Biológiailag aktív konjugátumok szintézise módosított ösztrán vázon
Doktori (Ph.D.) értekezés
Zóka Johanna
Témavezetők:
Prof. Dr. Wölfling János tanszékvezető egyetemi tanár
Dr. Mernyák Erzsébet egyetemi adjunktus
Kémia Doktori Iskola Szerves Kémiai Tanszék
SZTE TTIK
2017 Szeged
TARTALOMJEGYZÉK
1. Bevezetés ... 1
2. Irodalmi előzmények ... 3
2.1. Vázmódosított ösztron származékok és azok biológiai aktivitása ... 3
2.2. Azid-alkin „click” reakció szterán vázon ... 13
2.2.1. Cu(I)-ion katalizált azid-alkin „click”-reakció ... 13
2.2.2. „Click”-reakció szterán vázon ... 15
2.3. Sonogashira keresztkapcsolás szteroid modellen ... 21
2.3.1. A Sonogashira kapcsolás ... 21
2.3.2. Biológiailag aktív ösztron származékok előállítása Sonogashira-kapcsolással ... 24
2.3.3. Az ösztron aromás gyűrűjének jódozási reakciói ... 28
2.4. Átmenetifém-katalizált reakciók aktiválása mikrohulámmal... 32
2.5. Heterociklus kialakítása átmenetifém-katalizált „one-pot”reakcióval ... 36
3. Célkitűzés ... 39
4. Kísérleti eredmények tárgyalása ... 40
4.1. Kiindulási D-szeko vegyületek előállítása a 13- és 13-ösztron sorban ... 40
4.2. Ösztron-alkinek előállítása ... 44
4.3. Az újonnan előállított szteroid alkinek (90‒94, 102, 103) CuAAC reakciói ... 50
4.4. Palládium-katalizálta keresztkapcsolások D-szekoösztrán alapvázakon ... 59
4.4.1. A D-szekoösztronok aromás A-gyűrűjének jódozási reakciói ... 59
4.4.2. Jódszteroidok Sonogashira keresztkapcsolási reakciói ... 65
4.5. „One-pot” Sonogashira-„click” reakció ... 72
5. Az előállított vegyületek biológiai eredményei ... 78
5.1. A triazol konjugátumok és előanyagaik gyógyszer hatástani eredményei ... 78
5.2. A vegyületek 17-HSD1 enzimgátlási eredményei ... 84
6. Általános kísérleti rész ... 89
7. Részletes kísérleti rész ... 90
8. Összefoglalás ... 100
9. Summary ... 105
10. Irodalomjegyzék ... 110
11. Köszönetnyilvánítás ... 119
12. Mellékletek ... 120
RÖVIDÍTÉSEK JEGYZÉKE
ER ösztrogén receptor
17-HSD1 17-hidroxiszteroid-dehidrogenáz 1 enzim CuAAC réz-katalizált azid-alkin cikloaddíció DIPEA N,N-diizopropil-etilamin
PPh3 trifenilfoszfán
TFA trifluorecetsav
NIS N-jódszukcinimid
MW microwave (mikrohullám)
TMSA trimetilszilil-acetilén
DCC N,N-diciklohexil-karbodiimid
HBTU O-benzotriazol-N,N-tetrametil-urónium hexafluorofoszfát HOBt 1-hidroxi-benzotriazol
COSY Correlated Spectroscopy, kétdimenziós korrelációs NMR spektroszkópiai technika
NOESY Nuclear Overhauser Effect Spectroscopy, olyan kétdimenziós NMR spektroszkópiai technika, amely a magok téren keresztüli hatásán alapul HSQC Heteronuclear Single Quantum Coherence, olyan kétdimenziós NMR spektroszkópiai technika, amely két közvetlenül kötésben különböző magok közötti kölcsönhatást mutatja
HMBC Heteronuclear Multiple Bond Correlation, olyan kétdimenziós NMR spektroszkópiai technika, amely két nem közvetlenül kötésben lévő különböző magok közötti kölcsönhatást mutatja
NEt3 trietilamin
NH(iPr)2 N,N-diizopropil-amin TBAF tetrabutilammónium-fluorid DABCO 1,4-diazabiciklo[2.2.2]oktán
1 1. Bevezetés
Az ösztron és származékai a szteránvázas vegyületek csoportjába tartozó természetes, illetve szintetikus vegyületek. Hormonhatásuk mellett sokrétű biológiai funkció kifejtésére képesek.
A farmakológiai sajátságokat a szteroid vázának szerkezete és a különböző helyzetben lévő funkciós csoportok minősége, térállása határozza meg. Így az ösztrán váz módosításával befolyásolhatjuk a molekula magreceptoraihoz, ill. szteroidogén enzimekhez való kötődését.
Ennek köszönhetően alkalmas kiindulási pontjai lehetnek a gyógyszertervezésnek. A modern gyógyszerkutatás egyik fő iránya a szelektív hatású antitumor hatóanyagok kifejlesztése. A hormonális aktivitás visszaszorítása azonban nem könnyű feladat, ehhez az ösztrán váz szerkezetének célzott megváltoztatására van szükség. A 13-as szénatom epimerizálása, a D- gyűrű felnyitása vagy D-homologonokká alakítása megváltoztatja a vegyület konformációs viszonyait, ezzel a magreceptoraihoz való kötődését is. Az ösztrogén hatás visszaszorítása mellett azonban egyéb, kedvező biológiai aktivitások jelenhetnek meg. A természetes ösztron 13-as helyzetben történő epimerizálása ma már egy egyszerű, egylépéses folyamatban megvalósítható. Több 13-epimerről is bebizonyosodott, hogy ösztrogén aktivitás helyett egyéb kedvező biológiai funkcióval rendelkeznek.1,2 Egyes 13-ösztron származékok sejtosztódást gátló hatást fejtenek ki különböző sejtvonalakon, mások az ösztradiol bioszintézisében résztvevő enzimek gátlásán keresztül befolyásolják az ösztrogénfüggő elváltozásokat.
A vázmódosított származékok egy további csoportját a nyitott B-, C- vagy D-gyűrűs szeko-vegyületek alkotják. A szeko-ösztronok biológiai hatásáról kevés adat áll rendelkezésre az irodalomban, néhány képviselőjükről azonban ismert, hogy nem mutat affinitást magreceptoraihoz.3 A 13- vagy a D-szeko-ösztron, mint hormonálisan inaktív módosított vázak megfelelő alapot biztosítanak olyan hatóanyagok tervezéséhez, amelyek szelektív hatásúak lehetnek.
A szteroidok antitumor hatása megfelelő heterociklusok beépítésével fokozható.4 A heterociklusos szteroid konjugátum alkotórészeinek biológiai hatása lehet hasonló vagy különböző, de mindenkori cél a kívánt aktivitás fokozása. A hidrofób, viszonylag merev ösztránváz nemcsak a biológiai hatásért felelhet, hanem a sejtmembránon való átjutásért is.
Míg a heterociklus és a hozzá kapcsolódó egyéb molekularészlet általában a „targethez” való kötődésért felel, de önálló biológiai funkcióval is rendelkezhet. Heterociklusos ösztron konjugátumok előállítására többféle szintetikus stratégia is rendelkezésünkre áll. Az
2
átmenetifém-katalizált reakciók az utóbbi időben rendkívül nagy népszerűségre tettek szert, széleskörű alkalmazhatóságuknak köszönhetően. A megfelelő reakciópartnerek kialakítását követően a két egység összekapcsolása egyszerűen, rövid reakcióidők alatt, szelektíven valósítható meg.
A „zöld kémia” elveinek figyelembe vétele a preparatív szerves kémiának is fontos szempontja. A mikrohullámú technika bevezetésével a szteroidkémiában is új irányvonal nyílhat meg. Olyan átalakítások kivitelezése is lehetővé válik, amelyek hagyományos körülmények között csak nagyon nehezen valósíthatók meg.
Jelen munkában ezen szintetikus és farmokológiai célokat igyekeztünk ötvözni.
3 2. Irodalmi előzmények
2.1. Vázmódosított ösztron származékok és azok biológiai hatása
A szteránvázas vegyületek a természetes szénvegyületek családjába tartozó, az élővilágban meghatározó szerepet betöltő, rendkívül változatos szerkezetű szerves molekulák. Az emberi szervezetben megtalálható női nemi hormonok között három ösztrogén hatású vegyület fordul elő: az ösztron (1a), a 17β-ösztradiol (2a) és a 16α,17β-ösztriol (3a, 1. ábra). Az ösztrogének közös szerkezeti elemei az aromás A-gyűrű és a transz-gyűrűanellációk. A B-gyűrű általában félszék, míg a C-gyűrű szék konformációjú. A természetes ösztron (1a) két oxigénatomja egymástól meghatározott távolságban helyezkedik el, amely fontos szerepet játszik hormonhatásának kifejtésében.
