Versatile synthesis of novel tetrahydroquinolines as potentially active semicarbazide-sensitive amine oxidase (SSAO) inhibitors
via tert-amino effect
Ruth Deme,a Michele Schlich,b Zoltán Mucsi,c Gellért Karvaly,d Gergő Tóth,e and Péter Mátyus*a
aDepartment of Organic Chemistry, Semmelweis University, Hőgyes E. u. 7., H-1092 Budapest, Hungary
bDepartment of Life and Environmental Sciences, University of Cagliari, Via Ospedale 72, 09124 Cagliari, Italy
cFemtonics Ltd., Tűzoltó u. 59., H-1094, Budapest, Hungary
dDepartment of Laboratory Medicine, Semmelweis University, Szentkirályi u. 46., H-1088 Budapest, Hungary
eDepartment of Pharmaceutical Chemistry, Semmelweis University, Hőgyes E. u. 9., H-1092 Budapest, Hungary
E-mail: matyus.peter@pharma.semmelweis-univ.hu DOI: http://dx.doi.org/10.3998/ark.5550190.p009.692
Abstract
Several aminomethyl tetrahydroquinoline derivatives were synthesized in a facile three-step procedure, in order to develop a semicarbazide-sensitive amine oxidase (SSAO) inhibitor library, as proved by in vitro test on rat aorta microsomal fraction. The efficient microwave-assisted cis- diastereoselective cyclization of 2-dicyanovinyl-tert-anilines is based on tert-amino effect as a thermal isomerization reaction. The cyclized dicarbonitrile intermediates were subjected to a decyanation reaction and a subsequent reduction, providing the biologically active aminomethyl tetrahydroquinoline derivatives.
Keywords: Diastereoselective cyclization, microwave-assisted reaction, SSAO activity, tetrahydroquinolines, tert-amino effect
Introduction
Semicarbazide-sensitive amine oxidase (SSAO), also known as vascular adhesion protein-1 (VAP- 1), belongs to the family of copper-containing amine oxidases (CuAOs), with its name derived from its sensitivity to inhibition by the semicarbazide.1 SSAO is identical to primary amine oxidase (SSAO/PrAO),2 as well as circulating benzylamine amine oxidase (BzAO).3 The oxidative
General Papers ARKIVOC 2016 (v) 164-196
Page 165 ©ARKAT-USA, Inc.
deamination of primary aliphatic and aromatic amines, results in a corresponding aldehyde metabolite, hydrogen peroxide and ammonia. The pharmacological significance of SSAO/VAP-1 inhibitors are demonstrated by several studies: pathological angiogenesis,4 ocular diseases,5-7 neuroprotective effect8-10 and anti-inflammatory effect.11-14 Furtheremore, SSAO substrates might also be of therapeutic value in the treatment of diabetes due to their insulin-like effects (e.g., glucose uptake, lipogenesis stimulation and antilipolysis). Therefore, several potent substrates were investigated in human adipocytes compared with benzylamine as the reference substrate.15,16 Recently, new therapeutic aspects of SSAO inhibitors have been published associated with preventing the progress of cerebral amyloid angiopathy in Alzheimer’s disease;17 analgesic effects in traumatic neuropathy and neurogenic inflammation18 and expression of glucose transporters in chronic liver disease.19 Furtheremore, the role of SSAO/VAP-1 in physiopathology of several diseases and application as a biomarker20 have been highlighted as well (e.g. ischemic stroke,21 renal dysfunction and vascular inflammation in type 1 diabetes22). In addition to, new therapeutic targets were reported by Payrits et al. namely, they have described a dual antagonistic action of a known SSAO inhibitor on transient receptor potential ankyrin 1 and vanilloid 1 ion channels on primary sensory neurons.23 In order to understand the role of substrate and inhibitor selectivity and efficacy in CuAOs, Shepard and Dooley have summarized the factors which may play the role of these. This study has presented the significance of copper in the biogenesis of topaquinone and in the catalytic cycle as well.24 Regarding the importance of the field of SSAO in our days, the phase 1 clinical trial of PXS-4728A should be mentioned as well, which is a very potent and selective inhibitor for the treatment of liver-related disease Nonalcoholic Steatohepatitis (NASH).14 Until now, only few reversible inhibitors of that enzyme were reported in the literature,25-27 however, none of them based on tetrahydroquinoline scaffold. In the present work, we aimed to develop a facile, short synthesis for a novel, small presumably reversible inhibitor library designed for SSAO, represented by few relevant examples.
The tert-amino effect as a basis for a ring closure method has been known for more than forty years.28 So far seven types of this effect have been recognized, depending on ring size and mode of formation.29,30 Stereoselective aspects of the tert-amino effect along with explanations of the mechanism have been discussed in several papers earlier.31-36 The Reinhoudt group thoroughly investigated the influence of the steric and electronic effects of substituents on the regioselectivity33 and the ’self-reproduction of chirality’ - without any auxiliary reagent.31 They concluded that the [1,5]-hydrogen shift proceeds enantioselectively in a suprafacial manner.
Furthermore, the diastereoselective outcome of the reaction depends on the interchange of the position of the vinyl group, due to the steric hindrance of the vinyl substituents (Scheme 1).
According to the X-ray analysis, the authors suggested that the vinyl compound preferentially exists in the conformer A. From this conformer the hydrogen atom (Ha) migrates via an 1,5- hydrogen shift to the vinyl carbon through a transition state, forming finally the product molecule, where this Ha occupies the position cis relative to the substituent R2.
with the literature data. Reinhoudt and co-workers synthesized the same derivatives 5b,c refluxing 3b,c in n-BuOH for several hours, resulting exlusively in the cis isomer.33
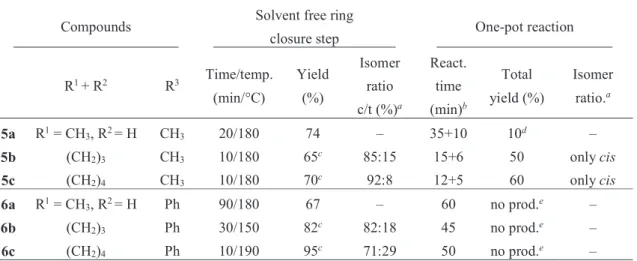
The two step procedure was managed to carry out under one-pot conditions in MW reactors in the case of 1 (R3 = CH3, Scheme 2) with relatively good yields (60%) and in a stereoselective manner, giving rise solely to the cis isomers (Table 1), previously employed by our group.46 The only exception was compound 5a, which was isolated with lower yield, due to the formation of the acid amide side product by the nucleophilic addition of the water to nitrile, which can be a result of the longer reaction time than that applied for compounds 5b, c. In contrast to methyl substituted compounds (1), compound 2 (R3 = Ph) could not be transform to the desired products under analogues conditions (Table 1).
