ContentslistsavailableatScienceDirect
Journal of Pharmaceutical and Biomedical Analysis
jou rn al h om e p a g e :w w w . e l s e v i e r . c o m / l o c a t e / j p b a
Simultaneous determination of chiral and achiral impurities of ivabradine on a cellulose tris(3-chloro-4-methylphenylcarbamate) chiral column using polar organic mode
Elek Ferencz
a,b, Béla Kovács
a,b, Francisc Boda
a,
Mohammadhassan Foroughbakhshfasaei
c, Éva Katalin Kelemen
b, Gerg ˝o Tóth
c,∗, Zoltán-István Szabó
a,b,∗∗aFacultyofPharmacy,UniversityofMedicine,Pharmacy,SciencesandTechnologyofTarguMures,Gh.Marinescu38,RO-540139,TîrguMures¸,Romania
bGedeonRichterRomaniaS.A.,RO-540306,TîrguMures¸,Romania
cDepartmentofPharmaceuticalChemistry,SemmelweisUniversity,H ˝ogyesE.u.9,Budapest,Hungary
a r t i c l e i n f o
Articlehistory:
Received14June2019
Receivedinrevisedform29August2019 Accepted30August2019
Availableonline30August2019
Keywords:
Chemoselectivity Polarorganicmode Chiralseparation Relatedsubstance HPLC
a b s t r a c t
A high performance liquid chromatographic method was developed for the simultaneous deter- mination of the related substances (R-ivabradine, dehydro-S-ivabradine, N-demethyl-S-ivabradine, ((S)-3,4-dimethoxy-bicyclo[4.2.0]octa-1,3,5-triene-7-yl-methyl)-methyl-amine)and1-(7,8-dimethoxy- 1,3,4,5-tetrahydro-2H-3-benzazepine-2-on-3-yl)-3-chloro-propane) ofthe heart-rate lowering drug, ivabradine. The separation capability of seven different polysaccharide-type chiral columns (Lux Amylose-1,Luxi-Amylose-1,LuxAmylose-2,LuxCellulose-1,LuxCellulose-2,LuxCellulose-3andLux Cellulose-4)wasinvestigatedwithamobilephaseconsistingof0.1%diethylamineinmethanol,2- propanolandacetonitrile.DuringthescrenningexperimentsthebestresultswereobtainedonLux Cellulose-2(basedoncellulosetris(3-chloro-4-methylphenylcarbamate)columnwithmethanolwith anidealcase,wherealltheimpuritieselutedbeforetheS-ivabradinepeak.Chromatographicparameters (flowrate, temperatureandmobilephase constituents)wereoptimized byafull factorialscreen- ingdesign. Using optimizedparameters(LuxCellulose-2columnwith 0.06%(v/v)diethylaminein methanol/acetonitrile98/2(v/v)with0.45mL/minflowrateat12◦C)baselineseparationswereachieved betweenallcompounds.TheoptimizedmethodwasvalidatedaccordingtotheInternationalCouncilon HarmonizationQ2(R1)guidelineandprovedtobereliable,linear,preciseandaccuratefordetermination ofatleast0.05%forallimpuritiesinS-ivabradinesamples.Methodapplicationwastestedonacommer- cialtabletformulationandprovedtobesuitableforroutinequalitycontrolofbothchiralandachiral relatedsubstancesofS-ivabradine.
©2019TheAuthors.PublishedbyElsevierB.V.ThisisanopenaccessarticleundertheCCBY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1. Introduction
Ivabradine 3-{3-[((S)-3,4-dimethoxy-bicyclo[4.2.0]octa-1,3,5- triene-7-ylmethyl)-methyl-amino]-propyl}-7,8-dimethoxy- 1,3,4,5-tetrahydro-2H-3-benzazepine-2-one; IVA) is an orally bioavailable,hyperpolarization-activated,cyclicnucleotide-gated
∗ Correspondingauthorat:DepartmentofPharmaceuticalChemistry,Semmel- weisUniversity,H-1092Budapest,H ˝ogyesE.u.9,Hungary.
∗∗ Correspondingauthorat:FacultyofPharmacy,UniversityofMedicine,Phar- macy,SciencesandTechnologyofTarguMures,Gh.Marinescu38,GedeonRichter RomaniaS.A.,RO-540306,TîrguMures¸,Romania.
E-mail addresses: toth.gergo@pharma.semmelweis-univ.hu (G. Tóth), zoltan.szabo@gedeon-richter.ro,zoltan.szabo@umfst.ro(Z.-I.Szabó).
channel inhibitor. It is used for chronic heart failure not fully managedby-blockers[1,2].IVAhasasingleasymmetriccarbon atom,resultingin twoopticalisomers.Thedrugismarketedas a single enantiomeric agent; the pharmaceutical formulations containonlytheS-enantiomer,becauseofitsimprovedelectro- physiological selectivity [3,4]. Nevertheless, to date, only one applicationnotefromDaicel Chiraltechnologiesisavailablefor chiral separation of IVA using Chiralpak IG(based on amylose tris(3-chloro-5-methylphenylcarbamate) column [5]. Moreover, thenumberofthedescribedmethodfordeterminationofachiral relatedsubstancesinIVAisalsolimited[6–9].Highperformance liquid chromatography (HPLC) using a chiral stationary phase (CSP)isthegoldenstandardinanalyticalenantioseparationdueto theseveraladvantagesitoffers,suchaseaseofuse,robustnessand
https://doi.org/10.1016/j.jpba.2019.112851
0731-7085/©2019TheAuthors.PublishedbyElsevierB.V.ThisisanopenaccessarticleundertheCCBY-NC-NDlicense(http://creativecommons.org/licenses/by-nc-nd/4.
