APPLICATION OF GRAPH MODELS IN BIOINFORMATICS
T
HESIS FOR THED
EGREE OFD
OCTOR OFP
HILOSOPHYB ALÁZS L IGETI
Roska Tamás Doctoral School of Sciences and Technology Pázmány Péter Catholic University,
Faculty of Information Technology and Bionics
Supervisor:
Prof. Sándor Pongor
Budapest, 2016
Abstract
Biomedical sciences use a variety of data sources on drug molecules, genes, proteins, complete genomes sequences, diseases and scientific publications, etc. This system can be best pictured as a giant data-network linked together by physical, functional, logical and similarity relationships. A new hypothesis or discovery can be considered as a new link that can be deduced from the existing connections. For instance, interactions of two pharmacons – if not already known – represent a testable novel hypothesis. Such implicit effects are especially important in complex diseases such as cancer. Currently huge amount of data is generated by experiments, such as whole genome sequencing of metagenomic data. Deriving new information, i.e. linking the experiments to microorganisms and supporting the new hypothesis with known data requires the proper analysis of a data-network.
The goal of the investigations carried out in this thesis is to predict novel drug combinations or novel biomarkers using the network of existing oncological and protein interaction databases and to interpret and analysis large metagenomic data using network principles.
I showed that the overlap of network neighborhoods is strongly correlated with the pairwise interaction strength of two pharmacons used in cancer therapy, and it is also well correlated with clinical data.
The strategy based on the hypothesis that novel, implicit links can be discovered between the network neighborhoods of data items lead to the discovery of novel biomarkers based on text analysis. In 2012 I prioritized ten potential biomarkers for ovarian cancers, two of which were in fact described as such in the subsequent years.
I showed that applying network principles and fast aligners in the evaluation of metagenomic whole genome sequencing experiments could improve the classification performance, and even sensitive detection of pathogens is possible.
The strategy seems to hold promises in several applications including prioritization of new drug combinations, discovering of novel biomarkers for experimental testing or sensitive detection of pathogens. Its use is naturally limited by the sparsity and the quality of experimental data; however,
Acknowledgements
I would like to express my deepest gratitude to my advisor, Professor Sándor Pongor, for his valuable guidance and support, and that he made me possible to work on a topic of great interest to me. This thesis would have not been finished without his endless encouragement. I am also grateful to Professor Tamás Roska for his inspiration and that he made me believe I have that certain marshal’s baton as well.
My sincere appreciation is extended to Professor Péter Szolgay for his unfailing support. This work would not exist without the support of the Faculty of Information Technology and Bionics, Pázmány Péter Catholic University.
I am also grateful to Balázs Győrffy and Gergely Lukács for their advices, comments and critics.
I also want to express my deepest appreciation to Roberto and Lőrinc: our work together provided the stimulating atmosphere (filled with a lot of fun) every scientist needs sometimes.
Here I have to mention my fellow PhD students and colleagues: Zsolt, Dóri, Endre, István, Zoltán, Norbi, Bence, Tamás, Miklós, Berci, Domokos, and many others. Having lunch together and discussing the big questions of life was always a refreshing moment of the day.
Last but not the least, I would like to thank my family: Noémi, for being my wife, my colleague, my secretary and my editor; my children, Emese, Alíz and Ágoston, for being the steady point in my life during these years.
A special thanks goes to Rufus: the one purring on my shoulders while writing this.
Table of Contents
Abstract ... ii
Acknowledgements ... iii
List of Abbreviations ... vi
List of Tables ... vii
List of Figures ... viii
1. Introduction ... 1
1.1. Biological background ... 2
1.1.1. Chemotherapy ... 2
1.1.2. Systemic therapy of recurrent or metastatic breast cancer ... 5
1.1.3. Targeted molecular therapy ... 6
1.1.4. HER2 positive breast cancer ... 6
1.1.5. Ovarian cancer ... 8
1.1.6. Future perspectives ... 9
1.2. Metagenomics concepts ... 10
1.3. Networks in biology ... 12
1.4. From databases to data networks ... 22
1.5. Graph – definitions and notations ... 23
1.6. Network analysis techniques ... 26
1.6.1. Random walk based algorithm: PageRank ... 26
1.6.2. Random walk and kernel methods ... 28
1.6.3. Kernels on graph ... 29
1.6.4. Methods for graph kernel computation ... 33
1.6.4.1. Krylov space methods ... 34
1.6.4.2. Arnoldi algorithm ... 35
1.6.4.3. Approximating the matrix exponential ... 36
1.6.5. Ranking in networks ... 36
1.6.5.1. Ranking by using PageRank with priors ... 37
1.6.5.2. Ranking based on kernels ... 37
1.6.5.3. Measuring ranking performance ... 38
1.6.6. Inference in ontologies ... 39
2. Databases and methods... 42
2.1. Databases ... 42
2.2. Data preprocessing ... 43
2.3. Methods: programs and environments ... 44
3. Results and discussion... 47
3.1. Discovering novel drug combinations... 47
3.1.4. TOS shows correlation with the outcome of clinical trials ... 56
3.1.5. Discussion ... 59
3.2. Prediction of cancer biomarkers by integrating text and data networks ... 60
3.2.1. Theory ... 61
3.2.2. Constructing a data-network with molecular and literature-based links ... 62
3.2.3. Principle of evaluation ... 63
3.2.4. Testing the methods on the rediscovery of known OC biomarker genes ... 64
3.2.5. Prediction of new OC biomarkers ... 65
3.2.6. Discussion ... 66
