ΔQ”-t sem. Az útfüggvények infinitezimális értéke nem teljes differenciál, azaz nem létezik olyan függvény, amely teljes differenciáljának képzésével megkapnánk egy útfüggvény infinitezimális értékét. Ez utóbbit ezért a δ szimbólummal jelöljük, pl. az infinitezimális munkára vagy a hőre a δW, ill. a δQ jelölések használatosak. Az útfüggvények értékének kiszámítása az infinitezimálisan kis értékek integrálásával történik, amelyhez ismerni kell a folyamathoz tartozó utat.
1.6. ábra. Az útfüggvények szemléltetése: ha egy testet eljuttatunk A-ból B-be, az 1-es és a 2-es úton végzett W1, ill. W2 munka nem egyenlő egymással
A termodinamikában használt mennyiségeket – mind az állapotjelzőket, mind az állapot- függvényeket – aszerint is osztályozhatjuk, hogy függenek-e a rendszer méretétől. Így megkülön- böztetünk extenzív és intenzív mennyiségeket. Az extenzív mennyiségek függenek a rendszer kiterjedésétől, azaz a rendszer méretével arányosan nőnek. Ilyen például a tömeg, a térfogat és a fent említett állapotfüggvények mindegyike. Nyilvánvaló, hogy az extenzív mennyiségek additívak, ami azt jelenti, hogyha a rendszert különálló részekre osztjuk, a rendszerre jellemző extenzív mennyiségek az alrendszerekre vonatkozó extenzív mennyiségek összegeként adódnak. Az intenzív mennyiségek függetlenek a rendszer kiterjedésétől és nem additívak. Intenzív mennyiség például a hőmérséklet, a nyomás és a koncentráció. Extenzív mennyiségeket könnyen intenzívekké alakíthatjuk, ha egységnyi tömegre, térfogatra, anyagmennyiségre stb. vonatkoztatjuk. Például az egységnyi térfogatra vonatkoz- tatott tömeg a sűrűség:
V
= m
ρ (1.1)
Gyakran használjuk a moláris mennyiségeket, amelyeket egy extenzív mennyiség és az anyagmennyiség hányadosaként kapunk. Jelölésükhöz az extenzív mennyiség jeléhez alsó indexben m betűt illesztünk. Így például a moláris térfogatot (más néven a móltérfogatot) a
Vm=V
n (1.2)
egyenlet definiálja, a moláris belső energiát pedig az Um= U
n (1.3)
alakban írjuk fel.
Egy rendszer állapotjelzői általában nem függetlenek egymástól. Egy egyensúlyban lévő rendszer állapotjelzői közötti kapcsolatot állapotegyenletnek nevezzük. Ennek klasszikus példája a tökéletes gázok állapotegyenlete
pV=nRT , (1.4)
ahol R az egyetemes gázállandó (Regnault-állandó), értéke 8,314 J mol-1 K-1. Ezt a törvényt egyetemes vagy általános gáztörvénynek is nevezzük. Valóságos anyagok állapotegyenletei empirikus függ- vények, és általában nem írhatók fel zárt formában, hanem hatványsor, diagram vagy táblázat for- májában adjuk meg őket.
1.2. A termodinamikai hőmérséklet és nyomás
A termodinamika legfontosabb intenzív állapotjelzői a hőmérséklet és a nyomás. Nemcsak jelen- tőségük miatt foglalkozunk kiemelten velük, hanem azért is, mert a hőmérséklet definiálása és mérése nem egyszerű, illetve mind a két állapotjelző esetében számos mértékegység van használatban.
A termodinamikában a nyomás nem új fogalom, a nyomást a fizika más területein is kiterjedten használják. A nyomás a felületegységre eső erő nagysága:
A
= F
p , (1.5)
ahol F az erő nagysága és A a felület. Általánosságban egy termodinamikai rendszerben F az A felületnek ütköző részecskék által a felületre kifejtett erő.
Érdemes bevezetnünk a hidrosztatikai nyomás fogalmát, amelyet többször alkalmazunk a későbbiekben. A hidrosztatikai nyomás a folyadékok súlyából származó nyomás. A Föld gravitációs terében lévő, h magasságú, ρ sűrűségű folyadékoszlop
ρgh
=
p (1.6)
hidrosztatikai nyomást fejt ki a folyadékoszlop alatt elhelyezkedő felületre, ahol g a nehézségi gyorsulás, g = 9,81 m/s2. Itt kihasználtuk a sűrűség definícióját, valamint hogy F = mg és A = V /h.
A nyomás SI egysége a pascal (Pa), ami az 1 m2-re ható 1 newton erőnek felel meg: 1 Pa = 1 N/m2. Tehát, ha azt mondjuk, hogy egy gáz nyomása 1 Pa, az azt jelenti, hogy egy 1 m2 felületű lapra, amit a gázba helyezünk, a lapnak folyamatosan ütköző gázrészecskék 1 N erővel hatnak. Elterjedten használják még a bár (bar) és az atmoszféra (atm) mértékegységet a nyomás számszerűsítésére: 1 bar
= 100 000 Pa és 1 atm = 101 325 Pa. Régies, de használatban lévő mértékegység a torr (Torr), definíció szerint 760 Torr = 1 atm. Alkalmazzák még a higanymilliméter (Hgmm vagy mmHg) egységet is, amit az 1 mm magas higanyoszlop által kifejtett hidrosztatikai nyomással definiálnak. A definíció alapján a higanymilliméterben mért nyomás elhanyagolhatóan kis mértékben ugyan, de függ a hőmérséklettől és a nehézségi gyorsulás értékétől. A gyakorlatban azonban a torrban és a
higanymilliméterben mért nyomás hibahatáron belül megegyezik, így a két mértékegység felcserélhető.