Table 1. Yield and reaction time of the ring closure products obtained by solvent free and one- pot reactions starting from compound 1a, b, c and 2a, b, c.
Compounds Solvent free ring
closure step One-pot reaction R1 + R2 R3 Time/temp.
(min/°C)
Yield (%)
Isomer ratio c/t (%)a
React.
time (min)b
Total yield (%)
Isomer ratio.a
5a R1 = CH3, R2 = H CH3 20/180 74 – 35+10 10d –
5b (CH2)3 CH3 10/180 65c 85:15 15+6 50 only cis
5c (CH2)4 CH3 10/180 70c 92:8 12+5 60 only cis
6a R1 = CH3, R2 = H Ph 90/180 67 – 60 no prod.e –
6b (CH2)3 Ph 30/150 82c 82:18 45 no prod.e –
6c (CH2)4 Ph 10/190 95c 71:29 50 no prod.e –
aRatio of the diastereomers in the crude product was determined by 1H NMR and/or HPLC.
bFirst: reaction time for the Knoevenagel condensation, second: reaction time for the cyclization
cTotal yield for the formed cis and trans isomers. dAcid amide side product was also formed in 10% yield. eAccording to the TLC a very little amount of the vinyl compound was formed, therefore the reaction was stopped.
The first example of decyanation of dialkylated malononitriles (geminal dinitriles) was published by Curran and Seong,50 proposed the geminal substituent effect. In this work, the decyanation reactions of compounds 5a, 5b-cis, 5c-cis and 6a, 6b-cis, 6c-cis were carried out by radical reduction with tributyltin hydride in the presence of azobisisobutyronitrile (AIBN) in toluene (Scheme 3), based on literature example.51 Only two diastereomers were formed during the reaction (5 9; 6 10), as proved by NMR, instead of the theoretically possible four isomers.
Column chromatography allowed the separation of the diastereomers, which were employed for the subsequent steps in pure form. The relative configuration of position 3a and 5 was identified
General Papers ARKIVOC 2016 (v) 164-196
Page 171 ©ARKAT-USA, Inc.
vinyl group, the [1,5] suprafacial migration of a hydrogen and the coordinated formation of a carbon-carbon bond. The formation of the trans product may attributed to steric factors, aligning the direction of the vinyl group in the starting compond.33
Our experiments verify that the rotamer of the vinyl substance which serves as a starting compound for the formation of the trans isomer via [1,5]-hydrogen migration is also obtained at higher temperatures. To confirm that no interconversion of the diastereomers takes place at the temperature of ring closure, the following experiments were performed. First, the temperature required for the ring closures of 4b, c was determined using differential scanning calorimetry (DSC).53 The obtained thermograms gave a good indication of the melting points (endothermic peaks) of the vinyl substances as well as the ring closure temperatures (exothermic peaks) (see Supplementary Material). Second, the pure cis isomers 5b, c and 6b, c, yielded in the ring closure reactions, were heated at the temperatures required for ring formation. The corresponding 1H NMR assays proved the exclusive presence of the cis isomers. Consequently, one may conclude from that experiment that there was no any transformation from cis to trans isomer, proving that the cis is not an intermediate of the reaction, but one of the final product. In order to determine undoubtedly the relative configuration of the trans isomer, the diastereomers were separated by chromatography, and elucidated by using 1D and 2D NMR spectroscopy.
Examining the substituent effect of the ring closure, one may conclude that there was no significant difference between the effects of the two R3 substituents (CH3 or Ph) on the ratio of the diastereomers formed, except in the case of compound 6c, where more of the trans isomer was produced than in the rest of the reactions. My assumption is that the simultaneous presence of the phenyl and piperidino groups on the vinyl compound force the molecule into a defined sterical position, allowing the migrating hydrogen to attack the α carbon atom of the vinyl group from both sides of the molecule, unlike in structures substituted with methyl and piperidino groups which exert a smaller grade of steric hindrance (compound 5c). Based on the published results it seems likely that the hydrogen migration takes place suprafacially and the formation of the trans isomer is controlled by the interchange of the conformation of the vinyl compound in addition to energy relations. The rate of the ring closure reaction is greater in structures containing the more reactive cyclic secondary amino groups (pyrrolidino and piperidino) than in those containing aliphatic (eg.
dimethylamino) substituents (Table 1).
Considering Reinhoudt’s and our results, we were led to the assumption that the formation of cis and trans diastereomers (5 and 6) depends not only on the steric effect, but also the difference between the activation energies of the cis and the trans transition states (see Scheme 1), in agreement with the Curtin-Hammett principle.
Pharmacological investigation. The SSAO activity of compounds 11a cis, 11b cis-trans, 11c cis- trans, 12a cis, 12b cis-trans and 12c cis-cis was tested on the microsomal fraction of rat aorta (Table 5). 4-Phenylbutylamine (4-PBA) was used as the reference substrate,15,54 while 2-bromoethylamine (2-BEA) was used as selective, irreversible inhibitor.55 According to the hydrogen peroxide formation assay, compounds 11a cis and 12b cis-trans act as inhibitors of
moderate potency as compared to 2-BEA, while compound 11b cis-trans behave as a substrate (Km = 0.94 ± 0.01 µM; dose response curves see Supplementary Material). Based on the Km data, we can conclude that the affinity of compound 11b cis-trans is higher than that of the substrate 4- PBA. The rest of the studied compounds showed only a weak, but promising enzyme inhibitory effect. The structure activity relationship analysis may suggest that pyrrolidine fused ring structure with Ph substituent (12b cis-trans) exhibits the best combination in the light of the inhibitory activity.