3.3. Inference on hierarchical graphs: fast and sensitive alignment of microbial whole metagenome sequencing reads ... 68
3.3.1. Taxoner algorithm ... 68
3.3.2. Execution times ... 70
3.3.3. Compatibility with various sequencing platforms ... 71
3.3.4. Detecting an unknown anthrax strain ... 72
3.3.5. Detection of very low abundance reads ... 74
3.3.6. Analyzing metagenomic datasets ... 74
3.3.7. Discussion ... 76
4. Conclusions and new scientific results ... 78
4.1. Network neighborhood analysis – revealing unexpected relationships ... 79
4.1.1. Prediction of efficient drug combinations ... 80
4.1.2. Prediction of cancer biomarkers by integrating text and data networks ... 81
4.2. Fast and sensitive characterization of microbial studies ... 82
5. Publications ... 84
6. References ... 85
List of Abbreviations
AC Drug regimen consisting of doxorubicin and cyclophosphamide
Bp Base pair
BWT Burrows-Wheeler transforms
CAF Drug regimen consisting of 5-fluorouracil, doxorubicin and cyclophosphamide CLI Command line interface
CMF Drug regimen consisting of 5-fluorouracil, methotrexate and cyclophosphamide DBMS Database management system
EC Drug regimen consisting of epirubicin and cyclophosphamide EGFR Epidermal growth factor receptor
ER Entity–relationship (model)
FAC Drug regimen consisting of 5-fluorouracil, doxorubicin and cyclophosphamide FEC Drug regimen consisting of 5-fluorouracil, epirubicin and cyclophosphamide
GO Gene ontology
HER2 Erb-b2 receptor tyrosine kinase 2
LCA Lowest common ancestor
NCCN National comprehensive cancer network
OC Ovarian cancer
TAC Drug regimen consisting of 5-fluorouracil, doxorubicin and cyclophosphamide TOS Target overlap score
WGS Whole-genome shotgun
XML Extensible markup language
List of Tables
Table 1.1. Chemotherapy combinations for recurrent or metastatic breast cancer adapted from the Guidelines of the US National Comprehensive Cancer
Network ... 5
Table 1.2. Cancer-related databases and resources ... 17
Table 1.3. Representative examples of molecular and molecular interaction databases relevant to cancer therapy ... 20
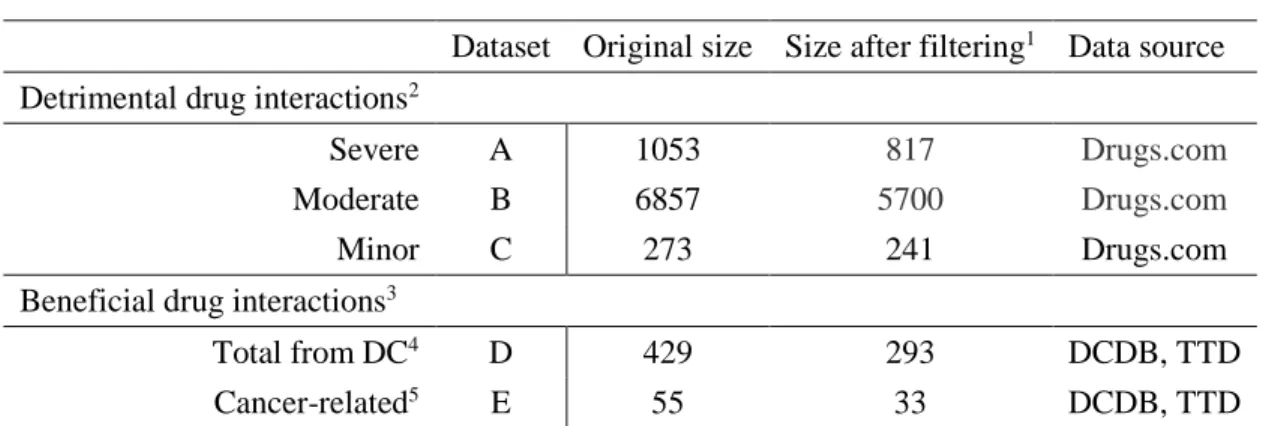
Table 3.1. Datasets ... 54
Table 3.2. TOS scores of binary and multicomponent combinations ... 58
Table 3.3. List of ovarian cancer biomarker genes published before May 2012 ... 63
Table 3.4. List rediscovery of genes suggested as OC biomarkers ... 65
Table 3.5. Predicted OC biomarker genes ... 66
Table 3.6. Benchmark datasets ... 70
Table 3.7. Average execution times of alignments ... 71
Table 3.8. Read assignment for Staphylococcus aureus genome sequencing data ... 72
Table 3.9. Identification of taxa in even MOCK community ... 75
List of Figures
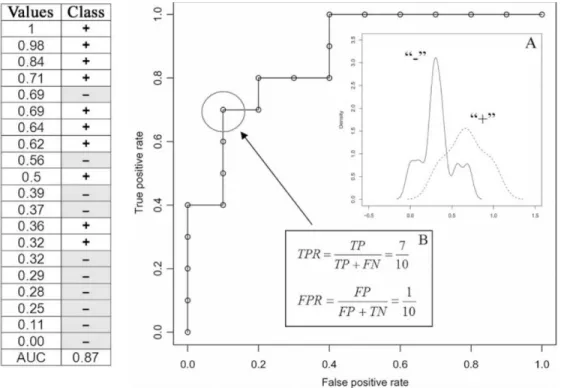
Figure 1.1. Constructing a ROC curve from ranked data (taken from Sonego et al.
[1]) 40
Figure 3.1. The network interaction hypothesis ... 49
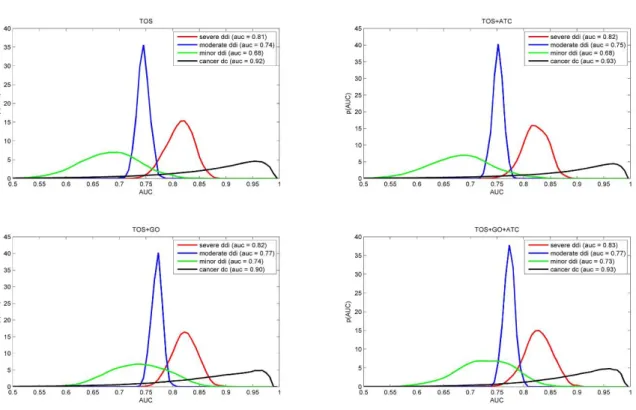
Figure 3.2. Prediction performance on known drug interactions and combinations ... 53
Figure 3.3. Flow chart of the training and the prediction procedure ... 55
Figure 3.4. Performance of combined predictors on different training sets ... 57
Figure 3.5 Scatter plot of prediction scores and Overall Response ... 58
Figure 3.6. The principle of biomarker prediction using terms rarely associated with cancer and a set of validated genes ... 62
Figure 3.7. The Taxoner algorithm ... 69
Figure 3.8. Analysis of anthracis strain not included in the database ... 73
Figure 3.9. Detection of low abundance strains ... 75
Ad maiorem Dei gloriam
1. Introduction
The network view on biological data has profoundly influenced the ways we are looking at problems of diagnosis and therapy in life sciences today. In traditional paradigms, we used to look at data as isolated entities stored in organized databases. Today, we increasingly consider data as an interconnected network. There are many kinds of connections – for instance, drugs can be connected to diseases, to their protein targets, to genes producing the targets, or to drugs they can replace or antagonize. In a similar manner, proteins can be linked to other proteins they physically contact, to genes they regulate, to diseases they play a role in, etc. This is a very complex picture, because we have many types of entities and relationships that are defined in separate ontologies that in turn can be considered as networks of terms. The storage and manipulation of such a large body of data is clearly too demanding for current computers. In addition, such data networks are both incomplete and noisy. Namely, we have a seemingly large number of proteins, but the knowledge on proteins is rarely validated by experiment, and a large part of the annotations is just taken over from homologous proteins of various organisms. In addition, we cannot be sure whether two proteins are linked in all tissues and/or in all phases of the cell cycle. The solution of these problems is to break down the hypothetical data-network into specific - disease-specific, tissue- specific, pathogen-specific, etc. - manually curated parts that contain reliable information on a given problem. This tedious and labor-intensive solution is justified only in very important fields.
Cancer-specific data networks are an example of this approach. In addition, two major information sources can help data-sparsity problems. On the one hand, various high-throughput experimental methods (two hybrid systems, DNA sequencing, Chip-seq, etc.) provide novel kinds of molecular interaction data that in principle can be easily added to the existing databases. However, high throughput data are most often laden with noise, which has to be handled. In such cases, hierarchical data networks (i.e. ontologies) may offer a good framework to balance between the reduction of noise and sensitivity to discover novel data links from the experiments.