1. ábra: Az ösztrogének (1a−3a) és 3-védett származékaik szerkezete (1b, c−3b, c)
Ezen megállapítás óta számos kutatás foglalkozott a természetes ösztron (1a) szerkezetének arra irányuló célzott kémiai megváltoztatásával, amely hormonálisan inaktív, biológiailag kedvező hatású szintetikus származékokat eredményezhet.5 Számtalan különbözőképpen módosított citosztatikus ösztron származék ismert,6−11 amelyek többféle mechanizmus révén fejtik ki hatásukat. Támadáspontjaik lehetnek az ösztron bioszintézisében részt vevő enzimek, vagy saját magreceptoraikon keresztül hatva, antiösztrogénként gátolhatják a tumorsejtek osztódását. Az irodalomban ismert, hogy a 2- és/vagy 17- szubsztituált ösztron-3-szulfamátok a szteroid-szulfatáz (STS) enzim inhibitorai12−14, a 2- haloösztronok aromatáz-gátlók,15 míg az ösztron számos szubsztituált, homológ vagy konjugátum származéka a 17β-hidroxiszteroid-dehidrogenáz 1-es típusú izozim (17-HSD1) inhibitoraként viselkedik.16−25 Ezen szteroidogén enzimek gátlásával új lehetőséget
4
teremthetünk a hormon függő tumoros megbetegedések kezelésében.26 A 17-ösztradiol (2a) a sejtosztódást serkentő növekedési faktorok aktiválásán keresztül fokozza az ösztrogén-függő tumorok növekedését. Ennek visszaszorítására célszerű az ösztradiol (2a, E2) bioszintézisébe beavatkoznunk, amelynek utolsó, kulcslépését a 17-HSD1 enzim katalizálja (2. ábra). A 17-HSD1 enzim szelektív gátlásával az ösztradiol (E2) bioszintézist az utolsó pontján állíthatjuk le. Ilyen szelektíven ható enzimgátlók alkalmazásával a mellékhatások csökkenését és a hatás szelektivitásának növekedését érhetjük el.
2. ábra A 17-HSD1 enzim szerepe az ösztrogén hatás kialakulásában
Számos szteroid- és nem szteroid-jellegű 17-HSD1 inhibitort fejlesztettek már ki, azonban egy sem jutott el a klinikai alkalmazásig. Ennek oka, hogy a leghatékonyabb vegyületek szinte mindegyike az ösztrogének magreceptoraihoz is affinitást mutat, azaz az enzimgátlással egyidejűleg hormonális hatást is kifejtenek. Mindezek alapján elmondható, hogy a hormonálisan inaktív ösztron-alapú antiproliferatív hatóanyagok kifejlesztéséhez nem elegendő csupán az ösztránváz szubsztituálása. A hormonális aktivitás megszüntetéséhez
5
magát a vázat célszerű módosítani. Ennek egyik lehetősége a 13-as szénatom konfigurációjának megváltoztatása, ami a konformáció megváltozását vonja maga után. A 13α-ösztront (4) elsőként Butenandt és munkatársai állították elő, fotokémiai izomerizációval.27 Munkájuk során 2 g ösztronból kiindulva, mindössze 700 mg, úgynevezett lumi-ösztront (4) nyertek. 1994-ben Yaremenko és Khvat igen hatékony, egylépéses izomerizációs módszert fejlesztett ki androsztánvázas vegyületekre. Az epimerizációt o-feniléndiaminnal, jégecetben végezték, amely 56‒58%-os hozammal szolgáltatta a várt terméket.28 A fenti eljárást Schönecker és munkatársai kiterjesztették ösztránvázas vegyületekre, és a hozamot 78%-ra növelték (3. ábra).29 Tanszékünk Szteroidkémiai Kutatócsoportja az epimerizálást követő oszlopkromatográfiás tisztítást Girard-reagenssel való elválasztással is összekötötte. Ez azért kulcsfontosságú, mert a reakció körülményei között az említett reagenssel csak a kiindulási anyag (1) reagál, így annak kis mennyiségű maradékától is hatékonyan megtisztítható a termék 13-epi-származék (4). Az ösztron 3-benzil-éterét (1c) hasonló körülmények között reagáltatva, 98 %-os hozammal állították elő annak 13α-megfelelőjét (4c).
3. ábra Egylépéses epimerizáció az ösztránváz 13-as szénatomján
A közelmúltban kanadai kutatók elvégezték a két 13-epimer négy lehetséges 17-hidroxi származékának (2a, 5‒7, 4. ábra) gyógyszerhatástani tesztelését.30 Az in vitro és in vivo vizsgálatok eredményei azt mutatták, hogy a 6-os jelű vegyület mutatja a legkisebb affinitást az ösztrogén receptorokhoz, legalacsonyabb az ösztrogén hatása az ösztrogénfüggő sejtvonalakon, és in vivo nincs hormonhatása. Az izomerek egymáshoz képest mutatott ösztrogén aktivitása a következő tendenciát mutatta: 6 < 7 < 5 << 2a. Ezek az eredmények arra utalnak, hogy az anguláris metilcsoport inverziójával az ösztrogén hatás jelentősen csökkenthető. Az ösztradiolokat (2a, 5–7) nagyobb koncentrációban (>5μM) alkalmazva, sejtosztódás gátló hatást figyeltek meg MCF-7 (ER-t kifejező) és T47D (ER-t, progeszteron és
6
HER2 receptort kifejező, ösztrogénfüggő) emlő karcinóma sejtvonalakon. A leghatékonyabb antiproliferatív vegyületnek a 7-es jelű, 17-13-epi-ösztradiol bizonyult. Poirier és munkatársai munkája alapján tehát a 13-ösztrán váz megfelelő modellként szolgálhat potenciálisan antitumor hatással rendelkező, hormonálisan inaktív konjugátumok képzéséhez.
4. ábra A négy ösztradiol-izomer (2a, 5‒7) szerkezeti képlete
A 2000-es évek eleje óta kutatócsoportunk is sokat foglalkozott 13-epi-ösztron származékok vizsgálatával, mind szintetikus, mind farmakológiai szempontból. Egy, a közelmúltban a 13α- ösztron 16-oximjából (8, 5. ábra) előállított vegyület, jelentős sejtosztódás gátlást mutatott HeLa (méhnyakrák), MCF-7 és A2780 (petefészek karcinóma) sejtvonalakon. Az irodalomban ez volt az első antiproliferatív 13-ösztron származék, amely a referenciavegyületnél (ciszplatin) alacsonyabb IC50 értékeket (3,5‒4,5 M) adott.31
5. ábra A 13-ösztron-3-benzil-éter 16-oxim-származékának (8) szerkezete
7
A vegyület (8) hatásmechanizmusa még nem tisztázott, de az eddigi irodalmi és a Kutatócsoportunk saját eredményei alapján feltételezhetően nem receptoriális úton fejti ki antitumor hatását. Áramlási citometriás mérésekkel sikerült igazolni, hogy a 16-oxim (8) a sejtek osztódási ciklusát a G1/S kontrollponton állíta meg. A sejtciklus G1/S ellenőrző pontja a G1 fázisból a szintézis fázisba történő eljutásért felelős, amelynek blokádja apoptózishoz (programozott sejthalál) vezet.
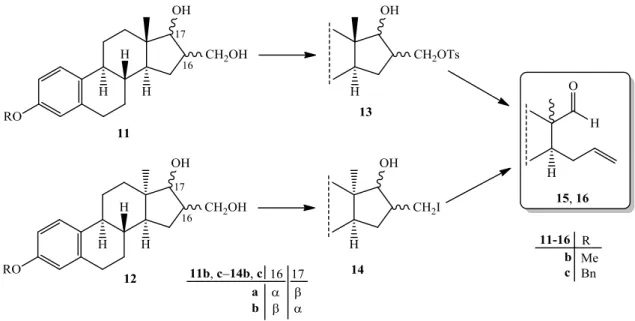
Az epimerizáláson kívül vagy azzal egyidejűleg egy másik vázmódosítási lehetőség az ösztron D-gyűrűjének felnyitása.3 Tanszékünk Szteroidkémiai Kutatócsoportjában korábban már vizsgálták ösztránvázas 16-hidroximetilidén származékok (9, 10) bázikus közegű szolvolitikus reakcióit, és hatékony eljárást dolgoztak ki a D-gyűrű felnyitására.32 Az ösztron védett származékaiból (1b, c) kiindulva, formilezési reakcióval 16-hidroximetilidén- vegyületeket (9b, c) állítottak elő, majd azokat redukálva nyerték a lehetséges cisz- és transz- szerkezetű 1,3-diolok keverékét (11ba‒11bd, 11ca‒11cd 6. ábra).33−35 A 13-epi-ösztron sorban az anguláris metilcsoport sztérikus gátlása miatt, hasonló reakciókörülmények között, csak a két transz diol 12bb, 12cb : 12ba, 12ca képződött, 6 : 1 arányban (6. ábra).36
6. ábra Az 1,3-diolok (11, 12) előállítása a 13β- és 13-ösztron sorban
A D-gyűrű fragmentálódásának a feltétele, hogy a nukleofug csoport térben távol helyezkedjen el a lúgos körülmények között kialakuló alkoholát csoporttól.37,38 Ennek teljesítéséhez a transz-vegyületek (11ba, 11bb, 11ca, 11cb, 12ba, 12bb, 12ca, 12cb) primer hidroxilcsoportját a 13-ösztron sorban tozilezték (13ba, 13bb, 13ca, 13cb), míg a 13- ösztron származékoknál Appel-féle jódozásnak39 vetették alá (14ba, 14bb, 14ca, 14cb, 7.
ábra). Ezt követően a jól távozó funkciós csoportot és az alkoholos OH-t 1,3-helyzetben tartalmazó 13-epimereken (13ba, 13bb, 13ca, 13cb, 14ba, 14bb, 14ca, 14cb) Grob-
8
fragmentációt hajtottak végre, amely mindkét epimer sorban egy D-szekoaldehid (15b, c;
16b, c) képződését eredményezte (7. ábra).