Table 5. Various measured SSAO biological activities [inhibition (IC50) and substrate binding constant (Km)] of the prepared compounds 11a-c and 12a-c as well as the references 2-BEA and 4-PBA
Compound IC50 (µM) Km (µM) 2-BEAa 0.56 ± 0.12
4-PBAb – 740 ± 55
11a cis 5.0 ± 0.4
11b cis-trans – 0.94 ± 0.01 11c cis-trans 14.7 ± 1.0
12a cis 17.8 ± 5.2 12b cis-trans 5.1 ± 0.5
12c cis-cis 31.0 ± 6.0
a2-BEA = 2-bromoethylamine; b4-PBA = 4-phenylbutylamine;
Conclusions
A facile microwave-assisted diastereoselective cyclization reaction was developed in order to extend an SSAO inhibitor library, containing condensed aminomethyl tetrahydroquinoline derivatives. The first, diastereoselective cyclizations step based on the known tert-amino effect, providing selectively the cis isomers in all the cases, irrespectively to the two types of R3 substituents (CH3 or Ph). The rate of the ring closure reaction is greater in structures containing the more reactive cyclic secondary amino groups (pyrrolidino and piperidino) than in those containing aliphatic (e.g., dimethylamino) substituents. Diastereomers were separated by chromatography then the relative configuration was elaborated by NOE interactions and vicinal coupling constants. The further transformation of the dicyano intermediates included the chemoselective reductive elimination of the cyano group, resulted also two diastereomers, which were separated and fully characterized. The final nitrile reduction step afforded six SSAO active aminomethyl derivatives. One of them showed high enzyme substrate activity, while two other compounds inhibited this enzyme with a moderate potential in a presumably reversibly way, as
General Papers ARKIVOC 2016 (v) 164-196
Page 173 ©ARKAT-USA, Inc.
compared to literature references. These compounds proved to be hit molecules for the further biological experiments.
Experimental Section
General. All reaction solvents were purified in accordance with Purification of Laboratory Chemicals (Fourth Edition) prior to use. All reagents were used as purchased without further purification. The solvents were removed under reduced pressure using standard rotary evaporators.
All of the reactions were monitored by TLC using Merck’s silica gel 60 F254-precoated aluminum sheets. Visualization was accomplished with UV light (254 or 365 nm). Solvent mixtures used for chromatography are always given in a vol/vol ratio. Flash column chromatography was generally performed using Silica Gel 60 (Merck, spherical, 40-63 µm).
Melting points were determined on a Büchi-540 capillary melting point apparatus and are uncorrected. The high-resolution accurate masses (HRMS) were determined with an Agilent 6230 time-of-flight mass spectrometer. Samples were introduced by the Agilent 1260 Infinity LC system. The mass spectrometer was operated in conjunction with a Jet Stream electrospray ion source in positive ion mode. Reference masses of m/z 121.050873 and 922.009798 were used to calibrate the mass axis during analysis. Mass spectra were processed using Agilent MassHunter B.02.00 software.
High-performance liquid chromatography (HPLC) was performed on a Jasco 2080 Plus isocratic binary pump, using a Jasco 2075 Plus variable wavelength absorbance detector and Jasco ChromPass v.1.8.6.1 software. All samples were dissolved in the mobile phase used for the assay at a level of approximately 1 mg/mL. Stock solutions were diluted 1:20 using the mobile phase, resulting in a concentration of approximately 50 µg/mL. The diluted samples were injected without further manipulation. Stock solutions were kept at -20 °C overnight. Diluted solutions were prepared on each day of the analysis and were not stored. Chromatographic runs lasted 30 min typically. When all peaks were recovered, runs were terminated manually regardless of the time that had passed. Two stationary phases were employed with the following parameters: 1) Chiralcel® „OJ-H” cellulose tris-4-methylbenzoate, 250 mm x 4.6 mm, 5 µm dp. 2) Chiralpak®
„AD-H” amylose tris-(3,5-dimethylphenyl-carbamate), 250 mm x 4.6 mm, 5 µm dp. The mobile phase was a n-hexane/ethanol mixture in all cases. The ratio of the components is provided in the Supplementary Material.
The differential scanning calorimetry (DSC) examinations were carried out with a Pyris 6 DSC (Perkin Elmer) instrument. The DSC curves were evaluated with Pyris Software. The starting and final temperatures were 30 °C and 300 °C, respectively. Heating rate was 5 and 10 °C/min.
Nitrogen atmosphere was always used. Samples from 0.79 to 3.20 mg were used (in aluminium sample pans). Three parallel examinations were made in every case. The instrument was calibrated by using indium.
Elemental analyses were performed on an Elementar VarioEL III apparatus. MW irradiation
experiments were carried out in a monomode CEM-Discover MW reactor, using the standard configuration as delivered, including proprietary software. The experiments were executed in 10 or 80 mL MW process vials with control of the temperature by infrared detection. After completion of the reaction, the vial was cooled to 50 °C by air jet cooling.
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at ambient temperature, in the solvent indicated, on a Varian Mercury Plus 400 spectrometer at a frequency of 400 and 100 MHz or on a Varian Unity 600 spectrometer at a frequency of 600 and 150 MHz or on a Bruker Avance III 500 spectrometer at a frequency of 500 and 125 MHz respectively. Chemical shifts are given using the δ-scale (in ppm) relative to tetramethylsilane or the residual solvent signal as an internal reference. Coupling constants are indicated in Hertz (Hz). The following abbreviations are used for spin multiplicity: s singlet, d doublet, t triplet, q quartet, m multiplet, ovl. m overlapping multiplet, br broad, dd doublet doublet, dm doublet multiplet and tm triplet multiplet. The signification of the one star (*) in the 13C NMR means tentative assignments.
General procedure for the synthesis of 2-(dialkylamino)acetophenone and benzophenone derivetives (method A). A mixture of 2-fluoroacetophenone or 2-fluorobenzophenone (1.0 eq), the appropriate secondary amine (pyrrolidine or piperidine purified by redistillation (760 mmHg, 85-110 °C) or dimethylamine (40 wt.% in water) (1.0 eq) and K2CO3 (1.0 eq) in water was irradiated in a pressurized vessel in microwave reactor for the time and the temperature indicated below (at a maximum power level of 200 W). The vessel was subsequently cooled to ambient temperature. To the reaction mixture distilled water was added and it was extracted with diethyl ether. The organic layer was washed with saturated solution of NH4Cl, then with distilled water and then dried over MgSO4, filtered and evaporated under reduced pressure. The crude product was used for the one-pot reaction without further purification. Analytical results of compounds 1(a,b,56c33) and 2 (a,57b,58c56) are corresponding with the literature data. The atom numbering for NMR assignation follows the scheme below in Scheme 5:
7.00 (1H, m, H – 5’), 2.95 – 2.93 (4H, m, H – 2”, 6”), 2.67 (3H, s, H - 2), 1.73 – 1.69 (4H, m, H – 3”, 5”), 1.57 - 1.56 (2H, m, H – 4”).