Gázelegyek esetén az elegy teljes nyomása mellett az elegyet a komponensek parciális nyomá- sával is szokták jellemezni. Az i-edik komponens parciális nyomása a gázelegy p nyomásának és a komponens yi móltörtjének a szorzata:
pi=yip . (1.7)
Nyilvánvaló, hogy a komponensek parciális nyomásainak összege megegyezik a teljes nyomással, ugyanis
∑
ipi=∑
i yip=p∑
i yi= p . (1.8)Tökéletes gázok elegyére igaz Dalton törvénye, amelyet kísérleti tapasztalatok alapján fogalmazott meg Dalton a XIX. században. Eszerint a parciális nyomás az a nyomás, amelyet a gáz akkor fejtene ki, ha egyedül töltené ki a rendelkezésre álló teret. A Dalton-törvény abból következik, hogy ideális gázok részecskéi között nincs kölcsönhatás (lásd 3. fejezet). Reális gázok esetében ez nem igaz, és ezért a törvény nem érvényes reális gázok elegyeire. Ez azonban ne tévesszen meg bennünket, mert a definíció alapján természetesen reális gázok elegyeire is igaz, hogy a komponensek parciális nyomásainak összege egyenlő a teljes nyomással.
A hőmérséklet tipikus termodinamikai fogalom, értelmezése sokkal nehezebb, mint a nyomásé.
A hőmérséklet fogalma a hideg- és melegérzetből fejlődött ki. Mindannyian el tudjuk dönteni, hogy két test közül melyik a hidegebb, illetve a melegebb, vagy hogy körülbelül nincs közöttük különbség.
Tapasztalatból azt is tudjuk, hogyha egy test érintkezik egy hidegebb testtel, akkor a melegebb test lehűl, a hidegebb felmelegszik, azaz hő áramlik a hidegebb testből a melegebbe. Hogy a testek közötti különbséget számszerűsíteni tudjuk és képesek legyünk a folyamatok irányának értelmezésére, szükségünk van a hőmérséklet fogalmára, illetve valamilyen hőmérsékleti skálára. A hőmérséklet definiálása nem könnyű feladat. A meglévő definíciók valamilyen anyag fizikai tulajdonságát használják ki – pl. a víz olvadás-, forrás- vagy hármaspontját – és ehhez mint vonatkoztatási ponthoz képest adják meg a hőmérséklet értékét egy mérési utasítással. Ez utóbbi régebben valamely anyag fizikai tulajdonságán alapult, pl. egy folyadék hőtágulásán. Ennek hátránya, hogy így a definíció függ az anyagtól, másrészt az adott fizikai tulajdonság mérése problémákba ütközhet, pl. függ a nyomástól, nem lineárisan változik a hőmérséklettel. Ezeknek a problémáknak az elkerülése végett vezették be a termodinamikai hőmérséklet fogalmát, amely ma az általánosan elfogadott hőmérsékletdefiníció.
A termodinamikai (abszolút) hőmérséklet (jele: T) definíciója a tökéletes gázok pV szorzatára épül, ami nyilvánvalóan anyagfüggetlen. A tökéletes gázok állapotegyenlete alapján bármely tökéletes gázra igaz, hogy T=pVm/R. Ezt az összefüggést közvetlenül alkalmazhatnánk a hőmérséklet definiálására, azonban fontos, hogy olyan definíciót találjunk, amely a gyakorlatban is alkalmazható.
Mivel a valóságban egy gáz csak a végtelen kis nyomás határesetében viselkedik tökéletes gázként, célszerű az előbbi kifejezés p=0 nyomásra vonatkozó határértékét venni és a hőmérsékletet a
m
p pV
= R
T 1lim 0
(1.9)
egyenlőséggel definiálni. A gyakorlatban a pVm szorzatot fokozatosan csökkenő nyomásértékeknél mérik, és az értékeket nulla nyomásra extrapolálják. Ahhoz, hogy egy hőmérsékleti skálát is definiáljunk, szükségünk van egy vonatkoztatási pontra. A termodinamikai hőmérsékletskála vonatkoztatási pontja a víz hármaspontja. A hármaspontban egy anyag mindhárom halmazállapota egyszerre van jelen (lásd 7. fejezet), a hármaspont hőmérséklete és nyomása anyagi állandók. A víz hármaspontjához definíció szerint a 273,16 értéket rendeljük. Ennek megfelelően a termodinamikai hőmérsékletskála mértékegysége a víz hármasponti hőmérsékletének 1/273,16 része, ezt kelvinnek nevezzük és K-val jelöljük. A definíció alapján tetszőleges T hőmérséklethez tartozó érték a
hármaspont m
p
T m p
pV T pV
) ( lim
) ( 16 lim
, 273
0 0
(1.10)
képlet szerint számítható, ahol (pVm)T és (pVm)hármaspont az adott hőmérsékleten, illetve a víz hármaspontján mért pVm érték.