0/).
CSPsavailableonthemarket,polysaccharide-type CSPsarethe most frequently applied, due to their high enantiorecognition capability for most of the compounds and multimodal nature [10–12].Thesecolumnscanbeoperatedinnormalphase,reversed phase and polar organicmobile phase modes. In polarorganic modeonlypolarorganicsolvents,neatalcohols(methanol,ethanol and2-propanol),neatacetonitrile(ACN)ortheircombinationsare usedasmobilephase.Polarorganicmodehasseveraladvantages, suchasshorterruntimes,highefficiency,andusuallyhighersolu- bilityoftheanalytesinthemobilephase[12–15].Apartfromtheir abilitytodiscriminatebetweenenantiomers,polysaccharide-type CSPsmayalsopresentexcellentchemoselectivityforstructurally relatedcompounds,thereforetheycanbeusedforsimultaneous determinationofchiralandchemicalimpurities[14,16,17].
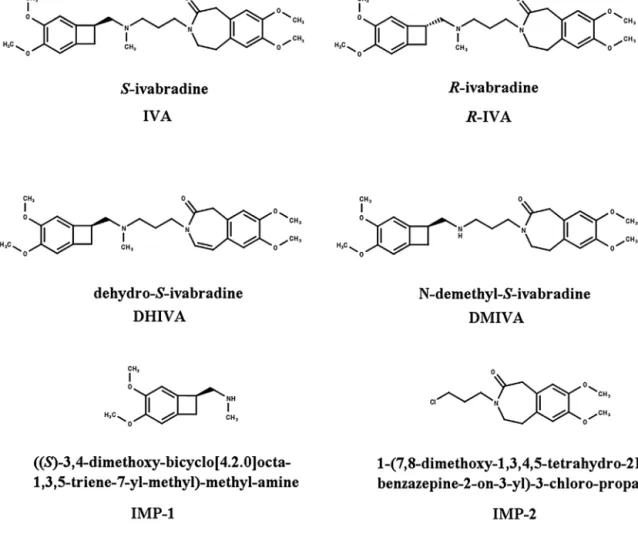
Allspecifiedimpurities, includingR-enantiomerareofinter- estin the analysis of S-IVA. Therefore, the aim of the present studywastodevelopamethodforthesimultaneousdetermination ofR-IVA,dehydro-S-ivabradine(DHIVA),N-demethyl-S-ivabradine (DMIVA),((S)-3,4-dimethoxy-bicyclo[4.2.0]octa-1,3,5-triene-7-yl- methyl)-methyl-amine (IMP-1) and 1-(7,8-dimethoxy-1,3,4,5- tetrahydro-2H-3-benzazepine-2-on-3-yl)-3-chloro-propane(IMP- 2)(Fig.1)inS-IVAsampleswithalimitofquantificationofatleast 0.05%orbelow.
2. Materialsandmethods
2.1. Materials
S-IVA (as a hydrobromide salt), racemic IVA, N-demethyl-S-ivabradine (DMIVA, 3-{3-[((S)-3,4-dimethoxy- bicyclo[4.2.0]octa-1,3,5-triene-7-yl)-methyl-amino]-propyl}- 7,8-dimethoxy-1,3,4,5-tetrahydro-2H-3-benzazepine-2-one), dehydro-S-ivabradine (DHIVA, 3-{3-[((S)-3,4-dimethoxy- bicyclo[4.2.0]octa-1,3,5-triene-7-ylmethyl)-methyl-amino]- propyl}-7,8-dimethoxy-1,3-dihydro-2H-3-benzazepine-2-one), ((S)-3,4-dimethoxy-bicyclo[4.2.0]octa-1,3,5-
triene-7-yl-methyl)-methyl-amine (IMP-1) and 1-(7,8-dimethoxy-1,3,4,5-tetrahydro-2H-3-benzazepine-2-on- 3-yl)-3-chloro-propane (IMP-2) (Fig. 1) were obtained from a pharmaceutical company in Târgu Mures¸. Diethylamine (DEA)
≥99.5%waspurchasedfromSigma-Aldrich(Budapest,Hungary).
Gradientgrademethanol(MeOH),2-propanol(IPA)andacetoni- trile(ACN)werepurchased fromThomaskerFinechemicals Ltd.
(Budapest,Hungary).
Ultrapure,deionizedwater waspreparedbyaMilli-QDirect 8 Millipore system (Milford, MA, USA). S-IVA 5mgfilm-coated tabletswereobtainedfromCentralPharmacyofSemmelweisUni- versity (Budapest, Hungary). All chiral columns with identical dimensions(4.6x150mm,5mparticles)wereorderedfromPhe- nomenex(Torrance,CA,USA):LuxCellulose-1[cellulosetris(3,5- dimethylphenylcarbamate)], Lux Cellulose-2 [cellulose tris(3- chloro-4-methylphenylcarbamate)], Lux Cellulose-3 [cellulose tris(4-methylbenzoate)],LuxCellulose-4[cellulosetris(4-chloro- 3-methylphenylcarbamate)], Lux Amylose-1 [amylose tris(3,5- dimethylphenylcarbamate)],Lux i-Amylose-1 [amylose tris(3,5- dimethylphenylcarbamate)], immobilized and Lux Amylose-2 [amylosetris(5-chloro-2-methylphenylcarbamate)] (Supplemen- taryInformationFigureS1).