On the other hand, literature databases that contain abstracts or full text of scientific papers provide a large body of new knowledge that can in principle be linked to molecular data. Again,
Disease-specific databases and tools represent a current approach where the above problems are tackled by large communities of scientists. Cancer databases and tools are a typical example, since cancer is one of the most important complex diseases which is responsible for ~15%
of all human deaths, and which has >100 more-or-less well-characterized types and >500 human genes associated with it [2]. Oncologists use a variety of traditional databases, but there are a number of data-collection efforts dedicated to the gathering of data on various cancer types. All this provides a solid knowledge base for designing integrated data-networks in which novel questions related to cancer therapy can be answered.
Here I am concerned with three types of questions that can be addressed via integrated data networks: i) finding drug combinations potentially useful for cancer therapy. I tackle this problem by using a simple network overlap measure applied to data networks; ii) finding novel gene-disease associations in ovarian cancer for generating a list of potential biomarkers. I approach this problem using a text mining approach applied to MEDLINE abstracts [3] as well as the STRING database [4]. iii) Finally, I present a practical application by testing a dedicated, data subnetwork in accelerating and improving the taxonomic identification. Here I take advantage of the fact that the taxonomic and even the functional subnetworks are hierarchical graphs, which allows a substantial speedup with respect to current algorithms. Section 1 is an introduction to the problem of biological topics discussed in this work, and it covers the mathematical and computational background as well. Section 2 presents the main database types used in this project. Section 3 describes the principle of hypothesis generation via network overlap analysis, the identification of drug combinations via a network overlap measure and the prediction of pathogenic species. Section 4 discusses conclusions and future trends while summarizing the scientific results.
1.1. Biological background
1.1.1. Chemotherapy
Chemotherapy is the most frequently used first-line treatment of cancer. Chemotherapeutic agents target all dividing cells in the body either by killing them (cytotoxic agents) or by blocking proliferation without cell elimination (cytostatic agents), regardless of their status as normal or neoplastic. Tumor cells proliferate rapidly, thus agents selectively damaging dividing cells exhibit a selective advantage. Victims of such a universal destruction are the fast-growing normal cells,
accounting for the side effects of chemotherapy such as damaged hair follicles, irritated epithelium of the mouth and digestive tract, and suppression of myelopoietic precursors in the bone marrow.
Chemotherapeutic agents can be classified according to the mechanisms of their action.
Drugs can destruct the structure of DNA, stop metabolic processes, and obstruct protein structures of the mitotic spindle. Cell cycle consists of four different phases: G1 (protein synthesis and cell growth), S (DNA replication), G2 (further protein synthesis and cell growth) and M (mitosis) – some agents are cell-cycle-phase-specific, while other agents require cell proliferation for action but are not linked to any given phases of the cell cycle [5]. Chemotherapeutic agents can be classified into five main categories.
1) Alkylating agents are not cell-cycle-phase specific, and their effects are dose-dependent, thus cell killing is a linear function of the applied dose of the medication. They form covalent bond with amino, sulfhydryl, phosphate and carboxyl groups to alkylate biologically active molecules and block the function of DNA, but also RNA and proteins [5]. The group consists of nitrogen mustards, platinum agents, nitrosoureas and cyclophosphamides. Nitrogen mustards are similar to mustard gas and are mainly effective in the hematopoietic system [6], while the lipid soluble nitrosoureas used to target brain tumors penetrate through the blood- brain barrier [5]. Carboplatin is a standard agent of care for ovarian cancer [7-9].
2) Antitumor antibiotics have been isolated from natural sources, such as plants, bacteria and fungi. Antibiotics intercalate between DNA base pairs, thus inhibit transcription and RNA synthesis. Their effectiveness is limited by dose-dependent cardiotoxicity as a main adverse effect [10]. Frequently used antibiotics are actinomycin-D, mitoxantron, and anthracyclines such as doxorubicin. Anthracyclines also inhibit topoisomerases I and II.
3) Antimetabolites are structurally similar analogues of naturally occurring molecules. They interfere with metabolic processes by either competing for key enzymes or substituting components of DNA during synthesis, thus block cell cycle in the S phase. Antimetabolites show a nonlinear dose-response, thus after a given concentration no further cells are eliminated. Methotrexate inhibits folate biosynthesis, ultimately leading to purine and pyrimidine depletion within the cell [11]. Nucleoside analog 5-fluorouracil and cytarabin
4) Vinca alkaloids and taxanes consist of cell-cycle-phase specific antimicrotubule blocking chemotherapy agents. During the S phase vinca alkaloids bind to tubulin, prevent polymerization and eventually mitotic spindle formation. Taxanes on the other hand, such as paclitaxel and docetaxel, stabilize tubulin inhibiting depolymerization and cell division [12].
5) Topoisomerase inhibitors, such as camptothecin analogs (irinotecan) inhibit DNA elongation by blocking topoisomerase I in the S phase of the cell cycle [13].
Response to chemotherapy is classified as complete (tumor is untraceable), partial (50%
shrinkage) or minimal (stable disease). When chemotherapy fails, tumor progression continues.
Chemoresistance is a complex multifactorial phenomenon [14, 15]. Mechanisms of resistance include pharmacological factors such as inadequate drug concentrations due to low accessibility of the tumor. Cellular resistance factors include detoxifying or transport mechanisms reducing drug concentrations in the target cell, altered drug-target interactions including the ability of the cells to repair damaged DNA, tolerate stress and evade apoptotic death [16-20]. Inherited genetic variability also influences susceptibility to chemotherapeutic agents. Single nucleotide polymorphisms (SNPs) have also been linked to altered drug response [21]. The one-gene one-drug approach with relevance to cancer chemotherapy has been gradually replaced by studying genetic variation on entire biological or pharmacological pathways, such as the complex network underlying folate metabolism [22] or enzymes responsible for detoxification [23].
Combination chemotherapy blends cytotoxic drugs with different mechanisms of action.
The goal is to eliminate a broader range of resistant cells in the heterogeneous population of cancerous cells, to prevent or slow the emergence of resistant clones, and to maximize the additive or synergistic effects of drugs on cell kill. Compelling evidence support combination treatments over sequential monotherapy [24]. Preferable combinations include drugs with different mechanisms of action, such as paclitaxel with cisplatin, and different pattern of resistance [5].
When applied sequentially, the order of combined agents influences responses. For example, carboplatin followed by docetaxel in advanced non-small-cell lung cancer patients suggested higher response rate when compared to reverse arrangements [25].