A számos további hőmérsékletskála közül Európában ma legelterjedtebb a Celsius-féle, amelyet 1742-ben javasolt Andres Celsius svéd csillagász. A Celsius-skála két alappontja 1 atm nyomáson az olvadó jég hőmérséklete, ezt tekintjük 0 egységnek, illetve a forrásban lévő víz hőmérséklete, ez a 100 egység. A skála egysége ennek az intervallumnak a század része, ezt Celsius-foknak nevezzük, jele
○C. A Celsius-skálán mért hőmérsékletet t-vel jelöljük. A Celsius-féle skála történetileg sokkal előbb született, mint a termodinamikai hőmérsékletskála (ez utóbbit 1954-ben vezették be). A termodinamikai skálát úgy alkották meg, hogy egy egységnyi változás ezen a skálán ugyancsak egy egységnyi változásnak feleljen meg a Celsius-skálán. Ezért rendelték a 273,16-os értéket a víz hármaspontjához, ugyanis a Celsius-skálán a hármaspont hőmérséklete 0,01○C. Ekkor a víz olvadás- és forráspontja 273,15 K, illetve 373,15 K az abszolút skálán, és így különbségük pontosan 100 K. A két hőmérsékleti skála a
T/K = 273,15 + t/○C (1.11)
egyenlőség szerint számítható át egymásba. Megjegyezzük, hogy elvileg a víz olvadás- és forráspontját is választhatnánk a termodinamikai hőmérsékletskála alappontjainak. A hármaspont választásának azonban két előnye is van. Egyrészt így csak egy alappont van, és a hőmérséklet meg- határozásához csak egy pontban kell méréseket végezni, ami csökkenti a mérési hibát. Másrészről a hármaspont – ellentétben az olvadás- és forrásponttal – független a nyomástól, ami szintén növeli a mérés reprodukálhatóságát.
A gyakorlatban nehézkes lenne közvetlenül a definíciót követve mérni a hőmérsékletet, azaz a hármaspontban lévő vizet alkalmazva kis nyomású gázok nyomását és térfogatát mérni. Ezért valamilyen más fizikai tulajdonságon alapuló hőmérőket használunk a hőmérséklet mérésére. A hőmérséklet mérése azon a felismerésen alapul, hogy ha egy test hőmérséklete megegyezik egy másik test hőmérsékletével, és ez utóbbi hőmérséklete megegyezik egy harmadikéval, akkor az első és a harmadik test hőmérséklete is megegyezik. Ez a megfigyelés, amelyet gyakran a termodinamika nulladik főtételének is neveznek, szolgáltatja az alapot a hőmérők készítéséhez. Ha összeállítunk egy olyan készüléket, melynek valamilyen tulajdonságát – pl. a higanyoszlop magasságát egy kapillárisban – a hőmérséklet definíciójának megfelelően, a megadott mérési utasítás szerint kalibráljuk, akkor a készülék ugyanazt az értéket fogja mutatni, mint ha a hőmérsékletet közvetlenül a definíció alapján határoznánk meg. A mindennapi életben és a laboratóriumban legelterjedtebbek a folyadékok (pl.
higany vagy alkohol) hőtágulásán alapuló hőmérők. Hátrányuk, hogy csak abban a hőmérséklet- tartományban használhatók, ahol az anyag folyadék halmazállapotban van. Pl. a higanyos hőmérő csak
−39 ○C-ig működik, mert ezalatt a hőmérséklet alatt megfagy a higany. Gyakran használunk még különböző anyagok elektromos tulajdonságainak hőmérsékletfüggésén alapuló hőmérőket.
2. A TERMODINAMIKA I. FŐTÉTELE
2.1. A belső energia, a termodinamika I. főtétele
A mechanikában egy test mozgását felbontjuk a tömegközéppont mozgására, amelyet egy külső vonatkoztatási rendszerhez képest írunk le, és a testet felépítő tömegpontok mozgására, amelyet a testhez rögzített vonatkoztatási rendszerben tárgyalhatunk. A test teljes energiáját három részre bonthatjuk:
U + E + E
=
E kin pot , (2.1)
ahol Ekin és Epot a test tömegközéppontjának kinetikus és potenciális energiája, U pedig a belső energia.
A belső energia a rendszert felépítő részecskék kinetikus és potenciális energiájának az összege. Nem foglalja magában az egész rendszernek mint makroszkopikus testnek a kinetikus és potenciális energiáját. A termodinamikában kizárólag a termodinamikai rendszer belső energiáját vizsgáljuk, és nem foglalkozunk a teljes rendszer kinetikus és potenciális energiájával egy külső vonatkoztatási rendszerhez képest.