2’-Benzoyl-N,N-dimethylaniline (2a). Following method A, the title compound was isolated. A mixture of 2-fluorobenzophenone (5.00 g, 24.97 mmol, 4.22 mL), dimethylamine (40 wt. % in water) (1.13 g, 24.97 mmol, 2.8 mL) and K2CO3 (3.45 g, 24.97 mmol) in 30 mL water was irradiated at 130 °C for 2 hours. Yellow dense oil (5.35 g, 95%). 1H NMR: (400 MHz, chloroform- d): 7.83 (2H, d, J 7.2 Hz, H – 2”, 6”), 7.56 – 7.37 (4H, m, H – 5’, 3”, 4”, 5”), 7.33 – 7.31 (1H, dm, J 7.6 Hz, H – 3’), 7.00 (1H, d, J 8.3 Hz, H – 6’), 6.90 (1H, m, H – 4’), 2.70 (6H, s, H – 2, 3).
1-(2’-Benzoylphenyl)pyrrolidine (2b). Following method A, the title compound was isolated. A mixture of 2-fluorobenzophenone (3.00 g, 14.98 mmol, 2.50 mL), pyrrolidine (1.07 g, 14.98 mmol, 1.24 mL) and K2CO3 (2.07 g, 14.98 mmol) in 30 mL water was irradiated at 150 °C for 2.5 hours. Yellow solid (2.51 g, 67%). mp 58-60 °C. 1H NMR: (400 MHz, chloroform-d): 7.95 – 7.93 (2H, m, H – 2”, 6”), 7.59 – 7.56 (1H, m, H – 4”), 7.47 – 7.43 (2H, m, H – 3”, 5”), 7.39 – 7.36 (1H, m, H – 5’), 7.27 – 7.25 (1H, dm, J 7.7 Hz, H – 3’), 6.85 – 6.85 (1H, dm, J 8.5 Hz, H – 6’), 6.69 – 6.67 (1H, m, H – 4’), 3.15 – 3.12 (4H, m, H – 2, 5), 1.90 – 1, 87 (4H, m, H – 3, 4).
1-(2’-Benzoylphenyl)piperidine (2c). Following method A, the title compound was isolated. A mixture of 2-fluorobenzophenone (5.00 g, 24.97 mmol, 4.22 mL), piperidine (2.10 g, 24.97 mmol, 2.5 mL) and K2CO3 (3.45 g, 24.97 mmol) in 30 mL water was irradiated at 150 °C for 1.5 hours.
Yellow solid (6.52 g, 98%). mp 90.1-91.0 °C. 1H NMR: (400 MHz, chloroform-d): 7.77 – 7.75 (2H, m, H – 2”, 6”), 7.53 – 7.37 (5H, m, H – 3’, 5’, 3”, 4”, 5”) 7.09 – 7.03 (2H, m, H – 4’, 6’), 2.83 (4H, m, H – 2, 6), 1.27 (2H, m, H – 4), 1.15 (4H, m, H – 3, 5).
General procedure for the synthesis of 2-vinyl-N,N-dialkylanilines from acetophenone derivatives (method B1). To a mixture of the appropriate acetophenone derivative (1.0 eq) in EtOH, malononitrile (1.0 eq) and 2 drops of piperidine were added. After several hours at room temperature, the yellow solution turned to orange. When the reaction was completed as followed by TLC, the solvent was removed under reduced pressure. The residue was purified by column chromatography to give the pure product. Analytical results of compound 3 (a, b, c33) are corresponding with the literature data.
General procedure for the synthesis of 2-vinyl-N,N-dialkylanilines from benzophenone derivatives (method B2). Malononitrile (1.1 eq) and i-PrOH (distilled from CaO, dried over 4 Å molecular sieves) were placed in a sealed tube under argon atmosphere. To this colorless solution, the appropriate benzophenone (1 eq) and Ti(O-i-Pr)4 (1.1 eq) were added and the mixture was heated at 70-80 °C. After completion of the reaction, the dark brown reaction mixture was poured into 1 N HCl and was vigorously stirred at 0-5 °C for 0.5 hours. It was extracted by ethyl acetate and the extract was washed with sodium bicarbonate solution and brine. The organic layer was dried over MgSO4 and evaporated. The crude product was purified by column chromatography and washed with diethyl ether to afford the pure product.
General Papers ARKIVOC 2016 (v) 164-196
Page 177 ©ARKAT-USA, Inc.
2-{1’-[2’-(Dimethylamino)phenyl]ethylidene}propanedinitrile (3a). Following method B1, the title compound was isolated. To a mixture of 1-[2-(dimethylamino)phenyl]ethanone (4.86 g, 29.80 mmol) in 50 mL EtOH, malononitrile (2.36 g, 35.76 mmol, 1.2 eq) and 2 drops piperidine were added. The orange reaction mixture was stirred at room temperature for 15 hours. The crude product was purified by column chromatography (n-hexane/EtOAc 10:1). Orange dense oil (5.52 g, 88%). 1H NMR (500 MHz, chloroform-d): 7.43-7.47 (1H, m, H-4’), 7.19 (1H, dm, J 7.7 Hz, H-3’), 7.09 (1H, dm, J 8.3 Hz, H-6’), 7.05-6.99 (1H, m, H-5’), 2.75 (6H, s, H-1”, 2”), 2.63 (3H, s, CH3); 13C NMR (125 MHz, chloroform-d): 179.2 (=CqCH3), 151.3 (C-2’), 132.2 (C-4’), 129.1 (C-1’), 128.9 (C-6’), 121.5 (C-5’), 118.8 (C-3’), 112.7* (C-1), 112.6* (C-3), 85.5 (C-2), 43.7 (C-1”, 2”), 22.8 (CH3). HRMS (ESI+) m/z calcd. for C13H14N3 [M+H]+ 212.1182, found 212.1190.