2.2. LC-UVanalysis
LC-UVexperimentswascarriedoutonaJASCOHPLCsystem (JASCOPU-2089Plusbinarygradientpump,AS-4050autosampler, MD-2010PlusdiodearraydetectorandCO2065Pluscolumnoven).
wasChromNAV.UVdetectionwasperformedat286nm.MeOHwas usedasasamplesolventforthepreparationofsolutionsthrough- outthestudy.Forthepreliminaryexperiments,stocksolutionsof 200g/mLS-IVAwerepreparedinMeOHandwerespikedwith impuritiesataround2%(around4g/mL).Thefinaltestsolution ofS-IVAusedforvalidationandmethodapplicabilitytestingwas about8000g/ml.Allimpuritylevelpercentagesarereportedrel- ativetothisconcentration.Aninjectionvolumeof5Lwasused andthreeparallelmeasurementswereperformedinallcases.
Forpreparationofsamplesolutions,tentabletswereweighted, thengroundand mixedina mortar.In a5mLvolumetric flask, MeOH was added to an accurately weighted portion of the tablet powder corresponding to about 40mg S-IVA. Then the suspensionwas sonicatedfor 30min and centrifugedfor 2min applying4000rpm(Sartorius2–16Pbenchtopcentrifuge,Goettin- gen,Germany).Theclearsupernatantwasfilteredthrough0.22m poresizesyringecontainingPVDFfilter(FilterBiomembraneCo., LTD,NantongCity,China).
Theexperimentaldesign andmultivariatemethodoptimiza- tionwasperformedwiththeaidofModde11software(Umetrics, Sweden).
3. Resultsanddiscussion
3.1. Methodscoutingphase
Inroutinequalitycontrolofsingleenantiomericpharmaceutical substances,the“classicalapproach”wouldbetodevelopseparate methodsforthequantificationofachiralandchiralrelatedsub- stances.Methodsthatenablesimultaneousquantificationofboth chiral and achiral impurities arethus highly welcome,because theycansignificantlyshortentheanalysistime.However,inthese cases,thedevelopedmethodsneedtodisplaybothenantioselec- tivity(chiralseparation)andchemoselectivity(achiralseparation) towardstheanalytes.Althoughthiscanalsobeachievedbytandem couplingofchiralandachiralcolumns,usingasinglechiralcolumn ismoredesirable.Thus,forthepresentmethod,basedonregula- toryconsiderations,theanalyticaltargetprofilewasdefinedasa methodthatwouldallowthepreciseandaccuratedetermination ofallselectedimpurities(chiralandchemical)atalevelofatleast 0.05%orbelow,inshortanalysistime(t≤30min).
Recently, wehave successfully appliedpolysaccharide-based CSPsinpolarorganicmodeforthechiralseparationofalargevari- etyofanalytes[18–21],howeverpolysaccharide-typeCSPshave alsoprovedtheircapabilityforsimultaneouschiral-achiralanaly- ses[14,16,17].
Polysaccharide-basedCSPscomeinawidevarietyofcommer- cially available columns and usually display a great variety of enantio-andchemoselectivity.Inthepresentstudy,sevendifferent polysaccharide-basedCSPs,includingamylose-basedLuxAmylose- 1,Luxi-Amylose-1andLuxAmylose-2,aswellascellulose-based LuxCellulose-1,LuxCellulose-2,LuxCellulose-3andLuxCellulose- 4weretestedinpolarorganicmodeusingamobilephaseconsisting of0.1%DEAinMeOH,IPAorACN,with0.5mL/minflow rateat 25◦.Inourscreeningthecombinationofpolarorganicsolvents wasnotused,howeveritshouldbenotedthatsolventmixturecan changetheenantiorecognitionmechanismandalsocanimprove theresolution.
Duringthescreeningstepinallcases,IMP-1andIMP-2were wellresolvedfromeachotherandthemainpeak,however,dif- ficultieswereobservedduringtheseparationofthestructurally verysimilarDHIVA,DMIVA,R-IVAfromthemainpeakforwhich co-elutionswereoftenobserved.Particularlychallengingwasthe resolutionofIVAenantiomers,whichwasidentifiedasthecritical
Fig.1.Name,chemicalstructureandabbreviationsofthecompoundsusedinthisstudy.
Table1
Summaryofthechromatographicdataobtainedduringthepreliminaryscreeningphase.
Column Mobilephase* Rs Enantiomerelution
order
tr,min Lastelutedcompound
LuxAmylose-1
MeOH 0.25 R<S 23.1 S-IVA
IPA 1.34 R<S 16.1 S-IVA
ACN 0.24 S<R 19.0 DMIVA
Luxi-Amylose-1
MeOH – – 11.2 IVA
IPA 0.86 R<S 20.3 S-IVA
ACN – – 10.8 IVA
LuxAmylose-2
MeOH 1.06 S<R 18.2 R-IVA
IPA – – 49.3 DMIVA
ACN 1.80 S<R 49.9 DMIVA
LuxCellulose-1
MeOH 2.15 S<R 10.6 R-IVA
IPA 1.64 S<R 19.6 DMIVA
ACN 2.60 S<R 8.9 DMIVA
LuxCellulose-2
MeOH 1.78 R<S 19.9 S-IVA
IPA – – 40.9 DMIVA
ACN 0.20 R<S 39.3 DMIVA
LuxCellulose-3
MeOH 2.73 S<R 9.3 R-IVA
IPA – – 11.7 DMIVA
ACN – – 5.0 DMIVA
LuxCellulose-4
MeOH 0.25 R<S 14.1 S-IVA
IPA – – 35.8 DMIVA
ACN 1.11 R<S 26.4 DMIVA
Rs,resolutionbetweenR-andS-IVA.
tr,retentiontimeofthelastelutedpeak.