7. ábra Grob-fragmentáció a 13β- és a 13-ösztron sorban
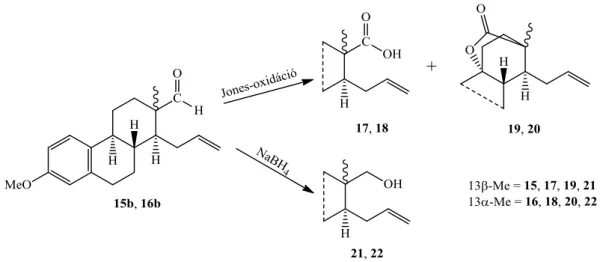
Ezek a D-szeko-származékok (15b, c; 16b, c) azért hasznos intermedierek, mert propenil oldalláncot és formil-funkciót tartalmaznak, amelyek számos átalakítási lehetőséget nyújtanak. A 3-metiléter sorban korábban már végrehajtották az aldehid-funkció Jones- oxidációját és redukcióját (8. ábra). A Jones-oxidáció mindkét epimer (15b, 16b) esetében hasonlóan játszódott le, főtermékként D-szekokarbonsav (17, 18), melléktermékként pedig egy áthidalt gyűrűs lakton (19, 20) képződött.40 Az oxocsoport (15b, 16b) fémhidrides redukciójával pedig 13-epimer D-szekoalkoholokhoz (21, 22) jutottak.
9
8. ábra D-Szekokarbonsavak (17, 18) és -alkoholok (21, 22) előállítása a 3-metiléter sorban
A D-szekoaldehid aldehidcsoportját egyéb nukleofil reagensekkel is átalakították.
Hidroxilamin-hidroklorid alkalmazásával, nátrium-hidroxid jelenlétében, kondenzációs reakcióban nyerték a 13-D-szekooxim 3-metiléterét (23, 9. ábra).41 A reakcióban melléktermékként dimetil-acetál (24) képződött. Ez utóbbi elkerülésére a későbbiekben a szervetlen bázist nátrium-acetátra cserélték. Így jutottak a D-szekooxim 3-metiléterének 13- epimereihez (23, 25, 9. ábra).42
9. ábra A D-szekooxim 3-metilétereinek (23, 25) előállítása
Az előállított nyitott D-gyűrűs, heteroatomot tartalmazó vegyületeket kiindulási anyagként szintetizálták különböző gyűrűzárási reakciókhoz. A D-szekoaldehidekből (15b, c; 16b, c), -karbonsavakból (17, 18) és -alkoholokból (21, 22) elektrofil reagensekkel D-homoösztron
10
származékokat állítottak elő (10. ábra). A hatástani vizsgálatok során kiderült, hogy a 26-os vegyület 5,5 M-os IC50 értékkel, szelektíven gátolja a HeLa sejtek osztódását. Továbbá a radioligand receptorkötődéses vizsgálatok és a patkány uterus mérések eredményeiből arra a következtetésre jutottak, hogy a D-homoösztron (26) sem in vitro, sem in vivo nem rendelkezik ösztrogén hatással.2,43 A D-szekooximokat (23) nitron dipólusokká alakítva 1,3- dipoláris cikloaddíciókat végeztek. Fenilizocianát dipolarofilek hatására olyan D- homoösztronokat nyertek, amelyek közül a feltüntetett 27-es vegyület, 2,2 M-os IC50
értékkel, szelektíven gátolta az A2780 sejtek osztódását (10. ábra).44
10. ábra Antiproliferatív hatású D-homoösztronok (26, 27)
A D-homoösztronok előanyagait, a D-szekoösztronokat gyógyszerhatástani szempontból nem vizsgálták. Az irodalomban csupán elvétve található példa ilyen típusú vegyületek biológiai vonatkozásait illetően. Az Újvidéki Egyetem kutatócsoportja Beckmann-fragmentációval állított elő D-szekoösztron származékokat és vizsgálta azok citotoxicitását, ill. affinitását az ösztrogén receptorokhoz.45−49 A dokkolási kísérletek arra utaltak, hogy a vizsgált D- szekoösztronok sem in vitro, sem in vivo nem rendelkeznek ösztrogén hatással. Az in vivo uterotróp és antiuterotróp módszerekkel50 kísérletesen is megállapították, hogy a 14- hidroxietilén oldalláncot tartalmazó D-szekoalkohol (28, 11. ábra) nem rendelkezik ösztrogén aktivitással. Továbbá a bisz-nitril (29) 3,9 és 7,3 M-os IC50 értékkel gátolja a HeLa és az MDA-MB-231 (emlő karcinóma, amely ösztrogén és progeszteron receptorokat nem fejez ki) sejtvonalak osztódását.51,52 Ezen eredmények gyógyszerkémiai szempontból ígéretessé teszik a D-szekoösztronok csoportját, ugyanis ez a kiaknázatlan terület még számos hatóanyag- fejlesztési lehetőséget nyújt.
11
11. ábra Az Újvidéki Egyetemen előállított D-szekoösztron származékok (28, 29)
A vegyületek magreceptoraikhoz való affinitását nemcsak az ösztrán váz módosításával és szubsztituálásával lehet csökkenteni, hanem különböző hibridek vagy konjugátumok képzésével is. Egy kanadai kutatócsoport olyan 17-ösztradiol homodimereket állított elő, amelyekben a két szteroid egység egy alkil vagy polietilénglikol (PEG) láncon keresztül, észter vagy éter kötéssel van összekapcsolva a 17-es szénatomon (12. ábra).8
12. ábra 17-ösztradiol homodimerek (30, 31)
A két monomert összekötő molekularész, a linker hossza jelentősen befolyásolhatja a vegyületek biológiai aktivitását. A Bérubé és csoportja által végzett in vitro sejtosztódás gátlási vizsgálataiból kiderült, hogy a 30a és a 30d jelű dimerek 5 M körüli IC50 értékkel gátolják a B16-F10 bőr karcinóma sejtek osztódását. Továbbá a fluoreszcens módszerrel meghatározott receptorkötődés53 alapján csekély affinitást mutatnak az ER-hoz. Ezzel szemben a 2 vagy 3 db (-CH2-O-CH2-)-egységet tartalmazó (30b, 30c), ill. 4 db (-CH2-)- egységet tartalmazó (31) dimerek a melanóma sejtvonalon kevésbé jó, míg az MCF-7 sejtvonalon hatékonyabb inhibitornak bizonyult. Az összekötő rész hossza és minősége tehát meghatározó szerepű a molekula szerkezetére és ezáltal biológiai hatására nézve. A linker és a
„beépítendő egység” kiválasztásánál ezért a szintetikus kémiai mellett farmakológiai szempontokat is figyelembe kell venni. Megfelelő szerkezeti elemek beépítésével ugyanis
12
növelhető a származékok biológiai aktivitása, stabilitása, oldhatósága és biohozzáférhetősége.
A szintézis oldaláról megközelítve pedig olyan reakciópartnerekre és kapcsolási módszerekre van szükség, amelyek gyorsan, szelektíven és az egyéb funkciós csoportok mellékreakciói nélkül szolgáltatják a kívánt konjugátumokat.
13 2.2. Azid-alkin „click” reakció szterán vázon
A 2000-es évek elején jelentős áttörés következett be a nitrogéntartalmú konjugátumok szintézisében, amikor először került bevezetésre a ″click″ reakció fogalma.54 A szintetikus elgondolás lényege, hogy a kívánt termékek kisebb szerkezeti egységek összekapcsolásával, enyhe reakciókörülmények között, kemo- és regioszelektíven, magas hozammal, melléktermék-képződés nélkül legyenek előállíthatóak. Ezen követelményeknek többek között megfelel a terminális alkinek és azidok [3+2] cikloaddíciója, amely már a 60-as évek óta ismert55, azonban népszerűségét és széles körű alkalmazását Cu(I)-katalizálta változatának köszönheti. A katalitikus módszert, amellyel 1,4-diszubsztituált triazolok regioszelektíven állíthatóak elő, Sharpless és Meldal kutatócsoportja egymástól független dolgozta ki.56,57
2.2.1. Cu(I)-ion katalizált azid-alkin „click”-reakció
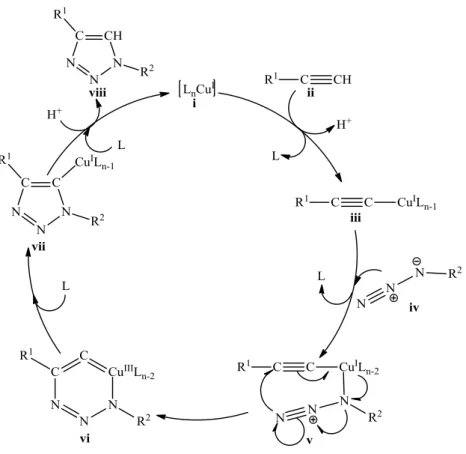
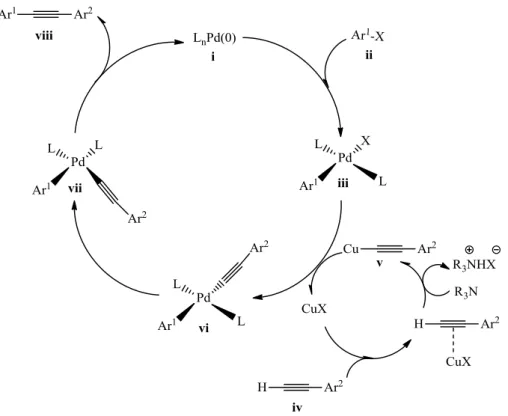
2002 óta több, mint 1000 közlemény jelent meg a Cu(I)-katalizált azid-alkin cikloaddíció (CuAAC) reakció vonatkozásában. A katalitikus körfolyamat mechanizmusának alapját DFT számítások adják, az egyes lépések kísérletesen még nem igazoltak (13. ábra).58Elsőként a komplexált réz(I)-ion (i) és a terminális acetilén között alakul ki egy π-komplex, majd egy ligandum kilépése mellett képződik a réz(I)-acetilid (iii). A π-komplex esetében a terminális alkin protonja elég savassá válik (~10 egység pKa csökkenés) ahhoz, hogy akár vizes közegben is disszociáljon, és σ-komplex képződjön. Elképzelhető lenne a csak π-koordinációt feltételező reakcióút is, de a modellszámítások szerint ez még a termikus reakcióutat is meghaladó aktiválási energiát igényelne. Az azid (iv) belépése a réz-acetilidre (iii) újabb ligandum távozásával jár, és a 13. ábrán negatív töltést hordozó nitrogén kapcsolódik a Cu(I)- ionhoz (v). Az azid terminális nitrogénje ezt követően nukleofil támadást hajt végre az alkin C-2 szénatomjára. A kialakuló hattagú Cu(III)-metallaciklus (vi) egy szokatlan szerkezetű intermedier, képződése endoterm folyamat, azonban az aktiválási energia jelentősen kisebb, mint a katalizálatlan reakció esetében. Gyűrűszűküléssel alakul ki a Cu(III)-metallaciklusból (vi) a Cu(I)-triazolil komplex (vii), amelyből a ciklus utolsó lépésében protonálódás útján létrejön a kívánt triazol (viii), és visszakaalakul a katalizátor aktív formája.