1.1.2. Systemic therapy of recurrent or metastatic breast cancer
In 2012 alone over 1.7 million women were diagnosed with breast cancer being the most common cancer in women [26]. High numbers pose economic burden and affect the quality of life of an enormous population. The universal goal to increase treatment efficiency is not trivial, as breast cancer is a heterogeneous disease. Based on molecular features breast cancers are grouped into subtypes with distinct gene expression pattern comprising luminal A, luminal B, basal like and HER2 positive subtypes [27]. Each of these phenotypes requires different management. The picture is further complicated with cancer stage and menopausal status. Local treatment of primary breast cancer differs from the systemic treatment of advanced or metastatic disease. Preoperative, so- called “neo-adjuvant” treatments, such as anthracyclines or endocrine agents given preoperatively are expected to downstage the disease. Advanced incurable malignancies require a sturdier cytotoxic treatment compared to a less serious disease. The guidelines of the US National Comprehensive Cancer Network suggest a list of preferred single agents for recurrent or metastatic breast cancer (that is not HER2-positive): doxorubicin or pegylated liposomal doxorubicin, paclitaxel, capecitabine or gemcitabine, vinorelbin or eribulin. Other single agent chemotherapies include cyclophosphamide, carboplatin, docetaxel, albumin-bound paclitaxel, cisplatin, epirubicin, ixabepilone. Chemotherapy combinations are listed in Table 1.1
Table 1.1. Chemotherapy combinations for recurrent or metastatic breast cancer adapted from the Guidelines of the US National Comprehensive Cancer Network
Regimen1 Component 12 Component 22 Component 32
CAF/FAC cyclophosphamide doxorubicin fluorouracil
FEC fluorouracil epirubicin cyclophosphamide
AC doxorubicin cyclophosphamide
EC epirubicin cyclophosphamide
CMF cyclophosphamide methotrexate fluorouracil
NA3 docetaxel capecitabine
GT gemcitabine paclitaxel
NA3 gemcitabine carboplatin
NA3 paclitaxel bevacizumab
1The abbreviation of the given chemotherapeutic regimen (i.e. CAF/FAC is a drug combination
1.1.3. Targeted molecular therapy
Unfolding the molecular mechanisms underlying neoplastic transformation [28] opened a new, “personalized” era in clinical practice. Identification of driver mutations [29] allowed the rational design of molecular-targeting agents (MTAs). MTAs as single or combination therapies aim at aberrations that appear in a broad range of cancers and can be targeted in many tumor cells simultaneously. Patients are eligible to a therapy with MTAs only if their cancer bears a driver mutation targeted by the given agent. Therapies include monoclonal antibodies (mAbs), that deplete growth factor supply for the cells or prevent receptor dimerization, and small-molecule inhibitors, that block the initiation of intracellular signal transduction or possess catalytic activities [30].
The efficacy of monotherapies using molecularly targeted agents is often inferior compared to combination strategies. The reason for this is that relatively few malignancies depend on only one unique pathway to achieve the malignant transformation. For instance, targeting the hyperactive ABL1 kinase with small molecule tyrosine kinase inhibitors, such as imatinib and nilotinib produced superior clinical outcome in chronic myeloid leukemia [31, 32]. The complexity of signaling pathways and the heterogeneity of tumors called forth the combination of MTAs and cytotoxic agents. In this, agents are selected based on biological considerations to alter complementary pathways of signal transduction or to inhibit multiple target molecules within the same pathway [33]. In general, MTAs are considered to be less toxic than conventional chemotherapies [34], but when combined, the crosstalk between pathways may result in unpredictable toxicities [35].
1.1.4. HER2 positive breast cancer
Evolution of treatment choice in HER2-positive breast cancer illustrates the difficulties in targeting complex biological systems. About 20% of breast cancer patients overexpress Epidermal Growth Factor Receptor 2 (HER2), facing aggressive tumor growth and inferior prognosis [36].
The first successful targeted therapy approved by FDA in 1998 was an anti-HER2 monoclonal antibody, trastuzumab, combined with chemotherapy. The treatment dramatically changed the clinical outcome of the aggressive HER2-positive metastatic breast cancer [37, 38]. Trastuzumab monotherapy was effective in about 15-26% of patients [39], and combining trastuzumab with chemotherapy provided significantly better outcomes [40].
HER2 (ERBB2/neu) belongs to the family of type I receptor tyrosine kinases (RTKs) including EGFR (ERBB1), HER3 (ERBB3), and HER4 (ERBB4). HER2 is overexpressed in tumor tissue but not in healthy cells, hence offers an ideal target for personalized therapy. Ligand binding of RTKs – except HER2 with no known ligand - induces receptor homodimerization or heterodimerization at the plasma membrane. Dimerization activates complex signal transduction involving the PI3K/Akt, Ras/MAPK, and JAK/STAT pathways, leading to cell transformation and cancer. Ligand and heterodimer compositions tightly regulate downstream signaling. With its permanently open conformation, HER2 is a favored dimerization partner of the other RTKs conferring lateral transmission to create a complex network of signaling pathways [41].
Redundant signaling cascades, as in the case of EGFR receptor family, facilitate bypassing the targeted node in the network [42]. Eventually, about 70% of patients develop resistance against trastuzumab. In addition, more superior patient stratification will be needed to improve initial clinical response. Despite constant evaluation of predictive biomarkers, the extent of HER2 expression remains the sole reliable trait for treatment decision [43]. Improved outcome can be obtained in case the inhibition involves other members of the EGFR receptor family. Preferred first line treatment includes simultaneous treatment with pertuzumab and trastuzumab assisted either by docetaxel or by paclitaxel [44, 45]. Pertuzumab targets the second extracellular domain of HER2 and prevents its dimerization with HER3.
Following the most current NCCN guidelines, in case the preferred first line treatment cannot be implemented, the subsequent regimes should include the antibody-drug conjugate trastuzumab emtansine (T-DM1), consisting of trastuzumab covalently linked to a microtubule inhibitor [46]. Trastuzumab is also suggested to be utilized in combination either with paclitaxel and carboplatin, or with one of the following: docetaxel, vinorelbine, or capecitabine. Lapatinib, a small-molecule tyrosine kinase inhibitor blocks EGFR and prevents its dimerization with HER2.
After trastuzumab failure, addition of lapatinib to chemotherapy improved post-progression free survival rates in metastatic breast cancer patients [47]. Trastuzumab combined with lapatinib offers a chemotherapy-free alternative to trastuzumab exposed HER2-positive breast cancer.
Trastuzumab exposed HER2-positive breast cancer may also be treated with the combination of lapatinib and capecitabine, or trastuzumab and capecitabine. Trastuzumab can be combined with
1.1.5. Ovarian cancer
Ovarian cancer is the fifth most common cancer in women worldwide and the deadliest gynecologic malignancy, as less than 30% of patients with advanced disease reach 5-year survival [48]. It is characterized with extreme heterogeneity, as a considerable proportion of tumors does not originate from the ovaries [49, 50]. Their common features are their shared location and dissemination to the pelvic organs. Histological subtypes display cellular and molecular diversity and distinct pathogenesis. Type I tumors progress from benign precursor lesions and consist of low- grade serous, low-grade endometrioid, clear cell, mucinous and Brenner carcinomas with a distinct genetic profile and a low malignant potential. Type II tumors including high-grade serous, high- grade endometrioid, undifferentiated and mixed-mesodermal tumors are highly aggressive, genetically unstable, lack precursor lesions, frequently harbor p53 mutations (<90%), and approximately 20% of them carry BRCA1/2 mutations [51, 52].