*allmobilephasescontained0.1%DEAasbasicmodifier.
-noenantiorecognitioncouldbeobserved.
parameter.Thus,thescreeningwasfocusedmainlyonselectingthe adequatecolumnforbaselineseparationoftheenantiomerswith thedesiredelutionorder(chiralimpurityandR-IVAelutingbefore
theeutomer).Someofthechromatogramsobtainedaredepicted inFig.2,whiletherelevantchromatographicdataarepresentedin Table1.Asitcanbeobserved,baselinechiralseparationbetween
Fig.2.Representativechromatogramsobtainedduringthepreliminarystudy.Chromatographicconditions:(A)LuxCellulose-2with0.1%DEAinMeOH;(B)LuxCellulose-3 with0.1%DEAinMeOH;(C)LuxCellulose-2with0.1%DEAinACN;(D)LuxCellulose-4with0.1%DEAinMeOH(flowrate:0.5mL/min,temperature25◦C,detectionat 286nm.)1.IMP-1,2.IMP-2,3.DHIVA,4.DMIVA,5.R-IVA,6.S-IVA.
enantiomerswasachieved ontheLuxCellulose-1 columnwith highresolutionvaluesforallthreemobilephases,buttheelution orderoftheenantiomerswasunfavorable.Distomer-firstelution orderwasobservedonLux-i-Amylose-1,LuxCellulose-2andLux Cellulose-4,butthebaselineseparationwasachievedonlyinthe caseofLuxCellulose-2using0.1%DEAinMeOHasmobilephase.
Mobilephasedependentenantiomerelutionorderwasrecorded onLuxAmylose-1column,whenchangingthemaincomponentof themobilephasefromalcoholstoACN.
Sincethe final goalwasto develop a singlemethodfor the separation of all related substances, not only enantioselectiv- ity, but chemoselectivity wasalso important. Thus, for further methodoptimizationthefollowingstarting-point wasselected:
LuxCellulose-2column,with0.1%DEAinMeOHwith0.5mL/min flowrateat25◦C.Byapplyingthismethod,allinvestigatedrelated substancescouldberesolved.Moreover,notonlytheenantiomeric impurity R-IVA, but allinvestigated impurities eluted beforeS- IVA(Fig.2A).InanattempttogainhigherresolutionMeOHwas changedtoEtOH,howeverusingtheethanolicmobilephaseDMIVA andS-IVAco-eluted,thereforeitwasnotsuitable(Supplementary InformationFigureS2).AdditionofsmallamountsofACNtoMEOH asasecondarysolvent,however,leadtoreducedretentiontimes withoutmodifyingtheresolutions significantly (Supplementary InformationFigureS3).Thus,thisoptionwasalsoevaluatedduring furthermethodoptimization.
3.2. Methodoptimizationbyexperimentaldesign
Conventionalmethodoptimizationisusuallyperformedbythe so-called OFATapproach (one factorat a time),which leadsto amultitudeofexperiments,whilstgainingonlylittleknowledge
abouttheprocessitself.Duetotheseshortcomings,experimental design-based,multivariateoptimizationofanalyticaltechniques havebecomewidespreadinrecentyears.Themainadvantageof thisapproachisthatitoffersmaximumprocessunderstandingwith aminimalnumberofexperiments[22,23].
Thus,amultivariatemethodologywasundertakenforarapid optimizationofnumerousparametersaffectingthesimultaneous separationoftheanalytes.Basedonthepreliminaryexperimental runs,fourcriticalqualityattributeswereselectedandmonitored asresponsesintheexperimentalruns.Theseincluded:Rsbetween DHIVAandDMIVA(abbreviatedRs1),Rs betweenDMIVAandR- IVA (Rs2), Rs betweenR-IVA and S-IVA (Rs3)and total analysis time(measuredasretentiontimeofS-IVA,tr,S-IVA).Criticalprocess parameterswereidentifiedbasedonourpreviousexperiencewith polarorganicmodeonpolysaccharide-typeCSPsandariskassess- mentofdifferentfactorsthatcouldinfluencetheperformanceof themethod.Thecriticalprocessparametersselectedasexperimen- talvariablesduringtheDoErunsweredefinedasDEAvolumeratio addedtothemobilephase(DEA%,range0.05%–0.15%),ACN/MeOH volumeratioofthemobilephase(ACN%,0–10%),flowrate(0.3– 0.7mL/min)and columntemperature(T,10–40◦C).Inourwork DEAwasusedasmodifierhoweverthenatureofthebasicand/or acidicmodifiermaydeeplyinfluencetheseparation[15].
Inordertoobtainthoroughinformationabouttheinfluenceof definedfactorsonselectedresponsesandtherebytoachievethe optimalchromatographicconditions,aimingtofulfilallresponse targetsettings,afullfactorialexperimentaldesignwiththreecen- terpointswasselected.
Thedesignreturnedatotalof19experimentalruns,including threereplicatecenterpointedexperiments.Thesampleconsisted of 8000g/mL S-IVA and 80g/mL of each impurity (impurity
Table2
Optimizedmethodconditionswithpredictedandexperimentallyobtainedresponsevalues.