14
13. ábra A CuAAC reakció katalitikus ciklusának feltételezett mechanizmusa58
A folyamat megfelelő lejátszódásához elengedhetetlenül szükséges a Cu(I)-ionok nagy koncentrációja. Ez három különböző eljárással érhető el.54 Az egyik lehetőség a prokatalizátor alkalmazása, amikor pl. CuSO4·5H2O-ból kiindulva, redukálószer (nátrium-aszkorbát) segítségével in situ állítják elő a Cu(I)-ionokat. A másik módszer a CuI, CuBr vagy CuOAc és bázis vagy amin ligandum alkalmazása redukálószerrel egyidejűleg. Ez utóbbi vegyület a Cu(I)-ionoknak Cu(II)-ionokká történő aerob-oxidációját akadályozza meg, az amin bázis pedig segíti a réz(I)-acetilid komplex képződését (a Cu(I)-sók ugyanis kezdetben stabilis klasztereket képeznek). Továbbá rézdrót, -huzal, -por vagy -nanorészecskék is felhasználhatók, amelyek felületén Cu(II)-sóval történő oxidációval alakul ki a Cu(I)- katalizátor.
A reagensek és körülmények megválasztásakor, pl. biokonjugációknál, fontos szempont, hogy a reakciók enyhébb körülmények között, akár szobahőmérsékleten vagy vízben is lejátszódjanak. A cikloaddíciókhoz használhatunk pl. poliligandumokat, ami nem szükségszerű, de mégis jelentősen növelheti a reakciósebességet. Szobahőmérsékleten a CuAAC lassú, amennyiben nincs jelen nagy koncentrációban a Cu(I)-katalizátor, ez azonban biokonjugációknál nem megengedett. Ilyenkor többfogú N-donor segédanyagokat
15
alkalmaznak (ismeretes, hogy az aminok pl. nagyobb mértékben növelik a reakciósebességet, mint a piridinek). Gyakran használt gyorsítók továbbá a trifenilfoszfán vagy az egyes karboxilátok. Előbbinek az alkalmazhatóságát azonban korlátozza a Staudinger-reakció, mint kísérő folyamat, amely során az azid aminná redukálódik, így az egyik reakciópartner
„elfogy” a reakcióelegyből. Ennek kiküszöbölésére a trifenilfoszfánt nem sztöchiometrikus, csupán katalitikus mennyiségben alkalmazzák.
A CuAAC reakció szubsztrátjaként azidok és alkinek széles skálája alkalmazható, a kapcsolás különböző funkciós csoportok mellett is elvégezhető.59 A felhasználható oldószerek köre az apoláristól a vízig terjed. Meldal és Tornøe a különböző oldószerek és oldószer- elegyek hatását vizsgálták a reakcióra.60 Az oldószer kulcsszerepet játszik a szubsztrátok és a Cu(I)-katalizátor szolvatálásában. A poláris oldószer alkalmazása kedvez a heterociklus képződésének, azonban egy erősen koordinálódó oldószer lassíthatja vagy gátolhatja a fém- szubsztrát koordinációt.
2.2.2. „Click”-reakció alkalmazása szterán vázon
A Fokin és Sharpless által kifejlesztett módszer a szteránvázas vegyületek átalakításában is alkalmazásra talált. Az azid vagy a terminális alkin funkció kialakítása különböző szteroidokon a származékok nagyfokú változatosságát teszi lehetővé, amely mind kémiai mind farmakológiai szempontból érdekes lehet. Számításos vizsgálatok arra utalnak, hogy a triazol-gyűrű nagyfokú hasonlóságot mutat a peptidkötéssel (H-híd kialakítása révén), azonban attól eltérően kémiailag és metabolikusan is stabil. Ezek alapján ezt a szerkezeti egységet a szteránvázas vegyületekre építve növelhető azok vízoldékonysága, biológiai hozzáférhetősége és metabolikus stabilitása.61,62
A Szerves Kémiai Tanszék Szteroidkémiai Kutatócsoportja számos alapvázra, különböző helyzetbe építette be a triazolil-egységet. A kolesztán váz 2α-, az androsztán váz 15β- és 1α-, valamint az ösztrán váz 16-os és 17-es helyzetében azid funkciót kialakítva vizsgálták azok nem-szteroid-típusú terminális alkinekkel való „click”-reakcióit (14.
ábra).63−71 A cél a CuAAC reakciók optimalizálása, a reakcióidők csökkentése, a magas hozamok elérése és a régiószelektív szintézisek kidolgozása volt. A Pannon Egyetem kutatócsoportja a 2-es, 6-os és 16-os helyzetben -térállású azidocsoportot tartalmazó androsztán származékok reaktivitását vizsgálta.72
16
14. ábra Triazolil egység beépítése szterán vázra, szteroid azidból (kék) vagy alkinből (rózsaszín) kiindulva
Ösztrán vázon a cikloaddíciók CuI katalizátor alkalmazásával, DIPEA és PPh3 jelenlétében, a diklórmetán vagy a toluol forráspontján játszódtak le. A 3-metoxiösztron 17- és 17- azidjainak CuAAC reakcióinál, amennyiben a PPh3 gyorsító ligandum helyett trietilamint adnak a rendszerhez, kétféle termék keletkezését tapasztalták.70 A várt 1,4-diszubsztituált triazolok mellett, kis mennyiségben a triazol gyűrűn jóddal szubsztituált származék is megjelent. Az ösztron sorban előállított egyes célvegyületek mérsékelt in vitro antiproliferatív hatást mutattak több humán adherens tumorsejtvonalon.64,70 Továbbá azt is megállapították, hogy ezt a hatást jelentősen befolyásolja az azid és az alkin reakciópartner szerkezete is. A 17-epimer konjugátumok közül a heteroaromás csoportot -térállásban tartalmazó vegyületek a HeLa, míg -megfelelőik az MDA-MB-231 és -365 (emlő karcinómák) sejtek osztódását gátolták. A legpotensebbek a triazol gyűrűn cikloalkil-csoporttal szubsztituált, nem jódozott származékok voltak.
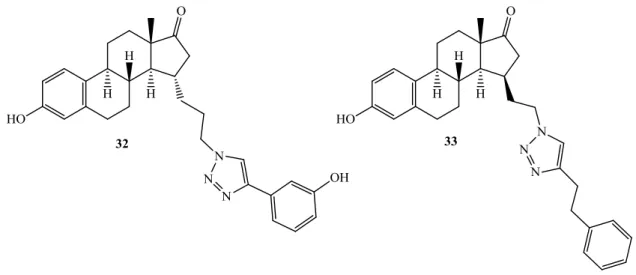
Az ösztron-triazoloknak a sejtosztódás gátlás mellett, szteroidogén enzimekre gyakorolt hatását is vizsgálták. Egyes 15-szubsztituált származékok (32, 33), a linker hosszától és térállásától függetlenül hatékony 17β-HSD1 inhibitornak bizonyultak.73,74
17
15. ábra 17β-HSD1 inhibitor hatású 15-szubsztituált triazolil-ösztron származékok (32, 33)
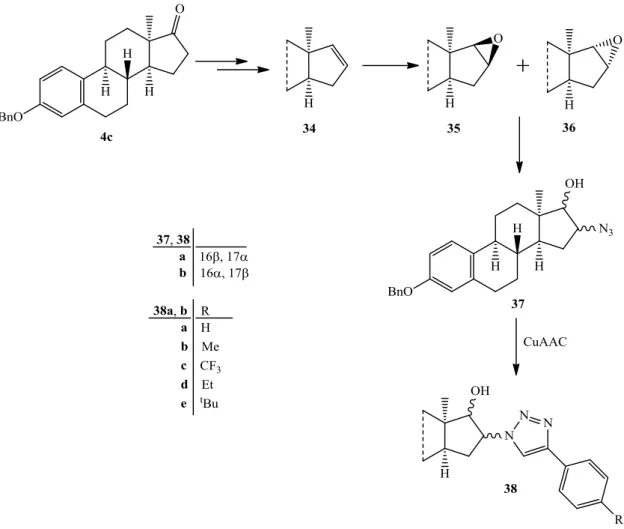
Kutatócsoportunk a közelmúltban 13α-ösztron modellvegyületen célozta meg a triazolil-funkció kiépítését. Az azid funkciót a 16-os helyzetben kívánták kialakítani, ahol korábban az oxim funkció beépítése előnyös volt (lsd. 8).31 A 13-epimer 3-benziléteréből (4c) kiindulva, a Schönecker és munkatársai által kidolgozott módszerrel,75 először egy 16-olefint (34) állítottak elő, majd a vegyületet szerves persavval epoxidálták (16. ábra). A β- és az α- epoxid 3:1 arányú keverékének (35 és 36) kromatográfiás szétválasztása után, az epoxidnyitást azid nukleofillel végezték. A reakció regioszelektíven eredményezte a transz 16-azido-17-alkoholokat (37a, b), amelyeket terminális etinilcsoportot tartalmazó kismolekulákkal konjugáltak.69
18
16. ábra Azidoalkoholok (37a, b) és konjugátumaik (38a, b) előállítása
A triazolok (38) hatástani tesztelése során fontos szerkezet-hatás összefüggést állapítottak meg: a konjugátumok közül csak azok az izomerek voltak hatásos apoptózis indukáló szerek (sejtciklus G2/M blokádja), amelyek a 16β-azido-17α-alkoholokból (37b) képződtek. Ezen eredmények összhangban vannak a 7-es jelű vegyülettel kapcsolatban korábban említettekkel,30 ugyanis Poirier és munkatársai is megállapították, hogy a 13-sorba tartozó 3,17-diolok közül a 17-izomer (7) rendelkezik a legerősebb citosztatikus hatással. A 38-as jelű vegyületek hatásának erősségét az újonnan beépített feniltriazolil-egység szubsztituense is befolyásolta, ugyanis a p-helyzetben alkilcsoporttal rendelkező származékok (38bd, 38be) voltak a legpotensebbek.