Egy termodinamikai rendszer belső energiája különböző járulékokból tevődik össze:
1. Termikus energia. Ez a rendszert alkotó részecskék (atomok, molekulák, ionok) mozgásához kapcsolódó kinetikus energia. Ez a járulék további részekre bontható. Egyik összetevője a részecskék tömegközéppontjának haladó mozgásából származó kinetikus energia. Emellett a nem pontszerű részecskék (molekulák, ill. ionjaik) még forgó mozgást is végeznek, valamint az őket alkotó atomok rezegnek egymáshoz képest. Ezeknek a mozgásoknak az energiája is idetartozik. A járulékot azért nevezzük termikus energiának, mert a hőmérséklet változta- tásával elsősorban ez változik. Például, ha hőt közlünk a rendszerrel, és növeljük a hőmérsékletet, a közölt energia elsősorban a részecskék haladó, forgó és rezgő mozgásának energiáját növeli.
2. Intermolekuláris energia. Ez a részecskék között ható másodrendű kölcsönhatások potenciális energiája. Elsősorban a részecskék közötti átlagos távolságtól függ, csak kevéssé változik a hőmérséklettel. A belső energia intermolekuláris része legnagyobb mértékben fazesátme- neteknél változik. Például egy folyadék elpárologtatásakor befektetett energia (a párolgáshő) a folyadék részecskéi között ható vonzó erők legyőzésére szükséges.
3. Kémiai energia. A kémiai energia az atomokat és molekulákat felépítő elektronok kinetikus energiájából és az elektronok, valamint a magok közötti kölcsönhatások potenciális ener- giájából tevődik össze. A járulék főleg a kémiai kötések létesítése és felbomlása, azaz kémiai reakciók esetén változik meg.
4. Magenergia. Az atommagokat felépítő nukleonok (protonok és neutronok) energiája.
Természetesen a felosztást tovább folytathatnánk, és figyelembe vehetnénk a nukleonokat felépítő elemi részecskék energiáját is. Ezt azonban nem tesszük, hiszen pontosan nem ismerjük ezen részecskék közötti kölcsönhatások természetét, sőt magukat a részecskéket sem. Ebből következik, hogy a belső energia pontos értékét nem tudjuk megadni. Ez nem okoz különösebb gondot, mivel a termodinamikában tárgyalt fizikai és kémiai változásokban a belső energiának csak a termikus, intermolekuláris és a kémiai része változik észrevehető mértékben, a magenergia, illetve a belső energia további nem ismert járulékai gyakorlatilag nem változnak, ezért a belső energia változását (ΔU) meg tudjuk adni.
Megjegyezzük, hogy a belső energia abszolút értékét elvileg mégis megadhatjuk valamilyen önkényesen választott vonatkoztatási ponthoz képest, amelyben a belső energia értékét rögzítjük (pl.
feltesszük, hogy 0). A 4. fejezetben látni fogjuk, hogy ezt meg is teszik, de nem a belső energia
vonatkoztatási pontját választják meg, hanem egy gyakorlati szempontból fontosabb állapotfüggvény, az entalpia értékét rögzítik egy bizonyos pontban. Ez a kikötés a belső energia értékét is meg- határozza.
A termodinamika I. főtétele az energiamegmaradás törvényének alkalmazása termodinamikai rendszerekre. Az energiamegmaradás törvénye szerint az energia átalakítható egyik formából a másikba, de nem lehet létrehozni vagy megsemmisíteni. Ez alapján a termodinamika I. főtétele azt mondja ki, hogy zárt rendszer belső energiája csak munkavégzés vagy hőcsere útján változtatható meg. Matematikai formában a főtételt a
Q + W
ΔU= (2.2)
alakba írhatjuk egy véges változásra. Infinitezimális változásra a főtétel a δQ
δW+
=
dU (2.3)
egyenlettel ekvivalens. Az első főtétel tehát azt mondja ki, hogy egy zárt rendszer belső energiája csak akkor változhat, ha a rendszer munkát végez a környezettel szemben (pl. egy gáz kitágul), vagy a környezet végez munkát a rendszeren (pl. összenyomjuk a gázt), illetve ha a rendszerrel hőt közlünk (pl. melegítjük a rendszert), vagy hőt vonunk el a rendszerből (pl. hűtjük). Elszigetelt rendszerek esetében az anyag- és energiaáramlás nem lehetséges a rendszer és a környezet között, így az I. főtétel zárt rendszerre vonatkozó alakjából triviálisan következik, hogy elszigetelt rendszerek belső energiája állandó. Egyenletekkel megfogalmazva: az I. főtétel véges és végtelenül kicsi változásokra a
0
ΔU= , (2.4)
illetve a
0
=
dU (2.5)
formában adható meg elszigetelt rendszerekre. Megjegyezzük, hogy az I. főtétel nyílt rendszerekre vonatkozó alakja szintén levezethető a zárt rendszerre vonatkozó alakból. Ezzel később, a 4. feje- zetben foglalkozunk. Az első főtétel triviális következménye, hogy a belső energia állapotfüggvény.
A fentiekből is látható, hogy a termodinamikában központi szerepe van a munkának és a hőnek, ezért a továbbiakban részletesebben foglalkozunk ezzel a két mennyiséggel.