Optimizedmethodconditions Rs1 Rs2 Rs3 tr,S-IVA
DEA% ACN% Flowrate
(ml/min)
T(oC) Predicted values
2.02 (1.85–2.19)
1.58 (1.54–1.62)
2.00 (1.95–2.05)
23.71 (21.98–25.57)
0.06 2 0.45 12 Experimental
obtainedvalues
2.13 1.55 1.98 23.20
levelsat1.0%relative toS-IVA)inMeOH. Allexperimentalruns wereperformedintriplicateandtheaveragevalueswereintro- ducedintheworksheet.Thedesignmatrixandtherecordedvalues forallresponsesaresummarizedinSupplementaryInformation TableS1.Theobtaineddatawasfittedbypartialleastsquares(PLS) regressionandthemodelpassedthereplicativeplotanalysisforall responses(SupplementaryInformationFigureS4),meaningthat replicate errors didnot interferewithdata analysis.Histogram plotsofresponsesindicatednormaldistributionforreponsesRs1, Rs3,butnotforRs2andtr,S-IVA.Forthelattertwo,thehistograms showedstrongpositiveskewness,implyingthatthefrequencyof lowervaluesinthedatasetwasmuchhigher.Inordertoachieve approximatelynormal distribution,logarithmic transformations wereemployed,whichresultedinasubstantialincreaseinmodel predictability (Q2) for both responses. The model was further adjustedbydeletingnon-significantfactorsandaddingdetected squarevaluestoincreasetheQ2values.Thefinalcoefficientplots obtained,withthesignificantfactorsaffectingeachresponseare presentedinSupplementaryInformationFigureS4.Forallofmon- itoredresponses,theexperimentalvariablesACN%,flowrateand Tpresentedanegativecorrelation,indicatingthatanincreasein anyofthesefactorsresultedinadecreaseinallRsvalues,butalso adecreaseintr,S-IVA.DEA%inmostcaseshadnosignificanteffect upontheresponses,apartfromRs2where,incomparisontoACN%, flowrateandTalsopresentsasignificantinverseproportionality withtheselectedresponse,althoughbeingtheleastinfluentialof thedefinedfactors.
Furthermodelrefinementimpliedtheappraisalof quadratic termsinsidethemodel.Thepresenceofquadratictermsindicatesa non-linearrelationshipin-betweenfactorsandresponsesandsug- geststheexistenceofcurvatureinsidethemodel.Quadraticterms werenotidentifiedincaseofRs2andRs3.RegardingRs1andtr,S-IVA
significantquadratictermswereobservedandincludedintheanal- ysisofmodelperformanceindicators.Astherefinedmodelallows onlyonequadratictermtobeincludedintheregressionequation, thiswasfulfilledbyselectingtheoneforwhichthecorresponding singularfactorhasthegreatestimpactontheselectedresponse.
Hence,incaseofRs1 andtr,S-IVA,ACN%xACN%andFlowxFlow wereretained,respectively.
Inallcases,significantregressionmodelswereobtained(p<
0.05)andapartfromthecaseoftr,S-IVA,nolackoffitwasdetected (p> 0.05).In thelattercase,thelackof fitwasalsoevidentby thenegativemodelvalidityvalue.Incontrasttothis,modelrepro- ducibilityshowedextremelyhighvalues( ˜0.99),sincethereplicate runsresultedinalmostidenticalretentiontimesofthemainpeak (seeSupplementaryInformationTableS1).Althoughretentiontime reproducibility is desirable and mostly expected in chromato- graphictechniques,thisalsomeans,thatthepureerrorinsidethe modeltendstozeroandthus,thelowmodelvalidityincaseof tr,S-IVAcanbeexplainedbyhighreproducibility.Themethodwas subsequentlyoptimizedusingthepredictionspreadsheetfunction ofthesoftware,applyingthefollowingcriteria:allRsvaluesmaxi- mized,withacriticalminimalvalue≤1.5,andtr,S-IVAminimized.
Theoptimizersetpoint resultsobtainedaftertheperformed experimentsduring screeningis basedona Monte Carlosimu- lation,where parameter settingswere chosen accordingtothe
lowest log(D) value, with DEA=0.058%, ACN=1.68%, flow rate
=0.46mL/minandcolumntemperature=12.0◦C.Designspacewas evaluatedaroundtheaforementionedparametersettings.The4D designspaceexplorermap(SupplementaryInformationFigureS5) indicatesthatthechromatographicsystemreturnsoptimalresults onlyatlowcolumntemperatureandflowratesettings.Atthelow- estsetpointsofboththeflowrateandcolumntemperature,the proportionofACNisallowedtobeusedinawiderrange,showinga shifttowardstighterspecificationlimitsashighersettingsforboth flowrateandcolumntemperaturearerequired.Thesetpointanaly- sisfunctionofthesoftwarerevealedthatthefrequencyhistograms oftheselectedfactorsanddefinedresponsesfollowanormaldis- tribution(SupplementaryInformationFigureS6andS7),andthe individualprobabilityoffailureforthedefinedresponsesissitu- atedbelow1%,withanoverallprobabilityoffailureof1.7%.Forease ofuse,thefinalproposedmethodusesslightlydifferentparame- tersettings(mostparametersettingsarerounded),whichstilllie inside thedesign spaceand returnsatisfactory resultsin terms ofselectedresponserequirements.Table2showstheoptimized parametersofthechromatographicsystemandtheclosenessof thepredictedandexperimentallyobtainedvaluesforallresponses, usingtherecommendedsettings.Sincetheanalyticaltargetprofile ofthemethodwasfulfilled,furthermethodoptimizationwasnot undertaken.Theparametersofthefinalmethodwerethefollow- ingLuxCellulose-2columnwith0.06%(v/v)DEAinMeOH/ACN98/2 (v/v)with0.45mL/minflowrateat12◦C.