About 80-85% of women are diagnosed with serous carcinoma, followed by endometrioid (10%), with clear cell and mucinous cancers being the least common subtypes. Clear cell and mucinous tumors appear in earlier stages more frequently than serous cancers, providing a generally good prognosis. Type I patients fare better after surgery and usually do not require chemotherapy [53]. However, the lack of symptoms at an early stage frequently results in a late diagnosis when the tumor has already reached an advanced state. The extent of surgical tumor- mass reduction is an important prognostic factor of survival. Complete resection of advanced tumors improves both progression-free and overall survival compared to suboptimal surgical outcomes [54]. Surgery complemented with adjuvant or neo-adjuvant carboplatin-paclitaxel treatment has been the standard of care in the past 15 years [55, 56]. In case of paclitaxel intolerance pegylated liposomal doxorubicin (PLD)-carboplatin or docetaxel-carboplatin treatments provide an alternative solution [57, 58]. Despite the good initial responses about 70% of patients relapse within the first three years. The relatively poor survival data compared to other types of solid tumors necessitated a more refined methodology. Ovarian histotypes are treated now as distinct diseases with different mutational profiles and treatment requirements, influencing early detection, clinical trial design and the identification of new drugable targets [59, 60].
Contrary to the good results associated with early stage mucinous cancers, women with advanced mucinous tumors do worse compared to other histological types of advanced disease related to the high frequency of platinum-resistance [61]. Mucinous tumors represent a distinct
spectrum of ovarian cancers ranging from benign to invasive with an individual molecular profile featuring frequent KRAS but infrequent p53 and BRCA mutations [62, 63]. Oxaliplatin combined with 5-fluorouracil represents a promising alternative treatment specific to mucinous tumors, validated in vitro and on xenografts [64].
Ovarian clear cell cancers (OCCC) in advanced stages are also particularly malignant, and refractory to platinum-based chemotherapy [65]. Clean cell cancers are characterized by high frequency mutations in the PIK3CA catalytic subunit of the PI3K gene [66] and mutations in the chromatin remodeling ARID1A gene [67]. The gene expression profiles resembling renal clean cell cancers, such as MET overexpression and overactivation of IL6-STAT3-HIF signaling pathway, suggest that antiangiogenic treatment used on renal clean cell tumors, such as the multi-kinase inhibitor sunitinib, may be applicable to OCCC [68].
The high-grade serous ovarian cancer (HGSOC) is of particular interest, as it accounts for 70-80% of ovarian cancer fatalities. Large portion of HGSOCs originate from outside of the ovaries, from the distal part of fallopian tubes [50]. Despite its sensitivity to platinum derived medications and other DNA damaging agents, patient survival has not been improved for years, as therapies targeting specific tumor biomarkers were lacking. Transcriptional profiling separated mesenchymal, immune, differentiated and proliferative subtypes associated with different prognosis, although the distinction has not yet been translated to clinical decisions [69]. Sequencing HGSOC revealed a frequent driver p53 missense or nonsense mutation indicating its role in tumor initiation [70], and the inactivation of tumor suppressor genes RB1, NF1, RAD51B, and PTEN [71]. CCNE1 encoding cyclin E1 for cell cycle progression is amplified in a large proportion of HGSOC that lacks defects in the HR pathways, likely representing an early event in tumor progression [51]. Drugs targeting cells deficient in DNA repair, such as poly (ADP) ribose polymerase (PARP)-inhibitors, selectively kill tumor cells with dysfunctional BRCA1/2. Olaparib, the first PARP-inhibitor have been approved for use as a maintenance therapy in Europe and for advanced recurrent disease in the USA, to the great advantage of BRCA-related ovarian cancer patients, particularly with a platinum-sensitive disease [72].
1.1.6. Future perspectives
term survival data have emerged with limited success. For example, initially well responding patients with cutaneous melanomas treated with the BRAF inhibitor vemurafenib relapsed shortly after treatment [74]. ALK-positive lung cancer patients treated with crizotinib showed a 65%
response rate – but the median duration of response was only 8 months [75]. In HER2-positive breast cancer, the majority of patients develop resistance within the first year of trastuzumab treatment [76-78]. Current guidelines suggest the simultaneous combination of molecularly targeted and immune checkpoint therapy [79]. The concept behind this approach is that T cells of the adaptive immune system show a remarkable ability to match the diversity and adaptability of tumors. Immune therapy can unleash T cells specific to many antigens present in the tumor by targeting a single immune checkpoint. In spite of promising ongoing studies, current results suggest durable tumor [80] inactivation only in a fraction of patients.
1.2. Metagenomics concepts
Metagenomics is the genomic study of microbial communities, i.e. microbes coexisting in the same space and time. The term metagenome was coined to designate the genome content of the whole community. The study of these communities is as old as microbiology itself, since at the beginning it was not yet possible to separate microbial samples into taxonomic units, and later studies relied mostly on morphological features. Without going into details of this long and impressive development, one can concentrate on the current trends of metagenomic analysis today as summarized by a recent review of Escobar-Zepeda et al. [81]. First, sequencing technologies allow us to investigate the species as well as functional (gene) variety present in microbial samples.
Species composition is easier to determine. We can analyze the 16S RNA genes of bacterial species according to the classical concept of Carl Woese. In this case, we can use amplicon sequencing of a short segment of bacterial genomes and compare it with 16S RNA databases. In this analysis, first step is assembling the whole sequence of the relatively short 16S RNAs present in the sample.
This analysis gives an estimate of the known bacterial species present in a bacterial community and estimates unknown taxonomic units. 16S RNA analysis carries out a search in a collection of short sequences with sensitive alignment techniques, like BLAST [82]. This category is considered as the standard method, although it has its limitations. For instance, there is an essential need for PCR amplification, which comes with extra overhead and experimental bias. Word-based techniques and artificial intelligence may offer a solution by making it possible to build databases of clade-
Another approach is to use whole genome shotgun sequencing, i.e. a full-scale sequencing of the entire sample. Complete assembly of the reads into full genomes is not possible – there are simply too many species in the sample. Partial assembly of genome fragment is possible with current, high coverage sequencing and in-depth data processing, but the analysis is never complete. Instead, the individual sequencing reads are simply mapped onto the genome of known bacterial species, a technique known as binning. This analysis often gives more detailed species composition as 16S RNA sequencing, but it tends to be limited to microbial species with known genomes. There are two main categories of computational approaches addressing the above problem. The first one is the group of marker-based methods, which aims to overcome the difficulties by reducing the search space, namely by using small, specified datasets. MetaPhlAn applies a small database of clade- specific sequence markers built from the genome sequences of the known taxa and then searching this with general-purpose aligners. Within large microbial communities, this kind of search can be exceptionally fast and accurate for identifying taxa and their approximate proportion. However, because of the fact that many strains often share identical markers, this approach many times lacks lower (e.g. strain-level) identification. This is a problem in the case of pathogenic strains of bacteria like E. coli.
Taxon assignment [84], which is one of the most critical points for all approaches, is mainly achieved by different kind of lowest common ancestor searches in a taxonomic hierarchy. This means that the reads are assigned to one taxon (e.g. an E. coli strain), if possible, or to more than one taxon (e.g. 100% identity with an E. coli strain and an E. fergusoni strain), but in this case the lowest common taxonomic ancestor (the genus Escherichia) is reported. Numerous programs use this approach, like MEGAN [83, 85], Mothur [86] and SOrt-ITEMS [87].