2.2. A munka
A mechanikai munka az erő és az elmozdulás skalárszorzata:
dl F
δW= (2.6)
A munka mértékegysége a joule, jele: J. A termodinamikában a legtöbbet a térfogati munkával találkozunk, ezért először foglalkozzunk ezzel! A térfogati munkát a Wtérf szimbólummal jelöljük.
Kiszámításához tekintsünk egy dugattyút, amelyben valamilyen gáz van (2.1. ábra). A gáz nyomása p, a külső nyomás pk. A dugattyúraFerő hat, és a dugattyú elmozdulásvektora adlvektor; a két vektor abszolút értékét F és dl jelöli, és feltesszük, hogy a két vektor ellentétes irányú. Mivel a két vektor párhuzamos, a mechanikai definíció és a skalárszorzat tulajdonságai alapján kapjuk, hogy
dl , F= dl F
δWtérf = (2.7)
ahol a negatív előjel a két vektor ellentétes irányából adódik. Tudjuk továbbá, hogy a dugattyúra ható nyomás a dugattyúra hatóFerő nagyságának és a dugattyú A felületének a hányadosa:
A
= F
pk , (2.8)
ezért
Adl p
δWtérf = k . (2.9)
Ha az A felületű dugattyú infinitezimális elmozdulása dl, akkor a dugattyú térfogata Adl-lel változik, ezt jelöljük dV-vel. Ezt behelyettesítve a fenti egyenletbe a térfogati munka definícióegyenletét kapjuk:
dV p
δWtérf = k (2.10)
Ez az egyenlet tehát megadja a végzett térfogati munka nagyságát, ha a rendszer térfogata végtelenül kis mértékben, dV-vel változik. Véges változás esetén az előbbi egyenlet mindkét oldalát integrálni kell a megfelelő határok között, a bal oldalt 0-tól Wtérf-ig, a jobb oldalt a kiindulási térfogattól (V1) a végső térfogatig (V2):
2
1
V
V k
térf p dV
W (2.11)
Ez a kifejezés a térfogati munka alternatív definíciójának tekinthető.
2.1. ábra. A térfogati munka értelmezése
A térfogati munka definíciója alapján nyilvánvaló, hogy amennyiben a térfogat nő (dV pozitív), a munka negatív, azaz a rendszer belső energiája csökken, a rendszer végez munkát a környezeten.
Fordítva, ha a térfogat csökken (dV negatív), a munka pozitív, tehát a rendszer belső energiája nő, a környezet végez munkát a rendszeren. Figyeljük meg, hogy a térfogati munka ezen tulajdonságai összhangban vannak a munkavégzésről alkotott szemléletes képünkkel: a rendszeren végzett munka növeli annak energiáját, és fordítva. Ez igazolja, hogy adlelmozdulásvektor irányának önkényes megválasztása helyes volt: ha a vektort azonos irányúnak vettük volna a dugattyúra ható erő vektorával, a fenti állítások ellenkezője teljesülne.
A térfogati munka kifejezésében a külső nyomás szerepel. Ez a legtöbb esetben előnytelen számunkra, hiszen a rendszerben végbemenő folyamat leírásához egy olyan mennyiség ismeretét feltételezi, amely általánosságban független a rendszer állapotjelzőitől. Azonban van egy kitüntetett eset, amikor a térfogati munka pusztán a rendszer állapothatározói ismeretében kiszámítható. Ha feltesszük, hogy a térfogati munkavégzés reverzibilis úton megy végbe, az azt jelenti, hogy a rendszer minden pillanatban egyensúlyban van a környezetével, és a külső nyomás és a rendszer nyomása csak végtelenül kis mértékben térnek el egymástól. Ekkor a térfogati munka kifejezésében a külső nyomás lecserélhető a rendszer nyomásával, azaz
pdV
δWtérf = , (2.12)
illetve véges folyamatra
2
1
V térf = V pdV
W . (2.13)
Ez az első pont, ahol nyilvánvalóvá válik számunkra a reverzibilitás feltételezése. Ha ezt kikötjük, lehetővé válik, hogy a térfogati munkát a rendszer nyomásával számítsuk, és – mint látni fogjuk a későbbiekben – ez nagyban egyszerűsíti majd a levezetéseket.
A másik fontos speciális eset, amikor a térfogati munkavégzés állandó nyomáson történik. Ekkor a térfogati munka számítása ismét egyszerűsödik. A nyomást az integráljelen kívülre vihetjük, és az integrálást triviálisan elvégezhetjük:
V p dV p pdV
W V
V V
térf
V12
12 , (2.14)ahol ΔV a folyamatot kísérő térfogatváltozás.