Az irodalomban csupán elvétve található példa olyan szteroid-triazolok szintézisére, amelyeket szteroid-alkinből kiindulva állítottak elő. Lipschutz és Alonso csoportjai alkin modellvegyületként a 17α-etinilösztradiolt választották. Olyan CuAAC reakciókat hajtottak végre, amelyekhez Cu/C,76 illetve nanoszemcsés réz77 katalizátort alkalmaztak. Montenegro és munkatársai az ösztradiol (2a) 17-etinil és -azidometil származékából, illetve a mestranol
19
17-etinil származékából állítottak elő hibrideket.78 A szteroid egységek közé különböző linkereket építettek, reakciópartnerként benzolt, heteroaromás és heterociklusos származékokat, ill. ferrocént alkalmazva.
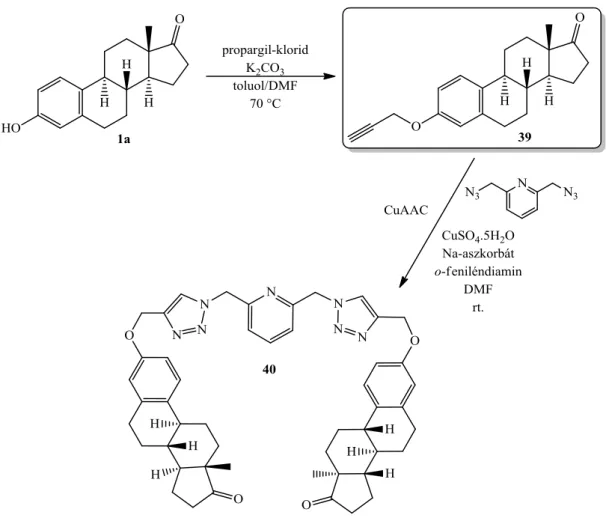
Az ösztron fenolos OH-jának éteresítésével is előállíthatók terminális alkinek. Az aromás A-gyűrű 3--alkinilétereinek előállítását már a 60-as években közölték.79,80 A szteroidot fém nátriummal etanolban forralva nyerték a kívánt vegyületeket. Később új módszereket dolgoztak ki az ösztron 3-propargil-éterének előállítására, bázisként kálium- karbonátot alkalmazva. A vegyületet többek között "click"-reakcióban alakították tovább, különböző célokkal: ösztron-alapú makrociklusok,81 gélek,82 vagy biológiailag aktív származékok83,84 előállítása. Jurasek és munkatársai propargil-kloriddal hajtotta végre az éteresítést toluol és dimetil-formamid elegyében (17. ábra).84 Az előállított szteroid alkint (39) bisz-azidometil-piridinnel CuAAC-reakcióban reagáltatva ösztron-homodimert (40) képeztek, majd MTT-módszerrel vizsgálták annak citotoxicitását. A korábbi tapasztalatoknak megfelelően, ez a triazol-származék is hatékony antiproliferatív hatású vegyületnek bizonyult, különböző leukémia sejtvonalakon.
20
17. ábra Ösztron homodimer (40) előállítása
A terminális alkin funkciót tartalmazó ösztron származékok számos továbbalakítási lehetőséget nyújtanak. A CuAAC reakciókon túlmenően, palládium-katalizált C-C keresztkapcsolási folyamatok kiindulási anyagaként is szolgálhatnak.
21
2.3. Sonogashira keresztkapcsolás szteroid modellen 2.3.1. A Sonogashira kapcsolás
A 2000-es években az átmenetifém-katalizált keresztkapcsolási reakciók forradalmasították a szintetikus kémiát. Olyan funkcionalizálási lehetőségeket nyitottak meg, amelyek a klasszikus módszerekkel nem, vagy csak nehézkesen voltak megvalósíthatók. A szén-szén kötés kialakulásával járó keresztkapcsolások közül kiemelkedő jelentőségűek a palládium- katalizálta reakciók, amelyek a transzmetallálási lépés nukleofil reagensében (fémorganikus reagens) különböznek egymástól. Az egyik legelterjedtebben alkalmazott típus a Sonogashira- kapcsolás, ahol a nukleofil szerepét rézorganikus vegyület tölti be.85 Az eljárással terminális acetilének és aril- vagy vinil-halogenidek közötti C(sp2)-C(sp) kötés alakul ki egy katalitikus körfolyamatban (18. ábra). A katalitikus ciklus első lépése a szerves elektrofil reagens (Ar1-X, leggyakrabban aril-halogenid, ii) oxidatív addíciója a nulla oxidációs számú palládiumra (i).
A palládium formálisan beékelődik a szén-halogén kötés közé (iii), ami egyfajta aromás nukleofil szubsztitúciónak is tekinthető. Ennek megfelelően a reaktivitás a távozó csoport minősége szerint a I ≥ OTf(trifluormetán-szulfonát) > Br > Cl sorban csökken.86 A jó távozó csoport mellett az átalakulást tovább gyorsítják az aril-vegyület elektronszívó szubsztituensei.87−92 A körfolyamat következő lépése a transzmetallálás, ami azt jelenti, hogy az acetilén reakciópartner rézorganikus nukleofilként kerül a palládium-komplexre. A folyamatot a rendszerben jelen levő bázis és az oldószer nagymértékben befolyásolhatja. A réz-acetilid (v) in situ keletkezik egy független katalitikus ciklusban, a jelenlévő bázis hatására. Ennek fontos szerepe van a ciklusban, mivel az acetilének (iv) pKa értéke 26 körüli, ami a réz koordinálódásának köszönhetően 10 egységgel csökken. A transzmetallálás hajtóereje a két fém elektronegativitása közötti különbség (ENCu > ENPd), ezért a fémhez koordinálódni képes arilcsoportok gyorsítani tudják a folyamatot.93,94 Ezt követően a kialakult transz-komplex (vi) izomerizáció révén cisz-komplexszé (vii) alakul, amit a poláris oldószerek segítenek. Végül a termék (viii) reduktív eliminációval hasad le a palládiumról és visszaalakul a palládium aktív formája (i). A lépést nagymértékben befolyásolja a fémhez kapcsolódó ligandum szerkezete, sztérikus és elektronikus sajátsága.95,96 Az 1. lépéssel ellentétben, az elektronhiányos, nagy térkitöltésű ligandumok gyorsítják a folyamatot, miközben a komplex stabilizálása révén megakadályozzák annak dezaktiválódását. Továbbá a
22
cisz-helyzetben lévő reakciópartnerek aril-, alkil- és alkinilcsoportjainak elektronküldő szubsztituensei is kedvezően hatnak a reduktív eliminációra.97−99
18. ábra A Sonogashira-kapcsolás általános mechanizmusa
A szerves kémiai átalakításokban leggyakrabban a 0-s oxidációs állapotú Pd(PPh3)4
komplexet vagy a palládium 2-es oxidációs állapotú sóit (PdCl2, Pd(OAc)2, PdCl2(PPh3)2) használják, mivel ezek levegőre és nedvességre kevésbé érzékenyek. A Pd(II) katalizátorok esetén a katalitikus körfolyamat beindítása a palládium redukciójával történik, ami foszfán ligandumok, valamint bázis együttes hatására könnyen végbemegy a reakcióelegyben.100 A fém katalitikus aktivitása és oldhatósága szerves oldószerekben befolyásolható az alkalmazott ligandumokkal. A nagyobb térkitöltésű ligandumok kedvezően hatnak az oxidatív addíciós és reduktív eliminációs lépésekre. Általában foszfán ligandumokat választanak, amelyekkel gyorsabban, enyhébb körülmények között mennek végbe a kapcsolási reakciók. Továbbá a foszfor elektronküldő szubsztituensei tovább növelik az átmenetifém elektronsűrűségét, ezáltal stabilizálják a kialakuló komplexet és elősegítik a folyamatot.101 Számos kísérleti bizonyíték van arra, hogy a reakciósebesség szempontjából a ligandum térkitöltésének is fontos szerepe van.102 Az ezredforduló környékén számos új típusú foszfán ligandumot állítottak elő, amelyek nagy térkitöltésű csoportokat és elektronban gazdag foszfor atomot
23
tartalmaznak. A belőlük képződő komplexek sztérikus zsúfoltsága megkönnyíti a ligandum disszociációját, ezáltal kedvez az oxidatív addíció lejátszódásának a koordinatíve telítetlen fém centrumon. A legújabb eredmények alapján a ligandumok ezen tulajdonsága a létrejött Pd(II)-komplex szerkezetére is hatással van. A sztérikusan kevésbé zsúfolt trifenilfoszfán (PPh3) jelenlétében a komplex [L2Pd(Ar)X] képlettel leírható formában van jelen, míg a nagyobb térkitöltésű terc-butil-csoporttal szubsztituált PtBu3 esetében a [LPd(Ar)X] formában létezik.103 Ennek eredményeként bifenil,104−106 terc-butil,107 binaftil és adamantil- szubsztituált108 foszfánok nyertek alkalmazást keresztkapcsolási reakciókban (19. ábra).
