Kaolinit-komplexek interkalációs és adszorpciós vizsgálata kvantumkémiai módszerekkel
TÁBOROSI Attila1, KURDI Róbert1, SZILÁGYI Róbert Károly2
1Pannon Egyetem, Környezetmérnöki Intézet, 8200 Veszprém, Egyetem Utca 10, Magyarország, a.taborosi@gmail.com, kurdir@uni-pannon.hu
2Montanai Állami Egyetem, Kémia és Biokémia Tanszék, MT 59718 Bozeman, USA, robertkszilagyi@gmail.com
Összefoglaló: Az interkalált rétegszilikátok az elmúlt években nagy érdeklődés övezte.
A kaolin típusú agyagásványok interkalációja fontos szerepet tölt be az új anyagok kutatásában, mivel az interkalált anyagásványokat katalizátorként vagy membrán komponensként, de akárt enzim helyettesítőként is lehet használni.
Kutatásom során vizsgáltam a kaolinit-komplexben létrejövő rétegközti intermolekuláris kölcsönhatásokat, illetve adszorpciós és interkalációs energiát számoltam a különböző reagensek esetében. Kvantumkémiai módszerekkel a Pannon Egyetem, Felületkémiai csoportban jelenleg kísérletileg tanulmányozott kaolin-reagens komplexeket vizsgáltam meg.
Kulcsszavak: kaolinit, kvantumkémia, adszorpció, interkaláció, kölcsönhatások
Bevezető
Az agyagásvány interkalációs kutatások jelenlegi tervezése túlnyomórészt kísérleti úton folyik, ami gazdaságossági problémákat vet fel. A laboratóriumi munkák idő és vegyszerköltség csökkentése, valamint a jobb tervezhetőség érdekében számításos módszerekkel célszerű kiegészíteni. Az elméleti kémiai módszerek arra is alkalmasak, hogy kísérletileg el nem érhető információkat nyújtsanak az interkaláció vagy az adszorpciós során lejátszódó mechaniz- musokról.
Az agyagásványok elméleti kémia vizsgálatai során kétfajta modellt használnak, az egyik a periodikus, ahol egy elemi cellát vizsgálnak a rétegekben [Berghout et al. 2010], a másik az úgynevezett „klaszter”, ahol pedig a rétegekből csonkolással hozzák létre a modellt [Michalková et al. 2002].
Erőforrás gazdaságosság miatt DFT (Density Functional Theory) módszert alkalmaznak a és ezen belül a leggyakrabban a B3LYP hibrid módszert [Becke et al. 1993]. A bázis készletek közül a 6-31G(d)-t [Francl et al. 1982] és Lanl2dz-t [Hay & Wadt 1985] használják.
Anyag és módszer
A vizsgálataim során kísérletileg meghatározott [Bish et al. 1993] elemi cellából indultam ki, ahol a nehéz atomok helyzete (Al, Si, O) ismert volt. Az elemi cellát sokszorosítva egy TO-TO rétegkomplexumot kaptam, melyből csonkolással megalkottam mind az adszorpciós, illetve interkalációs klaszter
modelleket. Végső lépésként a rendszerhez hozzáadtam a hiányzó hidrogén atomokat, illetve a reagenst is.
Az optimalizáció során a reagens atomjai,
az oktaéderes réteg hidas hidroxil csoportjai, melyek kölcsönhatásba lépnek a reagenssel,
a tetraéderes réteg oxid anionjai, és
minden hidrogén atom szabadon mozoghattak, míg a többi atomok pozíciója kötött volt.
A vizsgálat fő szempontjai a következőek voltak: a reagens elhelyezkedése a felületen és a rétegközti térben, az adszorpciós és interkalációs energia a különböző vegyületek esetében, illetve a H-híd számának és kötéstávolságának alakulása.
A számítások során DFT módszeren belül a B3LYP hibrid funkcionált használtam, LANL2DZ báziskészlettel. Entrópia és entalpia korrekciót, illetve bázis készlet átfedési hibát nem számoltam, mivel elsősorban szerkezeti modelleket vizsgáltam. A számítások elvégzéséhez a Gaussian 09 programot használtam.
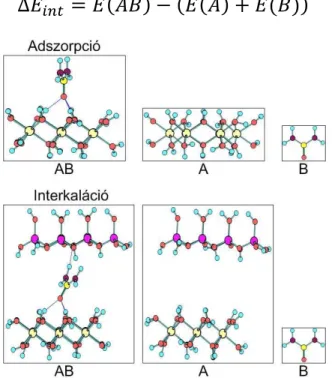
Az adszorpciós és interkalációs energiát a teljes (AB) és a részrendszerek (A, B) szerkezeti optimalizációval kapott energiáinak különbségéből állapítottam meg.
∆ܧ
௧= ܧሺܣܤሻ − ሺܧሺܣሻ + ܧሺܤሻሻ
1. ábra. Az adszorpciós és interkalációs energia megállapítása
Eredmények
Kaolinit-karbamid
2. ábra. A karbamid pozíciója a tetraéderes és oktaéderes réteg esetében
3. ábra. A karbamid elhelyezkedése a rétegközti térben
Az oktaéderes réteghez a karbamid az oxigén atomjával kapcsolódik.