2-{1’-[2’-(Pyrrolidin-1”-yl)phenyl]ethylidene}propanedinitrile (3b). Following method B1, the title compound was isolated. To a mixture of 2-(pyrrolidin-1-yl)acetophenone (1.00 g, 5.28 mmol) in 10 mL EtOH, malononitrile (0.35 g, 5.28 mmol) and 2 drops piperidine were added. The orange reaction mixture was stirred at room temperature for 24 hours. The crude product was purified by column chromatography (toluene) and washed with diethyl ether to afford the pure product. Orange crystals (0.94 g, 75%). mp 106.0-107.9 °C. 1H NMR: (400 MHz, chloroform-d):
7.36 – 7.29 (1H, m, H-4’), 7.14 (1H, dm, J 7.9 Hz, H-3’), 6.90 (1H, dm, J 8.5 Hz, H-6’), 6.87 – 6.80 (1H, m, H-5’), 3.22-3.05 (4H, m, H-2”, 5”), 2.59 (3H, s, CH3), 2.03 - 1.94 (4H, m, H-3”, 4”);
13C NMR: (100 MHz, chloroform-d): 178.6 (=CqCH3), 147.4 (C-2’), 132.0 (C-4’), 129.0 (C-1’), 123.4 (C-6’), 117.8 (C-5’), 115.0 (C-3’), 113.1* (C-1), 112.4* (C-3), 84.1 (C-2), 51.1 (C-2”, 5”), 25.8 (C-3”, 4”), 23.7 (CH3). Anal. calcd. for C15H15N3 (237.29): C, 75.92%; H, 6.37%; N, 17.71%.
Found: C, 75.59%; H, 6.18%; N, 17.30%. HRMS (ESI+) m/z calcd. for C15H16N3 [M+H]+ 238.1339, found 238.1343.
2-{1’-[2’-(Piperidin-1”-yl)phenyl]ethylidene}propanedinitrile (3c). Following method B1, the title compound was isolated. To a mixture of 2-(piperidin-1-yl)acetophenone (0.80 g, 3.94 mmol) in 10 mL EtOH, malononitrile (0.26 g, 3.94 mmol) and 2 drops piperidine were added. The yellow reaction mixture was stirred at room temperature for 21 hours. The crude product was purified by column chromatography (n-hexane/EtOAc 4:1). Yellow crystals (0.76 g, 77%). mp 100.6-101.9
°C. 1H NMR: (400 MHz, chloroform-d): 7.45 – 7.41 (1H, m, H-4’), 7.20 – 7.18 (1H, m, H-6’), 7.16 – 7.14 (1H, m, H-3’), 7.10 – 7.08 (1H, m, H-5’), 2.87 (4H, t, J 8.0 Hz, H-2”, 6”), 2.68 (3H, s, CH3), 1.73 - 1.69 (4H, m, H-3”, 5”), 1.59 – 1.57 (2H, m, H-4”); 13C NMR: (100 MHz, chloroform- d): 178.9 (=CqCH3), 151.9 (C-2’), 132.2 (C-4’), 131.2 (C-1’), 128.6 (C-6’), 122.8 (C-5’), 120.2 (C-3’), 112.7* (C-1), 112.6* (C-3), 86.1 (C-2), 53.8 (C-2”, 6”), 26.2 (C-3”, 5”), 23.9 (C-4”), 23.4 (CH3). Anal. calcd. for C16H17N3 (251.33): C, 76.46%; H, 6.82%; N, 16.72%. Found: C, 76.05%;
H, 6.62%; N, 16.37%. HRMS (ESI+) m/z calcd. for C15H18N3 [M+H]+ 252.1495, found 252.1504.
2-{[2”-(Dimethylamino)phenyl](phenyl)methylidene}propanedinitrile (4a). Following method B2, the title compound was isolated. To a mixture of malononitrile (0.55 g, 8.40 mmol) in i-PrOH (20 mL), [2-(dimethylamino)phenyl](phenyl)methanone (1.90 g, 8.40 mmol) and Ti(O- i-Pr)4 (2.39 g, 8.40 mmol, 2.50 mL) were added. The dark red reaction mixture was stirred at 70 °C
for 71 hours. The crude product was purified by column chromatography (toluene). Red crystals (1.66 g, 75%). mp 116-119 °C. 1H NMR: (400 MHz, chloroform-d): 7.52 -7.49 (1H, m, H-4’), 7.48 - 7.46 (2H, m, H-2’, 6’), 7.44 – 7.42 (3H, m, H-3’, 5’, 4”), 7.17 (1H, dd, J 7.7 and 1.4 Hz, H- 6”), 7.05 (1H, d, J 8.2 Hz, H-3”), 6.99 (1H, t, J 7.7 Hz, H-5”), 2.66 (6H, s, H-1”’, 2”’); 13C NMR:
(100 MHz, chloroform-d): 175.2 (=CqPh) 152.8 (C-2”), 135.9 (C-1’), 132.9 (C-4”), 132.4 (C-4’), 132.3 (C-6”), 129.6 (C-3’, 5’), 128.6 (C-2’, 6’), 127.6 (C-1”), 120.6 (C-5”), 118.6 (C-3”), 114.4*
(C-3), 114.1* (C-1), 81.4 (C-2), 43.1 (C-1”’, 2”’). Anal. calcd. for C18H15N3 (273.33): C, 79.10%;
H, 5.53%; N, 15.37%. Found: C, 78.95%; H, 5.49%; N, 15.21%. HRMS (ESI+) m/z calcd. for C18H16N3 [M+H]+ 274.1339, found 274.1345.
2-{Phenyl[2”-(pyrrolidin-1”’-yl)phenyl]methylidene}propanedinitrile (4b). Following method B2, the title compound was isolated, however the major product was the cyclized derivative (see below compound 6b). To a mixture of malononitrile (1.31 g, 19.89 mmol) in i- PrOH (70 mL), phenyl[2-(pyrrolidin-1-yl)phenyl]methanon (5.00 g, 19.89 mmol) and Ti(O-i-Pr)4
(5.65 g, 19.89 mmol, 5.88 mL) were added. The dark red reaction mixture was stirred at 80 °C for 96 hours. Dark red crystals (0.36 g, 6%). mp 148.2-151.1 °C. 1H NMR: (400 MHz, chloroform- d): 7.64 – 7.52 (3H, m, H-2’, 4’, 6’), 7.51 – 7.41 (2H, m, H-3’, 5’), 7.40 – 7.33 (1H, m, H-4”), 6.93 (1H, dm, J 8.5 Hz, H-3”), 6.87 (1H, dm, J 8.1 Hz, H-6”), 6.73 (1H, m, H-5”), 3.48 – 2.92 (4H, brm, H-2”’, 5”’), 2.13 - 1.81 (4H, brm, H-3”’, 4”’); 13C NMR: (100 MHz, chloroform-d): 174.7 (=CqPh), 148.8 (C-2”), 136.6 (C-1’), 133.0 (C-4”), 132.8 (C-4’), 132.5 (C-6”), 130.7 (C-1”), 129.0 (C-2’, 6’), 128.7 (C-3’, 5’), 121.4 (C-5”), 116.9 (C-3”), 114.9* (C-1), 113.7* (C-3), 78.7 (C-2), 51.0 (C-2”’, 5”’), 25.9 (C-3”’, 4”’). HRMS (ESI+) m/z calcd. for C20H18N3 [M+H]+ 300.1495, found 300.1497.