2.2. ábra. A térfogati munka szemléltetése: az indikátordiagram
A 2.2.-es ábrán egy gáz térfogatváltozása során végzett térfogati munka számítására látunk példákat. Megjegyezzük, hogy a hasonló diagramokat gyakran indikátordiagramnak nevezik, és kiterjedten alkalmazzák a hőerőgépek elméletében a térfogati munka meghatározására. Mindkét esetben a piros görbe a gáz nyomását adja meg a gáz térfogatának függvényében, állandó hőmérsékleten. Szintén mind a két folyamatra igaz, hogy a gáz ugyanabból a kiindulási állapotból (1) ugyanabba a végállapotba (2) jut el. Az a esetben nem teszünk egyebet, minthogy összenyomjuk a gázt az adott hőmérsékleten. A fentiek alapján a térfogati munka számításához integrálnunk kell a gáz nyomását leíró függvényt az 1-es pont térfogatától a 2-es pont térfogatáig. Az integrál értéke pontosan a görbe alatti terület. A b jelű ábrán kerülő úton jutunk el 1-ből 2-be. Először állandó térfogaton lehűtjük a gázt úgy, hogy a nyomása a végső nyomásra csökkenjen (I. szakasz). Második lépésben állandó nyomáson felmelegítjük a gázt a kívánt hőmérsékletre (II. szakasz). Az első lépésben nem végeztünk térfogati munkát, hiszen a térfogat nem változott. A második lépésben állandó nyomáson kiterjesztettük a gázt, így a végzett térfogati munka a nyomás és a térfogatváltozás szorzatából számítható, értéke az ábrán sötéttel jelzett négyszög területével azonos.
Mint korábban említettük, a munka útfüggvény, azaz értéke függ az úttól, amelyen a rendszer az egyik állapotból a másikba jut. Természetesen a térfogati munka sem kivétel ez alól, amit az előző példa is bizonyít. Két különböző úton jutottunk el 1-ből 2-be, és jól látható, hogy a két úton végzett munka nem egyenlő egymással. A térfogati munka tehát útfüggvény.
A térfogati munkán kívül sokféle más munka is előfordul a termodinamikában, néhány példát a 2.1. táblázatban találhatunk. Vegyük észre, hogy az elemi (infinitezimális) munka mindig egy intenzív mennyiség és egy extenzív mennyiség megváltozásának szorzata. A munkavégzés, azaz az energia- transzport mindig az intenzív mennyiség inhomogenitása miatt történik a rendszer és környezete között. A határfelületi munka egy folyadék felszínén új felület létrehozásakor végzett munka. Hajtó-
ereje a felületi feszültség (a folyadékfelület egységnyi hosszúságú vonalában ható erő) inhomogeni- tása. Az elektromos munkavégzés az elektromos potenciál inhomogenitása következtében történik. Ha a rendszernek és környezetének eltér az elektromos potenciálja, a pozitív töltések a nagyobb potenciálú helyről a kisebb potenciálú helyre áramlanak, a negatív töltések pedig az ellenkező irányba.
A folyamatban a rendszerrel közölt vagy a rendszerből kivont energia az elektromos munka.
2.1. táblázat. Néhány munka a termodinamikában
Munka Intenzív mennyiség Extenzív mennyiség Elemi munka
térfogati nyomás (-p) térfogat (V) δWtérf =pdV
határfelületi felületi feszültség (γ) felület (A) δWfel=γdA
elektromos elektromos potenciál (φ) töltés (q) δWel=φdq
A fentiek alapján megadhatjuk a munka általános termodinamikai definícióját. Eszerint a munkavégzés a rendszer határfelületén fellépő energiatranszport, amelyet a folyamathoz tartozó (hőmérséklettől különböző) intenzív állapotjelző inhomogenitása mint hajtóerő hoz létre. Az energiatranszport által átadott energiát munkának nevezzük.
2.3. A hő
A hő, ellentétben a munkával, tipikusan termodinamikai fogalom. A hő is egy energiatranszport- mennyiség, de itt az energiatranszport, azaz a hőcsere (hőátmenet) egyedüli hajtóereje a rendszer és a környezet közötti hőmérséklet-különbség. Másik fontos eltérés a munkához képest, hogy a hőt nem kíséri anyagtranszport, mint például az elektromos munkát, ahol a töltések áramlásához a töltéseket hordozó részecskéknek is mozogniuk kell. A hőcsere tehát a rendszer határfelületén fellépő anyag- transzport nélküli energiatranszport, amelyet a hőmérséklet inhomogenitása mint hajtóerő hoz létre.
Az energiatranszport által átadott energiát hőnek nevezzük. A hő mértékegysége szintén a joule (J).
A kémiai szempontból fontos hőátmenettel járó folyamatok a melegítés, illetve a hűtés, a fázisátalakulások és a kémiai reakciók. Itt most az első kettővel foglalkozunk, a kémiai reakciókat kísérő hőátmeneteket a 4. fejezetben fogjuk tárgyalni.
Ha melegítjük vagy hűtjük a rendszert, akkor hőt közlünk vele, illetve hőt vonunk el a rend- szerből, azaz növeljük vagy csökkentjük a rendszer energiáját. Mindkét folyamat hajtóereje a rendszer és a környezetének eltérő hőmérséklete. Az energiatranszport hatására a rendszer hőmérséklete nő vagy csökken. Adott mennyiségű hő hatására különböző anyagok esetében a hőmérséklet eltérő mértékben változik. Az arányossági tényezőt az átadott hő és az általa előidézett hőmérséklet-változás között hőkapacitásnak nevezzük:
CdT
δQ= (2.15)
A hőkapacitást általában C-vel jelöljük, és mértékegysége a J/K. A hőkapacitás általános esetben nem állandó, hanem a hőmérséklet függvénye, a hőmérsékletfüggést gyakran a C(T) jelöléssel fejezzük ki.