The above-discussed tools represent a high variability in the computational approaches meaning that there is a demand for further improvements. For instance, current tools being developed for general purposes means that there is a need for specified tools. Furthermore, qualitative and quantitative answers cannot always be clearly separated: e.g., E. coli reads in an output may express the presence of it in the sample; however, the abundance of the species can not be measured by the number of identified reads. At this moment, only MetaPhlAn is able to supply reliable quantitative results for species abundance [88].
the read to genome hits are evaluated in terms of gene groups, rather than in terms of strains and taxa. The analysis of gene function is based on the classification schemes such as COGSs (mentioned below).
1.3. Networks in biology
Biological databases, including the ones related to cancer therapy and metagenomics, contain annotated data items cross-referenced to each other. In the mathematical sense, such an entity can be pictured as a subgraph or subnetwork, in which some of the edges (cross references) point to other entities or subgraphs defined in other databases. For instance, a drug in the drug interaction database can be linked to another drug item within the same database, as well as to a disease defined in a medical ontology [89, 90], a protein defined in Uniprot, etc. In principle, there is no problem to represent all such subgraphs in one large network that we term here a data network.
The advantage of such a network is that it allows a large variety of queries to be answered within the same system. In practice, the construction of such a large network is prohibitively difficult. First it would be far too large, second it would contain a large number of heterogeneous and partly conflicting data types [91]. The current solution is to build partial networks that allow one to answer a few questions related to a given project.
From the practical point of view, cancer data networks consist of, on the one hand, dedicated cancer-related sequence databases, and on the other hand, molecular and molecular interaction databases that include drug and drug interaction databases. The former ones are collected by focused next generation sequencing projects carried out by an often large number of research groups (Table 1.2). Such projects contain data on cancer mutations, and are often divided into type-specific datasets or comprehensive datasets. Another subgroup of these databases are data resources that are made available via WWW interfaces and include dedicated search facilities.
Molecular and molecular interaction databases used to build cancer data network consist of those datasets that help one to describe and interpret cancer-related sequence information. These databases can be roughly categorized as 1) general-purpose sequence databases, 2) drug-related databases, 3) molecular interaction databases and 4) literature databases.
A wide range of experimental methods used to study molecular interactions fall into two
interactions and try to gather fine details by studying the interacting partners with methods like x- ray crystallography [92, 93], nuclear magnetic resonance [94, 95], often in conjunction with structural bioinformatics and/or conventional biochemical methods. Interaction data of a selected protein can be collected with methods such as affinity chromatography or co-immunoprecipitation [80, 96, 97]. These are typically “small-scale” (focusing only on very few molecules) and traditional biochemical methods. ii) Large-scale or system-level approaches can be used to collect a large number of interaction data in one experiment. One of the best known methods for detecting protein-protein interactions is the yeast two-hybrid system [98]. The underlying idea is that the expression of the reporter genes depends on two separate components, a binding domain (BD) and an activation domain (AD). If the two domains are indirectly connected via a protein-protein interaction, where one of the interaction partner is fused with BD and the other fused to the AD, then one can detect the reporter gene. This approach makes it possible to detect a large number of interactions by screening a certain protein against a DNA library representing all possible proteins the organism can have. Another system-level technique, proteomics, can be used to study post- translational modifications or protein-protein interactions via affinity purification coupled with mass spectrometry (AP-MS). This approach can also be useful for detecting strong connection between proteins, thus exploring protein complexes [99]. High-throughput methods are productive but there are several drawbacks and biases – among others, the number of erroneous interaction assignments can exceed 10 percent.
In addition to experimental methods, the body of databases available in other fields is also a source of information. While experiments provide data on the biological entities themselves, the databases provide information on a wide variety of concepts. In this way we broaden the scope of molecular interaction data to “data networks” that allow us to link biological data to the results of further scientific fields. For instance, a drug database such as Drugbank [100] provides information on chemical structures and their biological targets (proteins and genes) and/or the diseases. A database of scientific publications, on the other hand, provides information on a large class of descriptions (scientific abstracts) that are linked to each other by common keywords, authors, statements etc. Further examples for special data network are the ontologies. These are special, hierarchical knowledge representations; for example, the Anatomical Therapeutic Chemical Classification (ATC) System classifies drugs into groups at five levels in a hierarchical way. Thus
and the leaves are the full ATC codes (7 characters). There are 14 main groups at this level such as code A (Alimentary tract and metabolism), code B (Blood and blood forming organs),
General-purpose databases such as Uniprot [101], Ensemble [102] or RefSeq [103, 104], GenBank [105] hold high quality and reliable information about proteins and genes (focusing on the amino acid or nucleotide sequence, protein names or descriptions, and citation information).
Usually they provide data mining tools and APIs as well.
Drugbank database [100] is one of the most comprehensive and freely available, complex data source about drugs. Currently, it holds information about 2200 FDA approved and more than 6000 experimental drugs. It also provides detailed information about the food-drug and drug-drug interaction information. The information was manually curated from web resources and published papers and has been continuously developed [106, 107]. It also provides data about drug mechanism of action and drug labels and ADMET (drug metabolism, absorption, distribution, metabolism, excretion and toxicity) profile, thus the drug card of Drugbank could be a rich source of text mining.
TTD database [108] is tailored to peptide molecules and its target information. It also includes information about diseases and drug combinations, however the last one is only available as excel tables, but not in a structured format, such as XML. Both Drugbank and TTD contains manually curated data.
STICH [109-111] is an automatically created, integrated database. It was created by using similar concepts as those of the STRING network. The database focuses on small molecules and their relations to other small molecules and proteins. Similarly to the STRING database there are various types of associations between the molecular entities. It mainly contains protein-chemical and chemical-chemical links based on text mining and other complex predictions extended with chemical structure description strings.
The Drug Combination Database [112, 113] focuses on agents combined together to achieve some therapeutically advantage over single agent drugs. Drug regimens are typically used in treating cancer and other complex diseases. The database is partly based on the FDA orange book [114], clinical trials (https://clinicaltrials.gov/), and publications. It also holds information about the individual drug components, such as ATC codes, target and cross references.
Furthermore, it also provides annotations for drug combinations, such as possible mechanism of actions, interaction type, suggested doses, etc.
Drug side effects and drug interactions are often not covered in standard public databases.
These kinds of data are available, for instance, in the SIDER database [115, 116], where the side effects are extracted from the drug labels (using controlled vocabulary such as UMLS [90]). A well-maintained collection of drug side effects is provided by the Tatonetti Lab [117].
Experimental results of protein-protein interaction measurements are deposited in various primary databases such as the Database of Interacting Proteins (DIP) [118], Biomolecular Interaction Network Database (BIND) [119], Molecular Interactions Database (MINT) [120-123], Biological General Repository for Interaction Datasets (BioGRID), Human Protein Reference Database (HPRD), IntAct Molecular Interaction Database [124].
The DIP database contains large number of manually curated and reviewed interactions from numerous species [118, 125]. It also provides some services and visualization tools for the available data [126], and a cytoscape plugin (MiSink) [127]. Different evidences for the interactions were integrated and considered manually.
Human Protein Reference Database (HPRD) [128] contains various types of data about proteins such as post-translational modification, known or predicted disease associations, cellular localization, tissue expression, mainly from publications. The data have also been reviewed by scientific experts. The database contains information about 30047 proteins and 41327 interactions among them.