19. ábra Különböző szerkezetű foszfán ligandumok
A Sonogashira-kapcsolásoknál nemcsak a reagensek stabilitása és levegőre való érzékenysége okozhat problémát. A réz(I)-só az acetilidek transzmetallálása mellett azok dimerizációját, vagyis homokapcsolását is elősegíti.109 Ennek az ún. Glaser-típusú kapcsolásnak a visszaszorítása érdekében került kidolgozásra a rézmentes Sonogashira kapcsolás. Mivel ebben az esetben a katalitikus körfolyamatból kimarad a transzmetallálás lépése, a bázisnak kulcsfontosságú szerepe van az acetilén aktív formába hozatalában. Ehhez szerves amin bázisok jelenléte szükséges.110−112 Az irodalomban kevés példa található rézmentes körülmények között végrehajtott kapcsolásra. A reakciók erősen bázikus közegben (nagy mennyiségű NEt3) vagy piperidin alkalmazásával, több órás reakcióidő alatt, ill.
sokszor nagyon magas hőmérsékleten játszódnak le.112−115 A rézmentes kapcsolásnak két feltételezett mechanizmusa létezik (20. ábra). Az első katalitikus lépés (iii komplex kialakulása) mindkét mechanizmus szerint a rézzel történő kapcsolásnak megfelelően történik. A következő lépésben viszont az acetilén (iv) fém-centrumhoz való koordinálódása (v) valósul meg, amely kétféleképpen alakulhat tovább. Az I mechanizmus szerint116 a bázis deprotonálja az acetilént és kialakul a Pd-acetilid komplex (vi), melynek reduktív eliminációjával kapjuk a kapcsolt terméket (vii). A II mechanizmus alapján117 az acetilén
24
koordinációja után beékelődéssel alakul ki a Pd-(aril-alkenil)-komplex (vi), amelyből β- hidrogén eliminációval keletkezik a termék (vii). A Pd-(arilalkenil)-komplex (vi) jelenlétének feltételezése és annak továbbalakulása β-hidrogén eliminációval, analóg a Mizoroki-Heck reakció feltételezett mechanizmusával.118−121 A ciklus végén mindkét esetben visszaalakul a katalizátor aktív formája (i).
20. ábra A rézmentes Sonogashira kapcsolás feltételezett mechanizmusai
A mechanizmus vizsgálata során megállapították, hogy poláris oldószer választása és annak hidrogénkötések kialakítására való képessége stabilizálja a körfolyamat ionos intermediereit, ezáltal kedvező hatással van kapcsolási reakció lejátszódására.117,122,123
A Cu(I)-sóval elősegített Sonogashira kapcsolásokkal megegyezően, a rézmentes változat is megvalósítható szilárd hordozóra választott palládiummal124 és ionos folyadékokban115 is.
2.3.2. Biológiailag aktív ösztron származékok előállítása Sonogashira-kapcsolással
A C-C kötés kialakítására alkalmas kapcsolási módszer előnyeit hamar felismerték a szteroidkémiában is. A 2000-es évek eleje óta több kutatócsoport is foglalkozott ösztron származékok Sonogashira-kapcsolással történő előállításával, 17-etinil-ösztradiolból kiindulva. A szteroid (alifás) alkint változatos szerkezetű halogenidekkel vitték kapcsolási reakcióba különböző célokból. A mestranolt a Sonogashira-kapcsolás körülményei között
25
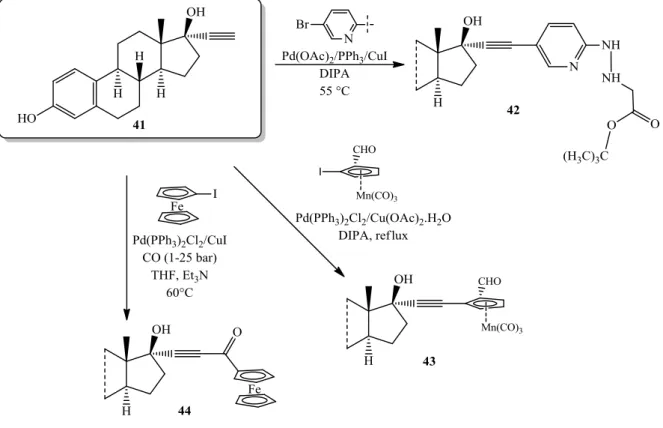
[11C]metil-jodiddal reagáltatva, olyan radioaktívan jelzett ösztron konjugátumokat nyertek, amelyek a PET (positron emission tomography) képalkotó technikánál nyomjelzőként alkalmazhatóak.125A 17-alkin (41) reakcióit különböző fémkomplexekkel is vizsgálták (21.
ábra). Egyes származékok (42) az ösztrogén receptor kötődési vizsgálatokban bizonyultak értékesnek, más vegyületeknek (43) pedig az antitumor hatásáról számoltak be.126−128
21. ábra 17-Etinil-ösztradiol származékok szintézise Sonogashira-kapcsolással
Egy orosz kutatócsoport az ösztron 3-propargil-éteréből (39) nukleozidokkal képzett konjugátumokat és vizsgálták azok antivirális hatását HSV-1 (herpes simplex virus type 1) ellen.129,130 Azt tapasztalták, hogy egy flexibilis linker, a nukleobázis (uracil) és az aromás alkin partner között növeli a vegyületek citotoxicitását, miközben csökkenti azok antivirális aktivitását. Bérubé és Poirier az ösztron 16-os szénatomján alakított ki terminális alkin funkciót (45), amelyet Sonogashira-keresztkapcsolással alakítottak tovább (22. ábra).131,132 Az előállított ösztron-adenozin hibrid hatékony 17-HSD1 enzimgátlónak bizonyult, azonban a szerkezetet egyszerűsíteni kellett (47) a vegyület biohozzáférhetőségének javítása miatt. Az 22-es ábrán feltüntetett IC50 értékekből látható, hogy a linkerben lévő CH2-egységek
26
számának növelése (m) kedvezőtlenül hat a konjugátumok 17-HSD1 enzimgátló képességére.
22. ábra 17-HSD1 enzimgátló ösztron konjugátumok (47)
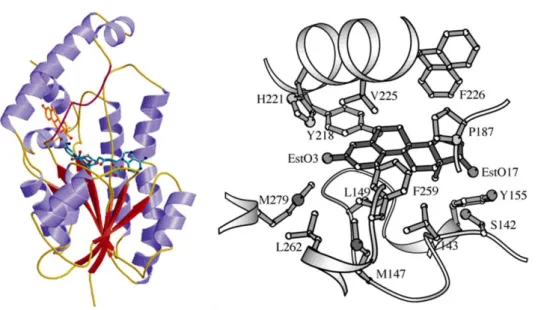
A 17β-HSD1 izozim kristályszerkezete már 1995 óta ismert,133 amely lehetőséget nyújt inhibitorok célzott tervezésére. Az enzim egy merev kofaktor- és egy keskeny, lipofil szubsztrátkötő régióból áll, amelyek együttesen alkotják a „szubsztrát felismerő domént” (23.
ábra).74 Az enzimreakció során hidrid anion addícionálódik a szubsztrát ösztron 17-es szénatomjára, amely az irodalom szerint többféle mechanizmussal is végbemehet. A redukciót a 142-es szerin (S142), a 155-ös tirozin (Y155) és a 159-es lizin (L159) által alkotott katalitikus triád végzi egy vízmolekula jelenlétében.
27
23. ábra A 17β-HSD1 izozim másodlagos szerkezete (bal) és a szubsztrátkötő domént alkotó aminosavak az ösztronnal (jobb)134
A szteroid-alapú inhibitorok között számos szubsztituált és heterociklusos ösztron-származék ismert.135,136 A vegyületek többségénél megtartották az ösztránvázra jellemző fenolos OH- és 17-oxocsoportot, amelyek jelenléte az irodalom szerint szükséges a vegyületnek az enzim szubsztrátkötő helyéhez való bekötődéséhez. A röntgendiffrakciós és számításos vizsgálatokból továbbá az is kitűnt, hogy a szubsztrátkötő csatorna lipofil ürege alkalmas olyan ösztron származékok megkötésére, amelyek a 2-es pozícióban lipofil szubsztituenst tartalmaznak. Ez alapján a 2-es helyzetbe többek között különböző halogéneket, aralkil- vagy aralkinil-csoportokat építettek be potenciális inhibitorok előállítása céljából.23,137 Möller és kutatócsoportja ösztron-3-acetát modellen, 2-jódvegyületből (48) kiindulva végeztek Sonogashira-kapcsolást. Így jutottak a természetes ösztron 2-feniletinil származékaihoz (49, 50, 24. ábra). A 2-es helyzetben halogénnel, alkilcsoportottal vagy fenilalkinil-csoporttal szubsztituált vegyületek 17β-HSD1 enzimgátló hatását vizsgálva kiderült, hogy a szabad fenolos OH-s 50-es vegyület az egyik legjobb inhibitor, 56 nM-os IC50 értékkel. A fenolos hidroxilcsoport észteresítése és/vagy acetilén helyett alkilcsoport beépítése a 2-es helyzetbe, kevésbé előnyös az inhibitor hatás szempontjából.
28
24. ábra 2-Fenilalkinil-ösztron származékok (49, 50) előállítása
Tudomásunk szerint, az irodalomban eddig ez az egyetlen példa Sonogashira-kapcsolás megvalósítására az ösztron 2-jódvegyületéből kiindulva.