3.3. Methodvalidationandapplication
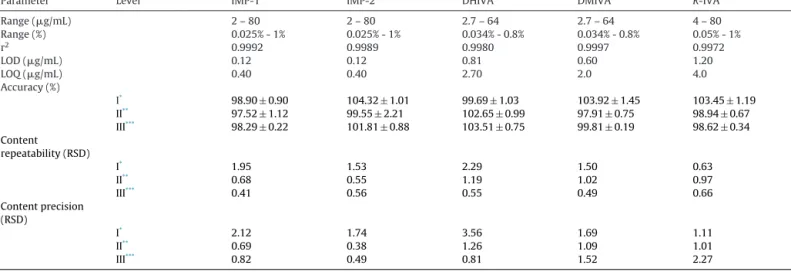
Validationoftheoptimizedmethodwasperformedaccording toInternationalCouncilforHarmonizationguidelineQ2(R1)forall relatedsubstancesandforR-IVAaschiralimpurity,withrespect ofsensitivity,linearity,accuracy,andprecision[24].Thelimitof detection(LOD) and thelimitof quantification(LOQ) werecal- culated based onsignal-to-noise ratiosof 3:1and 10:1 for the LOD and LOQ,respectively. (Baseline noise was measuredcon- sideringa peaktopeakwithin3minselectedinthree different partsofthechromatogramofthestandardsolution.)Theobtained valuesaresummarizedinTable3.Basedontheresults,thelin- earityofthemethodwasevaluatedateightconcentrationlevels forallimpuritiesandcalibrationplotswererepresentedbyplot- tingpeakareasagainstcorrespondingconcentrations(expressed ing/mL).Thecorrelationcoefficientwasdeterminedbylinear leastsquaresregressionanalysisanditishigherthan0.9972inall cases.Moreover,forallimpurities95%confidenceintervalsofthe y-interceptsincludedzeroandrandomdistributionoftheresiduals wasobserved.
Theaccuracyandprecisionwereanalyzedbyperformingintra- (repeatability)andinter-dayevaluation(twoconsecutivedays)of threeconcentrationlevelsforallimpurities,coveringthelinear- ity range,each solutionbeing injectedfive times.Theaccuracy (expressedinmeanrecovery%)rangedfrom97.52%to104.32%.The repeatability(expressedasRSD%)determinedbyfiveparallelinjec- tionsofthesolutionsonthesamedaywasbetween0.41%and2.29%.
Intermediate precisionof themethod(expressed in RSD%) was investigatedontwoconsecutivedaysandwaslowerthan3.56%.
Assayvalidationdata.
Parameter Level IMP-1 IMP-2 DHIVA DMIVA R-IVA
Range(g/mL) Range(%)
2–80 0.025%-1%
2–80 0.025%-1%
2.7–64 0.034%-0.8%
2.7–64 0.034%-0.8%
4–80 0.05%-1%
r2 0.9992 0.9989 0.9980 0.9997 0.9972
LOD(g/mL) 0.12 0.12 0.81 0.60 1.20
LOQ(g/mL) 0.40 0.40 2.70 2.0 4.0
Accuracy(%)
I* 98.90±0.90 104.32±1.01 99.69±1.03 103.92±1.45 103.45±1.19
II** 97.52±1.12 99.55±2.21 102.65±0.99 97.91±0.75 98.94±0.67
III*** 98.29±0.22 101.81±0.88 103.51±0.75 99.81±0.19 98.62±0.34
Content repeatability(RSD)
I* 1.95 1.53 2.29 1.50 0.63
II** 0.68 0.55 1.19 1.02 0.97
III*** 0.41 0.56 0.55 0.49 0.66
Contentprecision (RSD)
I* 2.12 1.74 3.56 1.69 1.11
II** 0.69 0.38 1.26 1.09 1.01
III*** 0.82 0.49 0.81 1.52 2.27
* LevelI:IMP-1=4g/ml,IMP-2=4g/ml,DHIVA=4g/ml,DMIVA=5.4g/ml,R-IVA=8g/ml.
** LevelII:IMP-1=16g/ml,IMP-2=16g/ml,DHIVA=12g/ml,DMIVA=10.8g/ml,R-IVA=16g/ml.
***LevelIII:IMP-1=56g/ml,IMP-2=56g/ml,DHIVA=40g/ml,DMIVA=54g/ml,R-IVA=56g/ml.
Fig.3.Chromatogramsof(A)Solution ofIvabradine5mgtablet.(B) Solution ofIvabradine 5mgfilm-coated tabletspiked with all impuritiesat the0.05%
level.Experimentalconditions:LuxCellulose-2columnwith0.06%(v/v)DEAin MeOH/ACN98/2(v/v)with0.45mL/minflowrateat12◦C.1.IMP-1,2.IMP-2,3.
DHIVA,4.DMIVA,5.R-IVA,6.S-IVA.
Basedontheobtainedresultstheoptimizedmethodprovedto besensitive,linear,accurateandpreciseforthedeterminationof fivedifferentimpurities,includingR-IVAaschiralimpurity.
Theoptimizedandvalidatedmethodwasappliedtotheanalysis ofrealsamples,intheformoffilm-coatedtabletswithanomi- nalcontentof5mgS-IVAbase.Therepresentativechromatograms recordedforthesamplesolutionandthesamplesolutionspiked withimpuritiesareshowninFig.3AandB,respectively.OnlyR-IVA ofthemonitoredimpuritiescouldbequantifiedinthecommercial tablet,theotherimpuritieswerebelow(orclose)theLOD.Thecon- tentofR-IVAwas0.032±0.001%.Inadditiontotheknownrelated substances,minorunidentifiedpeakscouldalsobeobservedinthe samplechromatogram,whichcouldbeindicatingunknownimpu- ritiesormaybetheresultsofdifferentexicipientsusedinthetablet formulation.Usingpeaknormalizationat286nm,thesumofthe totalimpuritieswascalculatedas0.14±0.01%.