Another important protein-protein interaction database is IntAct [124, 129-131], developed and maintained by the European Bioinformatics Institute (EBI), updated on regular basis. The interactions were partly curated from literature (14074 publication) in collaboration with the Swiss- Prot team, or the data were submitted directly. They also use controlled vocabularies [132] (PSI- MI [133, 134], gene ontology [135] and NCBI taxonomy terms [136]) for annotating the interactions and the proteins. The database contains information about the interacting domains as well.
representation and data integration. The MiNTAct [137], Imex [138], Mentha [139] consortial databases integrate the molecular interaction data collected from 11 databases.
STRING (Search Tool for the Retrieval of Interacting Genes) is one of the largest integrated protein interaction databases, which covers 66.9 Mio predicted and known interactions between proteins of 1100 organisms. The majority of the interactions (44.1 Mio) are predictions.
The links between the proteins are some kind of associations (among them several indirect ones) - not only physical interactions. The evidence types for the associations are neighborhood, gene fusion, co-occurrence, co-expression, experiments, databases, text mining, and homology. Each type of association has a confidence score, which is a probabilistic measure of the reliability of the link. The several types of links and their confidences can be combined into one association with one confidence score.
Transcription factor databases contain sequence motifs and genomic locations collected from genomic data using bioinformatics methods. In the network representation of the database the nodes are DNA motifs linked to genomic locations. A typical example of transcription factor databases is Transfac, first published by Edgar Wingender’s group in 1994 [140]. The database is manually and continuously updated. The current release contains 7915 sites assigned to 6133 transcription factors. Further examples of this database are given in Table 1.2.
Table 1.2. Cancer-related databases and resources
Database Description URL Refs.
Comprehensive databases and resources
TCGA The Cancer Genome Atlas http://cancergenome.nih.gov/ [141]
CGP Cancer Genome Project http://www.sanger.ac.uk/research/projects
/cancergenome/ [142]
CPTAC Clinical Proteomic Tumor Analysis Consortium
http://proteomics.cancer.gov/programs/cp tacnetwork
[143, 144]
ICGC International Cancer
Genome Consortium https://www.icgc.org/ [145]
Data mining resources
COSMICMart BioMart tool for COSMIC https://cancer.sanger.ac.uk/cosmic/login [146]
IntOGen
BioMart BioMart tool for IntOGen http://biomart.intogen.org/ [147]
UCSC Cancer Genomics Browser
A visualization and analysis tool specialized to cancer data
https://genome-cancer.ucsc.edu/ [148, 149]
ICPS An Integrative Cancer
Profiler System http://server.bioicps.org/ [143]
NCG 4.0 Network of Cancer Genes http://ncg.kcl.ac.uk/ [150,
151]
CGWB The Cancer Genome
WorkBench http://cgap.nci.nih.gov/cgap.html [152]
CancerMA A web-based tool for analyzing microarray data
http://www.cancerma.org.uk/information.
html [153]
ICPS
An Integrative Cancer Profiler System
http://server.bioicps.org/ [143]
Databases of genetic variations in cancer COSMIC Catalogue of Somatic
Mutations in Cancer
http://cancer.sanger.ac.uk/cancergenome/proj
ects/cosmic/ [154]
CaSNP Cancer SNP data on CNAs http://cistrome.dfci.harvard.edu/CaSNP/ [155]
DriverDB Cancer driver genes and mutation database
http://driverdb.ym.edu.tw/DriverDB/intranet/
init.do [156]
IntOGen Integrative Oncogenomics http://www.intogen.org/ [157]
MoKCa Mutations, Oncogenes,
Knowledge & Cancer http://strubiol.icr.ac.uk/extra/mokca/ [158]
CGAP Cancer Genome Anatomy
Project http://cgap.nci.nih.gov/ [159]
Mitelman Database of chromosome
http://cgap.nci.nih.gov/Chromosomes/Mitel
Table 1.2. (Continued)
Databases of epigenetic, proteomic and transcriptome variations in cancer CanProVar Human Cancer Proteome
Variation Database http://bioinfo.vanderbilt.edu/canprovar/ [162]
MethyCancer
A database of human DNA methylation and cancer
http://methycancer.psych.ac.cn/ [163]
CellLineNavigator Expression profiles of cancer cell lines
http://www.medicalgenomics.org/celllinenavig
ator/ [164]
ITTACA
Integrated Tumor Transcriptome Array and Clinical data Analysis
http://bioinfo.curie.fr/ittaca [147]
PubMeth
Cancer methylation database based on text- mining of PubMed
http://matrix.ugent.be/pubmeth/ [165]
OncomiRDB
A database of
experimentally verified oncomiRs
http://bioinfo.au.tsinghua.edu.cn/member/jgu/o
ncomirdb/ [166]
Cancer-specific clinical and drug resources CancerDR Cancer Drug Resistance
Database http://crdd.osdd.net/raghava/cancerdr/ [167]
HPtaa
The Human Potential Tumor Associated Antigen database
http://www.bioinfo.org.cn/hptaa/ [168]
CancerResource
A resource of cancer- relevant compound and protein interactions
http://bioinf-data.charite.de/cancerresource/ [169]
CanGEM Cancer Genome Mine http://www.cangem.org/ [170]
DTP Anti-cancer agent
database
http://dtp.nci.nih.gov/docs/cancer/searches/standa rd_mechanism.html
[171, 172]
ITTACA
Integrated Tumor Transcriptome Array and Clinical data Analysis
http://bioinfo.curie.fr/ittaca [147]
Cancer-type-specific resources and databases
RCDB RCDB http://www.juit.ac.in/attachments/jsr/rcdb/homen
ew.html [173]
curatedOvarianData
Clinically annotated data for the ovarian cancer transcriptome
http://bcb.dfci.harvard.edu/ovariancancer/ [174]
PED Pancreatic Expression
Database http://www.pancreasexpression.org/ [175]
HLungDB Human Lung Cancer
Database http://www.megabionet.org/bio/hlung/ [176]
Special types of molecular interactions are metabolic and signal transduction molecular interactions. One of the oldest pathway databases is KEGG [177]. However, the current version holds information related to pathways such as genome, diseases and related drugs. It provides a global map for each pathway.
Reactome [178], similarly to KEGG, is a comprehensive, manually curated, high quality pathway database with support of enrichment analysis and data visualization.
The Human Metabolome Database [179], however, concentrates on small molecule metabolites, and it is a rich source of biomarker discovery. It also provides enzymatic, biochemical, and clinical data.
The signaling and metabolic pathways are often handled as separate entities, however, crosstalks and regulatory coupling exist between the pathways [180]. The Signalink [181] and NDEx databases [182] not only offer manually curated and reviewed pathway information, but provide more context for pathway analysis such as transcriptional and post-transcriptional regulators.
Scientific literature databases contain data collected from scientific journals using increasingly automated electronic submission links. Medline/Pubmed [183] is perhaps the best known representative of public scientific literature databases, it collects scientific abstracts from the publishers and provides them with a unified system of keywords (mesh terms, reference [184]).
In the network representation of the database, the nodes are scientific abstracts; the edges correspond to shared keywords, citation links (X cites Y), etc. The Medline database was first published in 1971 and it gained a very wide acceptance as it became available via the PubMed search facility in 1997. For machine learning purposes, the database is downloaded, and word combinations are identified via natural language processing techniques in order to create new index tables. Further examples of this database are given in Table 1.3.