2-{Phenyl[2”-(piperidin-1”’-yl)phenyl]methylidene}propanedinitrile (4c). Following method B2, the title compound was isolated. To a mixture of malononitrile (2.50 g, 37.68 mmol) in i-PrOH (70 mL), 1-(2’-benzoylphenyl)piperidine (10.0 g, 37.68 mmol) and Ti(O-i-Pr)4 (10.72 g, 37.68 mmol, 11.2 mL) were added. The dark orange reaction mixture was stirred at 80 °C for 7 hours. The crude product was purified by column chromatography (n-hexane/EtOAc 9:1).
Orange crystals (46%). mp 166-168 °C. 1H NMR: (500 MHz, chloroform-d): 7.57 – 7.36 (5H, m, H-2’, 3’, 4’, 5’, 6’), 7.48 (1H, m, H-4”), 7.25 (1H, dm, J 6.5 Hz, H-6”), 7.13 (1H, dm, J 1.0 Hz, H-3”), 7.11 (1H, m, H-5”), 2.77 (4H, brs, H-2”’, 6”’), 1.50 – 1.25 (6H, brm, H-3”’, 4”’, 5”’);
13C NMR: (125 MHz, chloroform-d): 174.9 (=CqPh), 153.7 (C-2”), 136.0 (C-1’), 133.0 (C-4”), 132.4 (C-4’), 131.9 (C-6”), 131.1 (C-1”), 129.7 (C-2’, 6’), 128.5 (C-3’, 5’), 122.6 (C-5”), 120.8 (C-3”), 114.3* (C-1), 114.2* (C-3), 82.1 (C-2), 53.4 (C-2”’, 6”’), 25.9 (C-3”’, 5”’), 23.8 (C-4”’).
Anal. calcd. for C21H19N3 (313.40): C, 80.48%; H, 6.11%; N, 13.41%. Found: C, 80.74%; H, 6.18%; N, 13.33%. HRMS (ESI+) m/z calcd. for C21H19N3 [M+H]+ 314.1652, found 314.1640.
General procedure for the synthesis of pyrido-fused ring system – one-pot reaction (method C1). Step 1. To a mixture of the appropriate acetophenone (1.0 eq) in 30 mL of water, malononitrile (1.0 eq) was added. The reaction mixture was irradiated in a pressurized vessel for the time and the temperature indicated below (at a maximum power level of 200 W). The vessel
General Papers ARKIVOC 2016 (v) 164-196
Page 179 ©ARKAT-USA, Inc.
was subsequently cooled to ambient temperature, monitoring the completion of the reaction by TLC. The reaction mixture was used for the next step without work-up and purification.
Step 2.: To the reaction mixture trifluoroacetic acid was added in catalytic amount (3 drops). The vinyl precursor was irradiated in a pressurized vessel for the time and the temperature indicated below (at a maximum power level of 200 W). The vessel was subsequently cooled to ambient temperature, the crude product was taken for the analysis of the ratio of the diastereomers by NMR.
After transferring from the vial, the reaction mixture was extracted with dichloromethane (3x30 mL). The organic layer was dried over MgSO4 and evaporated under reduced pressure. The crude product was purified by crystallization or filtration.
General procedure for the synthesis of pyrido-fused ring system – solvent free reaction (method C2). The vinyl precursor was irradiated in a sealed vessel without solvent. When the reaction was completed as followed by TLC, the vessel was subsequently cooled to ambient temperature. The crude product was taken for the analysis of the ratio of the diastereomers by NMR. After transferring from the vial, to the reaction mixture dichloromethane (20 mL) and water (20 mL) were added, then the water phase was extracted with dichloromethane (2x30 mL). The organic layer was washed with saturated solution of NH4Cl (60 mL), then dried over MgSO4 and evaporated under reduced pressure. The crude product was purified by column chromatography and/or crystallization.
(±)-1,4-Dimethyl-1,2,3,4-tetrahydroquinoline-3,3-dicarbonitrile (5a). Following method C1, the title compound was isolated. (1. step) To a mixture of 2-(dimethylamino)acetophenone (5.50 g, 33.70 mmol) in 30 mL of water, malononitrile (2.22 g, 33.70 mmol) was added. The reaction mixture was irradiated at 100 °C for 35 min, then (2. step) to the reaction mixture trifluoroacetic acid was added and it was irradiated at 165 °C for 10 min. The crude product was purified by column chromatography (toluene), then was washed with n-hexane (in the reaction 10% side product was formed, see below). Pale yellow crystals (0.44 g, 6%). mp 86.0-86.8 °C. 1H NMR (500 MHz, chloroform-d): 7.22 – 7.21 (1H, m, H-7), 7.12 (1H, dm, J 8.0 Hz, H-6), 6.83 – 6.80 (1H, m, H-5), 6.71 (1H, dm, J 8.5 Hz, H-8), 3.78 (1H, dm, J 12.0 Hz, Hx-2), 3.74 (1H, dm, J 12.0 Hz, Hy-2), 3.49 (1H, q, J 7.0 Hz, H-4), 3.06 (3H, s, NCH3), 1.64 (3H, d, J 7.0 Hz, CH3); 13C NMR (125 MHz, chloroform-d): 142.9 (C-8a), 129.0 (C-7), 128.1 (C-5), 120.2 (C-4a), 118.6 (C-6), 114.8* (C-2’), 113.3* (C-1’), 112.3 (C-8), 54.1 (C-2), 39.6 (C-4), 39.4 (NCH3), 36.7 (C-3), 19.0 (CH3). Anal. calcd. for C13H13N3 (211.26): C, 73.91%; H, 6.20%; N, 19.89%. Found: C, 73.53%;
H, 6.15%; N, 19.86%. HRMS (ESI+) m/z calcd. for C13H14N3 [M+H]+ 212.1182, found 212.1188.
Following method C2, the title compound was isolated. The vinyl precursor (0.98 g, 4.62 mmol) was irradiated at 180 °C for 20 minutes. The crude product was washed with n-hexane. Pale yellow crystals (0.73 g, 74%).