4. Concludingremarks
Anovel,singleHPLCmethodusingLuxCellulose-2columnin polarorganicmodewasdevelopedusingDoEforthesimultane- ousdeterminationofDHIVA,DMIVA,IMP-1andIMP-2asrelated substances aswellas R-IVA asenantiomeric impurity in S-IVA.
ThemethodwasvalidatedaccordingtotheInternationalCoun- cilforHarmonizationguidelineQ2(R1)andprovedtobeprecise andaccuratefordeterminationofatleast0.05%orbelowforall impuritiesinS-IVAsamples.Applicationofthemethodwastested ona commercialtablet and showedthatthetablet contains R- IVAimpurity,butbelow0.05%.Ourmethodcouldbeappliedinan industrialenvironmentforsimultaneousquantificationofchemi- calandchiralrelatedsubstancesofS-IVAinoneruntosavetime andmoney.Furthermore,thepresentmethodisanotherexample thatrelatedsubstancesandenantiomericimpuritiescanbedeter- minedbyasinglemethodusingpolysaccharide-typeCSPinpolar organicmode.
Acknowledgements
ThisworkwassupportedbytheJánosBolyaiResearchScholar- shipoftheHungarianAcademyofSciencesandbytheSemmelweis Innovation Found STIA-M-17 and STIA-18-KF (to G.Tóth). The financialsupportfromBolyai+NewNationalExcellenceProgram (grant number: UNKP-19-4-SE-28) of the Ministry of Human Capacitiesishighlyappreciated(G.Tóth).
AppendixA. Supplementarydata
Supplementarymaterial relatedto this articlecanbe found, in theonline version, at doi:https://doi.org/10.1016/j.jpba.2019.
112851.
References
[1]K.Swedberg,M.Komajda,M.Böhm,J.S.Borer,I.Ford,A.Dubost-Brama,G.
Lerebours,L.Tavazzi,Ivabradineandoutcomesinchronicheartfailure (SHIFT):arandomisedplacebo-controlledstudy,Lancet376(2010)875–885, http://dx.doi.org/10.1016/S0140-6736(10)61198-1.
[2]C.W.Yancy,M.Jessup,B.Bozkurt,J.Butler,D.E.Casey,M.M.Colvin,M.H.
Drazner,G.Filippatos,G.C.Fonarow,M.M.Givertz,S.M.Hollenberg,J.
Lindenfeld,F.A.Masoudi,P.E.McBride,P.N.Peterson,L.W.Stevenson,C.
Westlake,C.Westlake,ACC/AHA/HFSAfocusedupdateonnew
pharmacologicaltherapyforheartfailure:anupdateofthe2013ACCF/AHA guidelineforthemanagementofheartfailure:areportoftheamerican collegeofCardiology/Americanheartassociationtaskforceonclinical practiceguidelinesandtheheartfailuresocietyofamerica,Circulation134 (2016)(2016),http://dx.doi.org/10.1161/CIR.0000000000000435.
[3]C.Thollon,J.P.Bidouard,C.Cambarrat,L.Lesage,H.Reure,I.Delescluse,J.
Vian,J.L.Peglion,J.P.Vilaine,Stereospecificinvitroandinvivoeffectsofthe newsinusnodeinhibitor(+)-S16257,Eur.J.Pharmacol.339(1997)43–51, http://dx.doi.org/10.1016/S0014-2999(97)01364-2.
[4]J.P.Vilaine,C.Thollon,N.Villeneuve,J.L.Peglion,Procoralan,anewselectiveI fcurrentinhibitor5(2003)G26–G36,http://dx.doi.org/10.1016/S1520- 765X(03)90005-8.
[5]chiraltech.com/wp-content/uploads/2017/04/Ivabradine-CHIRALPAK-IG.pdf (accessed08.29.2019).
[6]P.Pikul,M.Jamrógiewicz,J.Nowakowska,W.Hewelt-Belka,K.Ciura,Forced degradationstudiesofIvabradineandinsilicotoxicologypredictionsforits newdesignatedimpurities,Front.Pharmacol.7(2016),http://dx.doi.org/10.
3389/fphar.2016.00117.
[7]P.N.Patel,R.M.Borkar,P.D.Kalariya,R.P.Gangwal,A.T.Sangamwar,G.
Samanthula,S.Ragampeta,Characterizationofdegradationproductsof IvabradinebyLC-HR-MS/MS:atypicalcaseofexhibitionofdifferent degradationbehaviourinHClandH2SO4acidhydrolysis,J.MassSpectrom.50 (2015)344–353,http://dx.doi.org/10.1002/jms.3533.
[8]S.Maheshwari,A.Khandhar,A.J.-E.J.of,undefined,Quantitative determinationandvalidationofivabradineHClbystabilityindicating RP-HPLCmethodandspectrophotometricmethodinsoliddosageform, Eurasian,J.Anal.Chem.5(2010)(2010)53–62.
[9]N.P.Nadella,V.N.Ratnakaram,N.Srinivasu,Developmentandvalidationof UPLCmethodforsimultaneousquantificationofcarvedilolandivabradinein thepresenceofdegradationproductsusingDoEconcept,J.Liq.Chromatogr.
Relat.Technol.41(2018)143–153,http://dx.doi.org/10.1080/10826076.2018.
1427595.
[10]T.Ikai,Y.Okamoto,StructureControlofPolysaccharideDerivativesfor EfficientSeparationofEnantiomersbyChromatography,Chem.Rev.109 (2009)6077–6101,http://dx.doi.org/10.1021/cr8005558.