Table 1.3. Representative examples of molecular and molecular interaction databases relevant to cancer therapy
Contents URL Ref.
1. General-purpose databases Uniprot
Comprehensive database of protein sequences
http://www.uniprot.org/ [101]
RefSeq Genome sequences http://www.ncbi.nlm.nih.gov/projects/geno
me/assembly/grc/ [185]
GenBank
Comprehensive database of genetic sequences
http://www.ncbi.nlm.nih.gov/genbank/ [105]
Ensemble
Comprehensive database of sequences with data mining tools
http://www.ensembl.org/index.html [102]
2. Drug-related databases
DrugBank Drug data and drug-
drug interactions http://www.drugbank.ca/ [100]
Therapeutic Target Database (TTD)
Therapeutic Target Database
http://bidd.nus.edu.sg/group/cjttd/TTD_HOM
E.asp [108]
STITCH Drug molecular
interactions http://stitch.embl.de/ [109]
DCDB Drug Combination
Database http://www.cls.zju.edu.cn/dcdb/ [186]
Offsides, TwoSides
Drug adverse effects and Drug-Drug Interactions
http://tatonettilab.org/resources/tatonetti-
stm.html [117]
Drugs.com
FDA approved drugs linked to diseases and target proteins/genes
www.drugs.com
SIDER Drug adverse effects http://sideeffects.embl.de/ [115]
3. Protein /protein interaction databases DIP
Experimentally and manually validated molecular interactions
http://dip.doe-mbi.ucla.edu/dip/Main.cgi [118]
HPRD (Human Protein Reference Database)
Experimentally and manually validated molecular interactions
http://www.hprd.org/ [128]
Intact Manually curated
molecular interaction http://www.ebi.ac.uk/intact/ [124]
MIntAct Manually curated
integrated database http://www.ebi.ac.uk/intact [137]
STRING
Protein/protein interactions as well as connections derived from other databases
http://string-db.org/ [187]
Table 1.3. (Continued)
4. Transcription factor databases TRANSFAC Transcription factors
and binding sites
http://www.gene-
regulation.com/pub/databases.html [140]
JASPAR
Transcription factor binding profile database
http://jaspar.genereg.net/ [188]
DBD Transcription factor
prediction database http://www.transcriptionfactor.org/ [189]
5. Metabolic pathways KEGG
Kyoto
Encyclopedia of Genes and Genomes
http://www.genome.jp/kegg/ [177]
Reactome Curated pathway
database http://www.reactome.org/ [178]
MetaCyc Metabolic pathway
database http://metacyc.org/ [190]
HMDB
Human Metabolome Database
http://www.hmdb.ca/ [179]
6. Signal transduction databases NetPath
Manually curated signal transduction pathways
http://www.netpath.org/ [191]
Signalink
Manually curated signal transduction pathways
http://signalink.org/ [181]
NDEx Integrated network
database http://www.ndexbio.org [182]
7. Mutation databases COSMIC
Somatic mutations found in human cancer
http://cancer.sanger.ac.uk/cosmic [192]
OMIM
Disease gene and mutation database of humans
http://www.omim.org/ [193]
8. Literature databases
PubMed/Medline PubMed/Medline http://www.ncbi.nlm.nih.gov/pubmed [194]
EMBASE
Biomedical and pharmacological bibliographic database
http://store.elsevier.com/embase
Scopus
Bibliographic database of peer- reviewed literature
http://www.scopus.com/
1.4. From databases to data networks
One way to picture data network construction is to take a database of cancer genes or proteins, and then cross-reference it to general-purpose sequence databases, drug-related databases, etc. that will form a network among various types of entities allowing the cross querying of diverse biological databases in a unified manner. In practice, the construction of such a large network is prohibitively difficult, partly because of the incompatibility of ontologies, partly because of the sheer size of the network [91]. The design of these data networks is largely facilitated by database frameworks capable to handle an arbitrary set of biological entities and relationships [195].
Physical or structural connections rely on the well-known fact that molecules practically never function alone but rather in association with other molecules such as ligands, lipids, amino acids, proteins and nucleic acids. On the one hand, there are structural associations between the elements that can be “strong” such as covalent bonding and tight associations in the cytoskeleton (microfilaments are polymers of G-actin proteins), or “transient” such as in the case of receptor- ligand associations. Understanding the nature and the type of these relationships is crucial for interpreting complex biological phenomena such as disease mechanisms. As an example, the active forms of proteins are most often complexes assembled from various types of other proteins or other types of molecules such as RNA, DNA or small molecules. On the other hand, there are functional associations, such as between members of signaling pathways, transcriptional or metabolic networks. Functional associations may not even involve structural interactions, for instance distant members of a metabolic pathway are functionally related. The common motif in these widely different scenarios are the links between molecules that can involve various structural and functional aspects. For instance, a transient interaction act in catalyzing sequential steps within a metabolic network, or in a signaling pathway such as modifying the protein by adding phosphate group, etc.
Biological databases, including the above examples, contain annotated data items cross- referenced to each other. In the mathematical sense, such an entity can be pictured as a subgraph or subnetwork, in which some of the edges (cross-references) point to other entities or subgraphs defined in other databases. For instance, a drug in the drug interaction database can be linked to another drug item within the same database, as well as to a disease defined in a medical ontology, a protein defined in Uniprot, etc. In principle, there is no problem to represent all such subgraphs in one large network, which we term here a data network – but such a network would be
databases are stored as parallel items within the same computer, and integrated concepts and new data types take care of appropriate matching of underlying entities and attributes, including the resolution of conflicts. The result of such a common representation can be best pictured as a network of data, where the original data items (say drugs, target proteins, diseases, mutations) are represented in a common large network. For instance, a network combining drug targets and protein-protein interactions will contain links (network paths) between proteins that are targeted by the same (or similar) drugs, or drugs acting on proteins that are in physical contacts. In such a data network all data items (say drugs, target proteins, diseases, and mutations) are connected via a variety of different links, which makes processing complicated and time-consuming. As a practical workaround, one can construct a dedicated database tailored to a specific task, and that can be queried with simpler tools [195].
From the practical point of view, it is useful to distinguish comprehensive resources that aim to cover, for instance, all known genes and proteins and one selected type of interaction (say, regulatory connections). On the other hand, specialized resources concentrate on a selected species (Homo sapiens), or on a selected tissue type, or on a selected mechanism (signal transduction, or protein kinases). A few representative examples of databases are listed in Table 1.2.
1.5. Graph – definitions and notations
From the logical point of view, all interaction networks and data networks are graphs in which nodes are entities, such as molecules, diseases, i.e. biological, physical, as well as conceptual objects, while the edges or links between nodes are relationships, such as molecular interactions, drug-disease connections, drug compatibilities, etc.
A graph or network can be defined by a set of vertices and a set of edges. Two vertices are connected if they are linked to each other. For example, let the nodes be the cities and the edges be the roads. In this structure, there is an edge between two vertices if two cities are connected directly by a road. The graphs can be grouped by their different properties, such as weighted or unweighted edges, or by degree distribution, etc. [196].