Vegyük észre, hogy a hőmérsékletfüggés miatt használtunk a fenti összefüggésben infinitezimális mennyiségeket. Véges mennyiségű hőre és véges hőmérséklet-változásra nem lenne igaz az egyen- lőség, hiszen a hőmérséklet véges változása a hőkapacitás változását vonja maga után.
Beszélhetünk egy adott rendszer (pl. egy kaloriméter) hőkapacitásáról, de általában a hőkapacitást egységnyi tömegű vagy anyagmennyiségű anyagra vonatkoztatjuk. Ez előbbi a fajlagos hőkapacitás (fajhő), amely tehát 1 kg anyag hőkapacitása, jele c, mértékegysége J kg-1 K-1. Gyakrabban al- kalmazzuk az anyagmennyiségre vonatkoztatott hőkapacitást, ez a moláris hőkapacitás (mólhő), amely tehát 1 mol anyag hőkapacitása, jele Cm, mértékegysége J mol-1 K-1. A fajlagos és a moláris hőkapacitás segítségével m tömegű, illetve n anyagmennyiségű anyag esetén a hőkapacitás a C=mc, illetve a C=nCm formában írható. Felhasználva pl. az utóbbit az infinitezimális hő és az általa előidézett hőmérséklet-változás között, a
T dT nCδQ= m (2.16)
összefüggés áll fenn. Ahhoz, hogy egy véges változás hőjét kiszámítsuk, ezt a kifejezést integrálnunk kell a kiindulási (T1) és a végső (T2) hőmérséklet között:
2
1
) (
T
T Cm T dT n
Q (2.17)
Ha a hőkapacitás állandó vagy legalábbis jó közelítéssel állandónak tekinthető az adott hőmérséklet- tartományban, akkor Cm(T) kihozható az integráljel elé, és az integrálás triviálisan elvégezhető:
2
1
T
T m
m dT nC T
nC
Q (2.18)
Hasonló összefüggések igazak a fajlagos hőkapacitásra is.
A hő, mint a munka, útfüggvény. Ennek belátásához tekintsük ismét a 2.2. ábrát! A rendszer az egyes pontból két különböző úton jut el a kettesbe, és láttuk, hogy a két esetben eltérő a végzett munka. Viszont az I. főtétel alapján tudjuk, hogy a belső energia változása azonos a két úton, és egyenlő a végzett munka és a közölt hő összegével. Mivel a munkák különböznek, a hőknek is eltérőeknek kell lenniük, tehát a hő mennyisége függ az úttól, amelyen a folyamat végbement.
A fentiek értelmében a hő számításához mindig meg kell adnunk az utat, amelyen a hőközlés történt. Leggyakoribb az állandó nyomáson vagy az állandó térfogaton végbemenő hőcsere. Ezen folyamatok hőjére a Qp, illetve QV jelöléseket használjuk. Jelentőségük miatt általában a hőkapacitás- függvényeket is erre a két folyamatra adják meg. Ezeket rendre állandó nyomáson vett (moláris) hőkapacitásnak, illetve állandó térfogaton vett (moláris) hőkapacitásnak nevezzük, és Cmp-vel, illetve CmV-vel jelöljük. Mivel a nyomás, illetve a térfogat állandó, rögzítettük az utat, amelyen a folyamat végbemegy. Ekkor a hő megadható mint a hőmérséklet függvénye, és az infinitezimális hő teljes differenciál:
T dT nC= dQ
δQp= p mp (2.19)
T dT nC= dQ
δQV = V mV (2.20)
Ebből következik, hogy Cmp, illetve CmV megadható az alábbi deriváltakkal:
dT dQ= n T
Cmp 1 p
(2.21)
dT dQ= n T
CmV 1 V
(2.22) Ezek az egyenletek az állandó nyomáson, illetve térfogaton vett hőkapacitás definíciójának is tekinthetőek. Cmp, illetve CmV segítségével az állandó nyomáson, illetve állandó térfogaton lejátszódó véges folyamat hője a
2
1
) (
T
T mp
p n C T dT
Q (2.23)
és
2
1
)
T (
T mV
V n C T dT
Q (2.24)
képletek szerint számítható. Az állandó nyomáson vett hőkapacitás mindig nagyobb, mint az állandó térfogaton vett, mivel az állandó nyomáson végzett melegítésnél a rendszer kitágul, térfogati munkát végez a környezettel szemben, és a befektetett energia egy része erre fordítódik és nem a hőmérséklet emelésére.
Az anyagok hőkapacitás-függvényei általában nem írhatók valamilyen egyszerű alakba.
Leggyakrabban hatványsorok formájában adják meg a függvényeket, pl.
T =a+bT+cT +dT ,Cmp 2 2 (2.25)
ahol a, b, c és d valós számok, amelyeket kísérleti vagy számított hőkapacitás-értékekhez illesztenek.