[11]B.Chankvetadze,Polysaccharide-BasedChiralStationaryPhasesfor EnantioseparationsbyHigh-PerformanceLiquidChromatography:An Overview,2019,pp.93–126,http://dx.doi.org/10.1007/978-1-4939-9438-06.
[12]H.Ates,A.A.Younes,D.Mangelings,Y.VanderHeyden,Enantioselectivityof polysaccharide-basedchiralselectorsinpolarorganicsolvents
chromatography:implementationofchlorinatedselectorsinaseparation strategy,J.Pharm.Biomed.Anal.74(2013)1–13,http://dx.doi.org/10.1016/j.
jpba.2012.09.025.
[13]E.P.Sousa,M.E.Tiritan,R.V.Oliveira,C.M.M.Afonso,Q.B.Cass,M.M.M.Pinto, EnantiomericresolutionofkielcorinderivativesbyHPLConpolysaccharide stationaryphasesusingmultimodalelution,Chirality16(2004)279–285, http://dx.doi.org/10.1002/chir.20031.
[14]S.Niedermeier,I.Matarashvili,B.Chankvetadze,G.K.E.Scriba,Simultaneous determinationofdextromepromazineandrelatedsubstances
2-methoxyphenothiazineandlevomepromazinesulfoxidein
levomepromazineonacellulosetris(4-methylbenzoate)chiralcolumn,J.
Pharm.Biomed.Anal.158(2018)294–299,http://dx.doi.org/10.1016/j.jpba.
2018.06.012.
[15]L.Mosiashvili,L.Chankvetadze,T.Farkas,B.Chankvetadze,Ontheeffectof basicandacidicadditivesontheseparationoftheenantiomersofsomebasic drugswithpolysaccharide-basedchiralselectorsandpolarorganicmobile phases,J.Chromatogr.A1317(2013)167–174,http://dx.doi.org/10.1016/j.
chroma.2013.08.029.
[16]A.C.Servais,B.Janicot,A.Takam,J.Crommen,M.Fillet,Liquid
chromatographyseparationofthechiralprodrugeslicarbazepineacetateand itsmainmetabolitesinpolarorganicmode.Applicationtotheiranalysisafter invitrometabolism,J.Chromatogr.A1467(2016)306–311,http://dx.doi.org/
10.1016/j.chroma.2016.07.022.
[17]R.Ferretti,L.Zanitti,A.Casulli,R.Cirilli,Unusualretentionbehaviorof omeprazoleanditschiralimpuritiesBandEontheamylosetris
(3-chloro-5-methylphenylcarbamate)chiralstationaryphaseinpolarorganic mode,J.Pharm.Anal.8(2018)234–239,http://dx.doi.org/10.1016/j.jpha.
2018.04.001.
[18]M.Foroughbakhshfasaei,Z.-I.Szabó,G.Tóth,ValidatedLCmethodfor determinationofenantiomericpurityofapremilastusingpolysaccharide-type stationaryphasesinpolarorganicmode,Chromatographia81(2018) 1613–1621,http://dx.doi.org/10.1007/s10337-018-3546-9.
[19]Z.-I.Szabó,M.Foroughbakhshfasaei,R.Gál,P.Horváth,B.Komjáti,B.Noszál, G.Tóth,Chiralseparationoflenalidomidebyliquidchromatographyon polysaccharide-typestationaryphasesandbycapillaryelectrophoresisusing cyclodextrinselectors,J.Sep.Sci.41(2018)1414–1423,http://dx.doi.org/10.
1002/jssc.201701211.
[20]Z.-I.Szabó,M.Foroughbakhshfasaei,B.Noszál,G.Tóth,Enantioseparationof racecadotrilusingpolysaccharide-typechiralstationaryphasesinpolar organicmode,Chirality30(2018)95–105,http://dx.doi.org/10.1002/chir.
22772.
[21]M.Foroughbakhshfasaei,Z.-I.Szabó,A.Mirzahosseini,P.Horváth,G.Tóth, EnantiomericqualitycontrolofR-TofisopambyHPLCusing
polysaccharide-typechiralstationaryphasesinpolarorganicmode, Electrophoresis39(2018)2566–2574,http://dx.doi.org/10.1002/elps.
201800220.
[22]S.Orlandini,S.Pinzauti,S.Furlanetto,Applicationofqualitybydesigntothe developmentofanalyticalseparationmethods,Anal.Bioanal.Chem.405 (2013)443–450,http://dx.doi.org/10.1007/s00216-012-6302-2.
[23]P.K.Sahu,N.R.Ramisetti,T.Cecchi,S.Swain,C.S.Patro,J.Panda,Anoverview ofexperimentaldesignsinHPLCmethoddevelopmentandvalidation,J.
Pharm.Biomed.Anal.147(2018)590–611,http://dx.doi.org/10.1016/J.JPBA.
2017.05.006.
[24]InternationalCouncilforHarmonizationGuidelineQ2(R1),Validationof AnalyticalProcedures:TextandMethodology,2005(Accessed08.29.2019) https://www.ich.org/fileadmin/PublicWebSite/ICHProducts/Guidelines/
Quality/Q2R1/Step4/Q2R1 Guideline.pdf.

![structures.Suche ff ectsarepresentinalmostallintermolecularinteractionsincluding‘exotic’cases indrugdesign[ ].Thermodynamicquantitiescanbemeasuredbyexperimentalmethodssuchas isintroducedforatomicleveldeterminationofhydrationstructureandextractionofstructur](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)