3-Cyano-1,4-dimethyl-1,2,3,4-tetrahydroquinoline-3-carboxamide (side product). Following method C1, the title compound was isolated. Cream powder (0.69 g, 10%), mp 160.3-161.1 °C.
According to the NMR analysis the diastereomer ratio is 80:20 cis/trans. 1H NMR (500 MHz,
chloroform-d): 7.20 - 7.17 (1H, m, H-7), 7.08 (1H, dm, J 8.5 Hz, H-5), 6.75 (1H, dm, J 7.5 Hz, H- 6), 6.72 (1H, dm, J 8.0 Hz, H-8), 6.35 (1H, brs, NH2), 5.75 (1H, brs, NH2) 3.73 (1H, d, J 17.5 Hz, Hx-2), 3.46 (1H, d, J 14.0 Hz, Hy-2), 3.41 (1H, q, J 7.0 Hz, H-4), 3.03 (3H, s, H-NCH3), 1.32 (3H, d, J 7.5 Hz, CH3); 13C NMR (125 MHz, chloroform-d): 167.3 (C=O), 143.5 (C-8a), 128.9 (C-5), 128.5 (C-7), 122.8 (C-4a), 120.2 (CN), 117.8 (C-6), 111.9 (C-8), 50.1 (C-2), 45.8 (C-3), 40.1 (C- 4), 39.1 (NCH3), 20.0 (CH3). HRMS (ESI+) m/z calcd. for C14H16N2O [M+H]+ 229.1335, found:
229.1342.
cis-(±)-5-Methyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4,4-dicarbonitrile
(5b cis)33 and trans-(±)-5-Methyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4,4- dicarbonitrile (5b trans). Following method C1, the title compound (5b cis) was isolated. (1.
step) To a mixture of 2-(pyrrolidino)acetophenone (6.58 g, 34.80 mmol) in 40 mL water, malononitrile (2.30 g, 34.80 mmol) was added. The reaction mixture was irradiated at 100 °C for 15 minutes, then (2. step) to the reaction mixture trifluoroacetic acide was added and it was irradiated at 150 °C for 6 min. The ratio of the diastereomers in the crude product by NMR: only cis isomer was formed. The crude solid product was washed with Et2O and crystallized from EtOH (anhydrous, dried over 3 Å molecular sieves). White crystals (4.12 g, 50%). mp 134.0 – 134.5 °C.
1H NMR (500 MHz, chloroform-d): 7.25 – 7.16 (2H, m, H-6, 8), 6.82 – 6.75 (1H, m, H-7), 6.57 (1H, dm, J 8.0 Hz, H-9), 3.92 (1H, dd, J 9.0 and 6.0 Hz, H-3a), 3.56 – 3.40 (2H, m, H-1) 3.48 (1H, q, J 7.0 Hz, H-5), 2.60 – 2.50 (1H, m, Hx-3), 2.27 – 2.06 (3H, m, Hx,y-2, Hy-3), 1.77 (3H, d, J 7.0 Hz, H-CH3); 13C NMR (125 MHz, chloroform-d): 141.9 (C-9a), 129.1 (C-8), 126.8 (C-6), 118.7 (C-5a), 117.6 (C-7), 115.3* (C-1’), 112.0* (C-2’), 111.9 (C-9), 63.0 (C-3a), 48.1 (C-1), 42.3 (C-4), 41.1 (C-5), 30.3 (C-3), 22.7 (C-2), 16.1 (C-CH3). Anal. calcd. for C15H15N3 (237.30): C, 75.92%; H, 6.37%; N, 17.71%. Found: C, 75.90%; H, 6.41%; N, 17.87%. HRMS (ESI+) m/z calcd.
for C15H16N3 [M+H]+ 238.1339, found 238.1334.
Following method C2, the title compound (5b trans) was isolated. The vinyl precursor (0.20 g, 0.84 mmol) was irradiated at 180 °C for 10 minutes. The ratio of the diastereomers in the crude product by NMR: cis/trans 85:15. The crude product was purified by column chromatography (toluene). Pale yellow crystals (0.13 g, 65%). The two diastereomers were separated by HPLC. 5b trans isomer. mp 125.1 – 125.8 °C. 1H NMR (500 MHz, chloroform-d): 7.21 – 7.18 (1H, m, H-8), 7.09 (1H, dm, J 8.0 Hz, H-6), 6.76 – 6.73 (1H, m, H-7), 6.60 (1H, dm, J 8.0 Hz, H-9), 3.85 (1H, dd, J 9.0 and 6.0 Hz, H-3a), 3.60 (1H, m, Hx-1), 3.59 (1H, m, H-5), 3.42 (1H, m, Hy-1), 2.52 (1H, m, Hx-3), 2.28 (1H, m, Hx-2), 2.18 (1H, m, Hy-3), 2.10 (1H, m, Hy-2), 1.50 (3H, d, J 7.0 Hz, CH3);
13C NMR (125 MHz, chloroform-d):140.9 (C-9a), 129.1 (C-6, 8), 119.5 (C-5a), 117.5 (C-7), 114.3* (C-2’), 113.8* (C-1’), 112.3 (C-9), 56.9 (C-3a), 48.2 (C-1), 40.6 (C-5), 39.1 (C-4), 30.1 (C-3), 23.0 (C-2), 21.7 (CH3). HRMS (ESI+) m/z calcd. for C15H16N3 [M+H]+ 238.1339, found 238.1349.
cis-(±)-6-Methyl-1H-2,3,4,4a,5,6-hexahydropyrido[1,2-a]quinoline-5,5-dicarbonitrile (5c). 33 Following method C1, the title compound was isolated. (1. step) To a mixture of 2-(piperidino)acetophenone (6.55 g, 32.30 mmol) in 40 mL water, malononitrile (2.13 g, 32.30 mmol) was added. The reaction mixture was irradiated at 100 °C for 12 minutes, then (2.
General Papers ARKIVOC 2016 (v) 164-196
Page 181 ©ARKAT-USA, Inc.
step) to the reaction mixture trifluoroacetic acide was added and it was irradiated at 170 °C for 5 min. The ratio of the diastereomers in the crude product by NMR: only cis isomer was formed.