A melegítéskor vagy hűtéskor átadott vagy elvont hő a hőkapacitás-függvény integrálásával számolható:
2
1
) 3(
) (
) 2(
) (
)
( 2 2 2 1 22 12 21 11 23 13
T
p T d T T
T T c T b T
T T a n dT dT cT
bT a n Q
(2.26) Elterjedt még a hőkapacitás-függvények táblázatos megadása. Ebben az esetben a hőkapacitás- függvény értékeit adják meg különböző hőmérsékleteken, a hiányzó hőmérsékletekre vonatkozó hőkapacitás-értékeket interpolációval határozhatjuk meg.
Mint említettük, a másik fontos hőátmenettel járó folyamat a fázisátalakulás (fázisátmenet). A fázisátalakulás olyan izoterm és izobár folyamat, amely során az anyag átalakul az egyik fázisból egy másik fázisba. A folyamat hajtóereje (reverzibilitást feltételezve) a rendszer és a környezet közötti infinitezimális hőmérséklet-különbség. A legismertebb fázisátalakulás az olvadás, a forrás és a szublimáció, de emellett még számos más fázisátalakulást tartunk számon. Ilyen például egy anyag kristályszerkezetének átalakulása – gondoljunk pl. a monoklin kén rombos kénné való alakulására –, a folyadékkristályok különböző fázisai közötti átmenet vagy a szupravezető tulajdonságú anyagok átalakulása a vezetőből a szupravezető állapotba egy bizonyos hőmérsékleten. A fázisátmenet hőmér- séklete és nyomása nem független egymástól, egy tiszta anyag esetén csak az egyiket választhatjuk szabadon (lásd 7. fejezet). A legtöbb fázisátalakulás hőátmenettel járó folyamat. Az átadott hőt látens hőnek is nevezik utalva arra, hogy a hőközlés hatására nem változik a hőmérséklet. A legfontosabb fázisátmenetek, az olvadás, a forrás és a szublimáció látens hőjét olvadáshőnek, párolgáshőnek, illetve szublimációs hőnek nevezzük. Ezeket általában 1 mol anyagra vonatkoztatják, és ekkor a moláris olvadás-, párolgás- és szublimációs hő elnevezést is használják.
2.4. Az entalpia
Vizsgáljuk először az állandó térfogaton lejátszódó folyamatok belsőenergia-változását egy zárt rendszerre! Tegyük fel, hogy nincs egyéb munka. Mivel a térfogat állandó, térfogati munka sem lehetséges, így nincs semmilyen munkavégzés. Ekkor az I. főtétel alapján a belső energia meg- változása egyenlő a rendszerrel közölt hővel:
QV
=
U , (2.27)
illetve infinitezimális változásra:
δQV
=
dU (2.28)
A fenti egyenlőségek jelentősége abban áll, hogy az adott körülmények között egy útfüggvény, a hő értékét ki tudjuk fejezni egy állapotfüggvény, a belső energia megváltozásával. Az állandó térfogaton vett hőkapacitás definíciója alapján további fontos összefüggéseket tudunk levezetni. A QV
számítására vonatkozó képlet alapján a belső energia véges megváltozása a
2
1
) (
T
T CmV T dT n
U (2.29)
integrállal számolható. Ha a hőkapacitást állandónak tekintjük, kivihető az integráljel elé, és az integrál egyszerűen számítható, ekkor:
ΔT nC
= U mV
(2.30)
Az infinitezimális belsőenergia-változást szintén kifejezhetjük az állandó térfogaton vett hőkappa- citással:
T dT nC=
dU mV (2.31)
Ebből adódik, hogy a hőkapacitás felírható a belső energia hőmérséklet szerinti parciális derivált- jaként:
V
mV T
U
= n T
C
1 , (2.32)
ahol azért írhattunk parciális deriváltat, mert zárt rendszerben a belső energia megadható mint két változó – a hőmérséklet és a térfogat – függvénye, és a térfogatot rögzítettük.
Látjuk tehát, hogy az állandó térfogaton lejátszódó folyamatokat jól jellemzi a belső energia. A kémiában viszont nagyon gyakori az állandó nyomás. Ezért definiáltak egy olyan függvényt, amellyel az állandó nyomáson végbemenő folyamatokat jellemezhetjük. Ez a függvény az entalpia, amelynek definíciója:
pV + U
=
H (2.33)
Az entalpia állapotfüggvény, hiszen a belső energia állapotfüggvény, és a pV szorzat nyilvánvalóan független az úttól, amin a rendszer az adott állapotba került. A definíció alapján az entalpia energiadimenziójú, mértékegysége a joule (J).
Az entalpia fizikai értelemének vizsgálatához először képezzük a teljes differenciálját:
U+pV
=dU+d
pV =dU+pdV+Vdp d=
dH (2.34)
Tegyük fel, hogy kizárólag térfogati munka lehet, és a változás reverzibilis. Ekkor a belső energia infinitezimális megváltozása:
δQ + pdV δQ=
δW+
=
dU (2.35)
Ezt behelyettesítve az előbbi egyenletbe kapjuk:
Vdp δQ+
=
dH (2.36)
Ha a nyomás állandó, akkor dp=0, ezért az entalpia végtelenül kicsi megváltozása egyenlő az infinitezimális hővel:
dH=δQp (2.37)
Hasonló összefüggés igaz az entalpia véges változására is. Állandó nyomáson az entalpia definíciója alapján:
V p + U
=
H
(2.38)