2.3.3. Az ösztron aromás gyűrűjének jódozási reakciói
Szteroidok halogénezésére több példa is található az irodalomban biológiailag aktív származékok szintézise kapcsán.138−140 Az ösztron aromás A-gyűrűjének halogénezésével olyan származékok nyerhetők, amelyek változatos átalakítási lehetőségeket biztosítanak.
Kiindulási anyagként szolgálhatnak többek között a 2.3.2. fejezetben bemutatott Sonogashira keresztkapcsolási reakciókban. Továbbá egyes képviselőik az ösztradiol bioszintézisében szerepet játszó enzimek gátlásán keresztül antitumor hatást fejtenek ki.15,141−144 A halogenidek közül a jodid a legjobb távozó csoport és egyben legreaktívabb, ezért csak az aril-jodidok előállítását mutatom be.
Az ösztron aromás A-gyűrűjének jódozása már az 1950-es évek óta ismert. Hillmann- Elies és munkatársai az ösztron jódozását higany(II)-jodiddal valósították meg ecetsavban (25. ábra). Regioszelektíven nyerték a 2-jód-ösztront (51), 96%-os hozammal.145
29
25. ábra Hillmann-Elies és kutatócsoportja által kidolgozott jódozási eljárás
Azóta több kutatócsoport, más-más reakciókörülmények között, különböző arányban állította elő a 2- és 4-jód regioizomerek keverékét. 2003-ban szabadalmaztattak is egy eljárást, amelyben higany(II)-acetát és elemi jód segítségével szintetizáltak 2-jód-ösztront (51).146 A szerves higanyvegyületek alkalmazását toxicitásuk miatt később teljes mértékben kiszorította az elemi jód egyéb reagensekkel történő kombinálása. Horiuchi és kutatócsoportja közleményében réz(II)-acetát, ecetsav és elemi jód jelenlétében előállított 2-jódszármazékról számol be.147 Később ammónium-hidroxid és elemi jód metanolban vagy metanol/víz elegyben végrehajtott reakciójával kapták a 2-jódizomert.148,149 A napjainkban legszélesebb körben alkalmazott tallium(III)-trifluoracetát (TTFA) reagenst elsőként kanadai kutatók használták 1987-ben.150 Ösztron-3-acetátot (52) és ösztradiol-3,17-diacetátot reagáltattak szobahőmérsékleten, TFA (trifluorecetsav) oldószerben 1‒2 mol ekvivalens Tl(III)- reagenssel, elemi jód jelenlétében, és szelektíven nyerték a 2-jódizomert (48, ~95%).
Ösztradiolból kiindulva, a már említett higany(II)-acetátos módszerrel ugyan nagyobb hozammal kapták a jódszármazékokat, viszont a reakció szelektivitása csökkent: 60 %-ban a 2-, 30 %-ban a 4-, és 10 %-ban a 2,4-bisz-jódszármazékhoz jutottak. Megfigyelték továbbá azt is, hogy a szabad hidroxilcsoportok jelenléte oxidációs mellékreakciókhoz vezethet, míg a 3- metiléter védőcsoport megakadályozza a reakció lejátszódását. A 90-es években Bulman és munkatársai megismételték a tallium(III)-trifluoracetátos jódozást, 2 mol ekvivalens reagenssel, 0°C-on, N2 atmoszféra alatt, így 84 %-os hozammal jutottak a 3-acetoxi-2-jód- ösztronhoz (48).151 A közelmúltban Lawrence Woo és kutatócsoportja is megvalósította ez utóbbi szintézist,142 amelynek lépéseit és körülményeit a 26. ábra mutatja be.
30
26. ábra Jódszteroid regioszelektív szintézise tallium(III)-komplexszel
A nagyfokú regioszelektivitásra magyarázatot adhat egy hattagú kelátgyűrű (53) képződésének a feltételezése, amely térigénye miatt főként a 3-as helyzetű acetát karbonil oxigénje és a 2-es helyzetű tallium között alakulhat ki. Azonban a módszer gyógyszeripari felhasználásának gátat szab a tallium vegyületek mérgező hatása és magas ára. Egy alternatív eljárás kidolgozásához kutatócsoportunk a tallium-komplexeknél jóval olcsóbb N-jód- szukcinimidet (NIS) kívánta felhasználni. Az irodalomban az ösztron A-gyűrűjének jódozása ilyen körülmények között korábban nem volt ismert, azonban aromás gyűrűs alkaloidok jódszármazékait sikerrel állították elő ezzel a reagenssel.152 Kutatócsoportunk a 13-epi- ösztron 3-as helyzetben hidroxil- vagy metoxicsoportot tartalmazó származékainak (4a, 4b) jódozását valósította meg 1 mol ekvivalens NIS-del, trifluorecetsavat (TFA) alkalmazva oldószerként (27. ábra).153 A 4a-ból és annak 3-metil-éteréből (4b) kiindulva, 1 ekvivalens NIS-del a 2- és 4-jód izomerek (54a:54b, 55a:55b) 1:1 arányú keverékéhez jutottak. A reagens mennyiségét a duplájára emelve a 3-hidroxi sorban főtermékként megjelent a kétszeresen szubsztituált szteroid (54c), ill. a monojód izomerek aránya eltolódott 54a:54b = 1:4. Az előállított vegyületeknek (54, 55) vizsgálták a 17-HSD1 enzimre gyakorolt gátló hatását és azt tapasztalták, hogy az inhibitor jelleget nagymértékben befolyásolja a 3-as helyzetben lévő csoport minősége és a jód helyzete. A fenolos OH éteresítése kedvezőtlennek bizonyult a biológiai aktivitás szempontjából, a leghatékonyabb származék a 3-hidroxi-13- ösztron 2,4-bisz-jód izomere (54c) volt.
31
27. ábra A 13-ösztron (4a, b) aromás A-gyűrűjének jódozása NIS-del
32
2.4. Átmenetifém-katalizált reakciók aktiválása mikrohullámmal
A bemutatott átmenetifém-katalizált kapcsolási reakciók kézenfekvő eszközei a hetrociklusos vegyületek célzott szintézisének, valamint a természetes eredetű vegyületek és analogonjaik előállításának. A módszerek alkalmazhatóságát tovább növelte a mikrohullámú technika bevezetése a szintetikus szerves kémiába.154−158 Mikrohullámú besugárzással történő aktiváláskor hatékonyabb energiaközlés érhető el, így olyan kémiai átalakítások is megvalósíthatók, amelyek hagyományos úton nehezen. A MAOS (microwave-assisted organic synthesis) legfontosabb jellemzői a nagyobb szelektivitás és a lényegesen rövidebb reakcióidő. Lehetőség nyílik továbbá enyhébb reakciókörülmények alkalmazására, mint pl. az alacsonyabb hőmérséklet, kevesebb sav/bázis/katalizátor. A reakciók heterogén fázisban vagy akár oldószermentesen is lejátszódhatnak.
Ennek felismerése óta számos közlemény és több kutatócsoport is foglalkozott a Sonogashira-kapcsolás mikrohulámmal való hatékonyabbá tételével. A hatékonyabb energiaközlésnek köszönhetően lehetőség nyílik a réz, amin, vagy akár ligandum mentes körülmények között végrehajtható reakciók kidolgozására. Ez a mikrohullámú technika bevezetése előtt nem volt kivitelezhető, mivel ‒ ahogy azt a 2.3.1. fejezetben tárgyaltam ‒ ezekre a reagensekre az acetilén reaktivitásának növeléséhez van szükség a katalitikus körfolyamatban. A mikrohullámú besugárzással nemcsak az acetilént tudjuk reaktívabbá tenni, hanem az elektronszegény halogenid partnert is. Liu és munkatársai sztérikusan gátolt, elektronszegény aril-kloridokat vittek Sonogashira kapcsolási reakcióba mikrohullámú reaktorban.159 A Pd(II)/P(tBu) katalitikus rendszer alkalmazásával változatos szerkezetű klórvegyületek (56) és különböző terminális alkinek (57) között alakítottak ki C-C kötést. A mikrohullámú reaktorban a reakciók rövid idő alatt, jó-kiváló hozammal szolgáltatták a kívánt acetilén termékeket (28. ábra).
28. ábra Aril-kloridok (56) Sonogashira-kapcsolása mikrohullámú reaktorban
33
Nehézséget jelent továbbá a katalizátorok és a ligandumok magas ára, hogy nem újrahasznosíthatók, ill. a homogén fázisú reakciókból adódóan, a katalizátorok nehézkes elválaszthatósága a reakcióelegytől. Az utóbbi években ezért előtérbe került a heterogén rendszerek kidolgozása. A palládium katalizátort szilárd hordozóra választva (heterogén katalizátor), az könnyen újrahasznosíthatóvá válik. Ley és kutatócsoportja közleményében heterogén Pd-EnCatTM TPP30 katalizátor segítségével kivitelezett keresztkapcsolási reakciókról számolnak be.160 A különböző aromás brómvegyületek és vinil-kloridok mikrohullámú körülmények között, rövid idő alatt szolgáltatták a céltermékeket. Az eljárás további előnye, hogy a katalizátor szűréssel visszanyerhető a reakcióelegyből. A technika beépíthető különféle új farmakofórok szintézisének eljárásaiba. Luthman és munkatársai a kromán-váz szubsztituálását,161 míg Stevens és kutatócsoportja indol származékok (62) előállítását valósította meg mikrohullámmal aktivált Sonogashira kapcsolással.162 A több reakcióból álló szintézis egyik kulcslépése a kinon vázas terminális acetilén (60) Pd-katalizált keresztkapcsolása az o-amino-jódbenzolhoz (59). A kinol szerkezeti egységet tartalmazó célvegyületek (29. ábra) in vitro szelektív gátlást fejtettek ki vese és vastagbél tumorsejtek osztódására.