Három H-híd kötést alakít ki, ennek megfelelően az adszorpciós energia -123 kJ/mol. A tetraéderes réteghez a karbamid az amid csoportok hidrogén atomjaival kapcsolódik. Kétfajta pozíció figyelhető meg, az úgynevezett
„geminális”, amikor is a két amid egy-egy hidrogénje egy Si atom egy-egy oxigénjéhez kapcsolódik. Illetve a „vicinális”, amikor a két amid egy-egy hidrogénje két Si atom egy-egy oxigénjével alakít ki H-híd kötést. Mivel ennél az adszorpciónál csak két H-híd kötés alakul ki az adszorpciós energia is ennek megfelelően kisebb. „Geminális” esetben -74 kJ/mol, „vicinális” esetben -66 kJ/mol. Interkalációs esetben a karbamid oxigénje hasonló az oktaéderes réteg adszorpciójánál három H-híd kötést alakít ki. Viszont a tetraéderes rétegnél csak
egy H-híd alakul ki az amid hidrogénje és a Si atom oxid anionja között.
Továbbá a karbamid pozíciója már nem merőleges a réteg síkjára, hanem egyfajta „döntött” helyzetet vesz fel. Az interkalációs energia -126 kJ/mol, ami a várt közel -200 kJ/mol-nál jelentősen kisebb. Ez a rétegtávolság függvényében változhat.
Kaolinit-etilénglikol
Az oktaéderes rétegnél az etilénglikol két H-híd kötést alakít ki az oxigén atomjaival a hidas OH csoportok hidrogénjeivel. Az adszorpciós energia -102 kJ/mol. A tetraéderes réteg esetében az etilénglikol alkohol hidrogénjei kapcsolódnak a Si atom oxid anionjaihoz. Itt is kettő H-híd kötés alakul, azonban ezek gyengébbek, mint az oktaéderes rétegnél, mivel az adszorpciós energia -76 kJ/mol. Az etilénglikol ha a rétegközti térben helyezkedik el összesen hét H-híd kötést alakít ki. Hármat a tetraéderes és négyet az oktaéderes réteggel. A számolt energia 11 kJ/mol, mely annak tulajdonítható, hogy a két réteg közel helyezkedik el egymáshoz.
Kaolinit-kálium-acetát
A kálium-acetát az oktaéderes rétegnél az acetát az egyik hatos Al-„gyűrű” középpontjába helyezkedik el, míg a kálium ion a másik hatos Al-„gyűrű”-nél található. Az acetát az egyik oxigén atomjával alakít ki három H-híd kötést az oktaéderes réteg OH csoport hidrogénjeivel. Az adszorpciós energia ebben az esetben jelentős -202 kJ/mol. A kálium-acetát a tetraéderes rétegre nem tud adszorbeálni, még a kálium ion segítségével sem. A rétegközti térben hasonlóan helyezkedik el, mint az adszorpciónál viszont a nem kapcsolódó oxigén atom közelebb húzódik a kálium ionhoz. Az acetát másik oxigénje szintén három H- híd kötést alakít ki. Az interkalációs energia -137 kJ/mol.
1. Táblázat. A vizsgált reagensek összefoglaló eredményei Kaolinit-reagnes Mechanizmus Energia
(kJ/mol)
H-híd kötések száma Kaolint-
Karbamid
Adszorpció Al-réteg -123 3
Si-réteg -74/-66 2/2
Interkaláció -126 4
Kaolinit Etilénglikol
Adszorpció Al-réteg -102 2
Si-réteg -76 2
Interkaláció 11 7
Kaolinit- Kálium-acetát
Adszorpció Al-réteg -202 3
Si-réteg - -
Interkaláció -137 3
Következtetések
Az alkalmazott modell és módszer, illetve báziskészlet megfelelő arra, hogy a kialakult szerkezeteket és energiákat megállapítsam, de természetesen
szükséges a továbbiakban a modell pontosítása és más módszer-báziskészlet használata is.
Köszönetnyilvánítás
A kutatás a TÁMOP 4.2.4.A/2-11-1-2012-0001 azonosító számú „Nemzeti Kiválóság Program – Hazai hallgatói, illetve kutatói személyi támogatást biztosító rendszer kidolgozása és működtetése konvergencia program” című kiemelt projekt keretében zajlott.
Irodalom
Becke A. D. 1993 A new mixing of Hartree-Fock and local density-functional theories. The Journal of Chemical Physics 98: 1372-1377.
Berghout A., Tunega D., Zaoui A. 2010 Density Functional Theory (DFT) study of the hydration steps of Na+/Mg2+/Ca2+/Sr2+/Ba2+-exchanged montmorillonites. Clays and Clay Minerals 58: 174-187.
Bish D. L. 1993 Rietveld refinement of the kaolinite structure at 1.5 K. Clays and Clay Minerals 41: 738-744.
Francl M. M., Pietro W. J., Hehre W. J., Binkley J. S., DeFrees D. J., Pople J.
A., Gordon M. S. 1982 Self Consistent Molecular Orbitals Methods. A Polarization Basis Set for Second Row Elements. Journal of Chemical Physics 77: 3654-3665
Hay P. J. and Wadt W. R. 1985 Ab initio effective core potentials for molecular calculations – potentials for the transition-metal atoms Sc to Hg. Journal of Chemical Physics 82: 270-283
Michalková A., Tunega D., Nagy T. L. 2002 Theoretical study of interactions of dickite and kaolinite with small organic molecules. Journal of Molecular Structure 581: 37-49.
Investigation of intercalation and adsorption on kaolinite from quatum mechanical calculation
Abstract: An interest in intercalates of layered silicates has emerged in recent years.
Intercalation of kaolinite-type clay minerals represents an inportant research area in the development of new materials. Intercalated clay minerals can be used as catalyst membrane components, or even as enzyme mimics.
During the course of my research I examinated various intermolecular interaction in the kaolinite-organic reagents complexes. I also determined the adsorption and intercalation energy between the reagents and clay minerals from first-principle molecular simulation. With quantum chemical methods I investigated the complexes that are being characterized in parallel in the research laboratories of the Surface Science Group at University of Pannon.
Keywords: kaolinite, quatum chemical, adsoprtion, intercalation, interactions