29. ábra Indol származékok (62) Sonogashira kapcsoláson alapuló szintézise
A C-C kötés kialakítására alkalmas reakciók mellett a különböző cikloaddíciók mikrohullámú reaktorban történő kivitelezésére is számos összefoglaló közleményt találtunk az irodalomban. Elsőként van der Eycken és csoportja vizsgálta a mikrohullámú besugárzás hatását a Cu(I)-katalizált Huisgen-féle dipoláris cikloaddícióra (30. ábra).163 A szerves azid a
34
reakció körülményei között in situ képződött nátrium-azidból (NaN3) és szerves halogenidből (61). Azt tapasztalták, hogy a reakcióidő nagyságrendekkel (órákról percekre) csökkent, miközben az 1,4-regioszelektivitás nem változott, és a termékek (63) kristályosítással kinyerhetők voltak a reakcióelegyből.
30. ábra Az első mikrohullámú „click”-reakció
Ezen megállapítás óta több száz publikáció jelent meg a mikrohullámmal aktivált „click”- reakcióval kapcsolatban. A módszer alkalmas különböző biokonjugátumok: nukleozidok, szénhidrátok, peptidek, oligonukleotidok és analogonjaik előállítására,164−167 dendrimerek168−170 és polimerek szintézisére is akár szilárd hordozóhoz rögzítve171 is.
Napjainkban a dendrimerek mellett fullerének172 és nanocsövek funkcionalizására173,174 is használnak mikrohullámú cikoaddíciókat.
A Sonogashira keresztkapcsolás és különböző cikloaddíciós reakciók kombinálására is találunk példát az irodalomban. Egy belga kutatócsoport nukleozid analogonok szintézisét valósította meg 2-klórfuropirazinokból (64) kiindulva, mikrohullámú besugárzással (31.
ábra).175 A reakciósor első lépése a trimetilszilil-acetilén (TMSA) Sonogashira kapcsolása volt, amelyet a termék oszlopkromatográfiás tisztítása és a szilil-védőcsoport eltávolítása követett. A terminális acetilén (65) izolálása után a szénhidrát részt „click”-reakcióval, triazol linkeren keresztül kötötték a furopirazin egységhez (66, 31. ábra).
35
31. ábra Nukleozid analogon (66) szintézise átmenetifém-katalizált reakciók segítségével
36
2.5. Heterociklus kialakítása átmenetifém-katalizált „one-pot”reakcióval
Napjainkban az átmenetifém-katalizálta reakciók rendkívüli jelentőségét tovább növeli, hogy többlépéses kémiai átalakítások, egy vagy több katalizátor alkalmazásával, egyetlen reakcióedényben (one-pot) is megvalósíthatók. Az ily módon kivitelezett szintézisek hatékonyabbak, a kevesebb tisztítási lépésnek köszönhetően időtakarékosabbak, olcsóbbak és minimális környezetkárosító hatással járnak.176 Tietze és Beifuss a „one-pot” reakciókat két csoportra osztotta: domino és egymást követő (consecutive) reakciókra.177 A domino reakciók alatt tandem vagy kaszkád reakciósorokat értünk, amelyekben az egyes reakciók az előző lépésben kialakult funkciós csoport eredményeként követik egymást. Az egymást követő (consecutive) típusú reakciókról pedig akkor beszélünk, amikor az első átalakulást követően újabb reagenst vagy katalizátort adnak a rendszerhez, a köztes lépések termékeinek izolálása nélkül. Később Fogg és dos Santos egy újabb, specifikusabb terminológiát állított fel, elkülönítve a tandem és a nem tandem „one-pot” katalitikus eljárásokat (31. ábra).178 A nem tandem eljárások közé tartoznak:
azok a „one-pot” átalakítások, amelyeknél a katalitikus lépések egymástól elkülöníthetőek, és a katalizátorok az egyes lépések lejátszódása után egyesével kerülnek ugyanabba a reakcióelegybe.
azok a domino reakciók, amelyek egyetlen katalitikus és egy azt követő sztöchiometrikus átalakításból állnak, az összes reagens egyszerre van jelen a reakcióelegyben.
Tandem katalízisről akkor beszélünk, amikor a szubsztrát egymást követő átalakításai kettő vagy több elkülöníthető mechanizmuson keresztül mennek végbe. A katalizátor(ok)tól és a kémiai változástól függően megkülönböztethetünk ortogonális, auto-tandem és hozzáadott reagenssel (nem katalizátor) segített tandem katalízist.
37
31. ábra A „one-pot” eljárások osztályozásának folyamatábrája
A „one-pot” reakció kiválóan alkalmas heterociklusos gyűrűk kialakítására. A biológiailag aktív konjugátumok szintézisénél népszerű triazol-linker is felépíthető „one-pot”
reakciósorban is: a terminális alkin/azid kialakítása és a cikloaddíció külön reakciólépésekben, de a köztitermékek izolálása nélkül, ugyanabban a reakcióedényben megy végbe. Az irodalomban alig néhány példa található ilyen eljárás kidolgozására. 2010-ben Friscourt és Boons szubsztituált aril-jodidok (67) és benzilazidok között alakított ki triazol- gyűrűt (70), egy háromlépéses, „one-pot” reakciósorban (32. ábra).179 A mikrohullámmal aktivált reakciósor első lépése a TMSA Sonogashira kapcsolása az aromás jódvegyülethez (67). A reakcióelegyhez fluorid-tartalmú reagenst adva eltávolították a 68-as vegyület trimetilszilil-csoportját és az így nyert terminális alkint (69) benzilaziddal vitték „click”- reakcióba. A módszerrel egyszerűen, rövid idő alatt és hatékonyan kapcsolhatók össze a kívánt szerkezeti egységek, a terminális acetilén előzetes előállítása és tisztítása nélkül.
38
32. ábra „One-pot” háromlépéses triazol (70) szintézis
39 3. Célkitűzés
Munkánk célja olyan új, potenciálisan antitumor hatású ösztron származékok szintézise volt, amelyek hormonális aktivitással nem rendelkeznek. Mindezek eléréséhez a 13-epimer és/vagy D-szeko-alapvegyületekből kiindulva különböző konjugátumok előállítását terveztük, átmenetifém-katalizált reakciókkal:
1. A szteroidokból terminális alkineket előállítva, azok CuAAC reakcióit kívántuk megvalósítani kismolekula vagy szteroid azidokkal.
2. A D-szekoösztronok aromás A-gyűrűjét jódozva, szteroid-halogenidek és fenilacetilének Sonogashira kapcsolására mikrohullámú módszert terveztünk kidolgozni.
3. Továbbá a két reakció egymást követő, „one-pot” kivitelezését terveztük ösztrán vázas vegyületeken, mikrohullámú reaktorban.
Az új vegyületek szerkezetének nagyműszeres analitikai módszerekkel (NMR, MS) történő igazolása mellett célunk volt a folyamatok sztereo-, kemo- és regioszelektivitásának vizsgálata. Az előállított céltermékek és előanyagaik in vitro sejtosztódásgátló (humán adherens tumorsejtvonalakon) és 17-HSD1 enzimgátló hatását együttműködés keretében terveztük vizsgálni.
40 4. Kísérleti eredmények tárgyalása
4.1. Kiindulási D-szeko vegyületek előállítása a 13- és 13-ösztron sorban
Kísérleti munkánk első lépéseként olyan D-szeko-származékok előállítása volt a célunk a 13- és a 13-ösztron sorban, amelyek hormonális inaktivitásuknak köszönhetően potenciális antitumor hatóanyagok alapjai lehetnek. Kiindulási anyagként a D-szekoaldehid 3-benzil-éterének 13-epimereit (15c, 16c) választottuk, miután 17-oxocsoportjuk számos továbbalakítási lehetőséget biztosít. A kutatócsoportunk által 3-metil-éterekre (15b, 16b) kidolgozott eljárásokat követve,40,42 olyan intermedier D-szekoösztronok előállítását valósítottuk meg, amelyek számos átalakítás hasznos alapanyagai lehetnek. A kiindulási származékok (15c, 16c) további előnye, hogy a benzilcsoport egy könnyen, semleges körülmények között eltávolítható védőcsoport. Így a vegyületek több ponton történő módosításával – 3-as és 17-es funkciók, ill. 14-(prop-2-enil) oldallánc – egyszerűen nyerhetünk olyan származékokat, amelyek nagyban hozzájárulnak a hatás-szerkezet összefüggések felállításához. Ezen túlmenően lehetőségünk van az anguláris metilcsoport térállásának hatását vizsgálni mind kémiai, mind gyógyszerhatástani szempontból.
A 13- és a 13-D-szekoaldehid 3-benzil-éterét (15c, 16c) kálium-tetrahidrido- boráttal, metanolban reagáltatva kaptuk a megfelelő D-szekoalkoholokat (71, 72, 33. ábra).
Ezt követően hidrogenolízissel, palládium-csontszén katalizátor segítségével hasítottuk le a 3- as helyzetben lévő benzilcsoportot, amellyel egyidejűleg a propenil oldallánc is telítődött (73, 74, 33. ábra).
41
33. ábra A 13-epimer D-szekoalkoholok (71‒74) előállítása
Ezzel párhuzamosan a nyitott D-gyűrű oxocsoportját (15c, 16c) kondenzációs reakcióban is átalakítottuk. Nukleofil reagensként hidroxilamin-hidrokloridot, bázisként vízmentes nátrium- acetátot alkalmazva szelektíven nyertük a D-szekooximokat (75, 76, 34. ábra). A 75-ös és a 76-os jelű vegyületek 3-benzil-védőcsoportját utólag szintén eltávolítottuk (77, 78, 34. ábra).
34. ábra Oxim funkció kialakítása a nyitott D-gyűrűn