The crude product was crystallized from MeOH (distilled from Na and P2O5, dried over 3 Å molecular sieves). White crystals (4.88 g, 60%). mp 141.0 – 141.6 °C (MeOH). 1H NMR:
(600 MHz, chloroform-d): 7.23 – 7.18 (2H, m, H-7, 9), 6.93 (1H, dm, J 8 Hz, H-10), 6.87 – 6.85 (1H, m, H-8), 4.01-3.95 (1H, m, Hx-1), 3.53 – 3.52 (1H, m, H-6), 3.35 (1H, dm, J 11.5 and 3.0 Hz, H-4a), 2.70 – 2.68 (1H, m, Hy-1), 2.39 – 2.36 (1H, m, Hx-4), 2.02 – 2.00 (1H, m, Hx-3), 1.91 – 1.88 (1H, m, Hx-2), 1.81 – 1.79 (1H, m, Hy-4), 1.77 (3H, d, J 7.0 Hz, CH3), 1.75 – 1.73 (1H, m, Hy-2), 1.47 – 1.46 (1H, m, Hy-3); 13C NMR: (150 MHz, chloroform-d): 144.6 (C-10a), 128.9 (C- 9), 127.0 (C-7), 121.4 (C-6a), 119.6 (C-8), 115.1* (C-1’), 114.0 (C-10), 112.3* (C-2’), 60.2 (C- 4a), 48.1 (C-1), 46.2 (C-5), 40.0 (C-6), 30.0 (C-4), 24.8 (C-2), 22.8 (C-3), 16.6 (CH3). Anal. calcd.
for C16H17N3 (251.33): C, 76.46%; H, 6.82%; N, 16.72%. Found: C, 76.11%; H, 7.01%; N, 16.36%. HRMS (ESI+) m/z calcd. for C16H18N3 [M+H]+ 252.1495, found 252.1505.
Following method C2, the title compound was isolated. The vinyl precursor (0.20 g, 0.80 mmol) was irradiated at 180 °C for 10 minutes. The ratio of the diastereomers in the crude product by NMR: cis/trans 92:8. The crude product was purified by column chromatography (toluene). Pale yellow crystals (0.14 g, 70%). mp 131.3 – 133.1 °C. We were not able to isolate the trans isomer.
(±)-1-Methyl-4-phenyl-1,2,3,4-tetrahydroquinoline-3,3-dicarbonitrile (6a). Following method C2, the title compound was isolated. The vinyl precursor (1.33 g, 4.86 mmol) was irradiated at 180 °C for 1.5 hours. After transferring from the vial, to the reaction mixture ethyl acetate (20 mL) and water (20 mL) were added, then the water phase was extracted with ethyl acetate (2x20 mL). The organic phase was dried over MgSO4 and evaporated under reduced pressure. The crude product was purified by column chromatography (n-hexane/EtOAc 8:1).
Yellow crystals (0.87 g, 67%). mp 120.8-121.4 °C. 1H NMR: (400 MHz, chloroform-d): 7.40-7.38 (3H, m, H-3’, 4’, 5’), 7.29 – 7.22 (3H, m, 2’, 6’, 7), 6.84 – 6.79 (2H, m, H-5, 8), 6.74 – 6.70 (1H, m, H-6), 4.65 (1H, s, H-4), 3.81 (1H, d, J 11.5 Hz, Hx-2), 3.80 (1H, d, J 11.5 Hz, Hy-2), 3.14 (3H, s, NCH3); 13C NMR: (100 MHz, chloroform-d): 144.0 (C-8a), 137.4 (C-1’), 130.3 (C-5), 130.2 (C- 2’, 6’), 129.3 (C-7), 129.0 (C-4’), 128.8 (C-3’, 5’), 118.6 (C-6), 118.4 (C-4a), 114.5 (C-1”), 113.4 (C-2”), 112.4 (C-8), 54.6 (C-2), 51.3 (C-4), 39.7 (CH3), 37.5 (C-3). Anal. calcd. for C18H15N3
(273.33): C, 79.10%; H, 5.53%; N, 15.37%. Found: C, 78.71%; H, 5.52%; N, 15.17%. HRMS:
calcd for C18H16N3 [M+H]+ 274.1339, found 274.1333.
cis-(±)-5-Phenyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4,4-dicarbonitrile (6b cis) and trans-(±)-5-Phenyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4,4-dicarbonitrile (6b trans). Following method B2, the title compound (6b cis) was isolated. To a mixture of malononitrile (1.31 g, 19.89 mmol) in i-PrOH (70 mL), phenyl[2-(pyrrolidin-1- yl)phenyl]methanone (5.00 g, 19.89 mmol) and Ti(O-i-Pr)4 (5.65 g, 19.89 mmol, 5.88 mL) were added. The dark red reaction mixture was stirred at 80 °C for 96 hours. The ratio of the diastereomers in the crude product was determined by NMR: cis/trans 92:8. The crude product was purified by column chromatography (toluene) and washed with diethyl ether to afford the pure cis isomer. Beige crystals (1.36 g, 35%). mp 164.7-167.2 °C. 1H NMR: (600 MHz, chloroform-d):
7.48 – 7.41 (5H, m, H-2’, 3’, 4’, 5’, 6’), 7.25 – 7.20 (1H, m, H-8), 6.71 (1H, dm, J 8.1 Hz, H-6), 4.59 (1H, s, H-5), 4.08 (1H, dd, J 8.3, 6.2 Hz, H-3a), 3.58 (1H, m, Hx-1) 3.52 (1H, m, Hy-1), 2.55 (1H, m, Hx-3), 2.31 (1H, m, Hx-2), 2.28 (1H, m, Hy-3), 2.10 (1H, m, Hy-2); 13C NMR: (150 MHz, chloroform-d): 142.7 (C-9a), 136.2 (C-1’), 129.6 (C-6), 129.3 (C-8), 129.2 (C-2’, 6’), 128.9 (C- 4’), 128.5 (C-3’, 5’), 118.1 (C-5a), 117.3 (C-7), 114.5* (C-2”), 112.6* (C-1”), 112.2 (C-9), 63.7 (C-3a), 53.5 (C-5), 47.9 (C-1), 43.3 (C-4), 30.2 (C-3), 22.7 (C-2). Anal. calcd. for C20H17N3
(299.37): C, 80.24%; H, 5.72%; N, 14.04%. Found: C, 80.40%; H, 5.68%; N, 14.16%. HRMS (ESI+) m/z calcd. for C20H18N3 [M+H]+ 300.1501, found 300.1501.
Following method C2, the title compound (6b trans) was isolated. The vinyl precursor (0.20 g, 0.67 mmol) was irradiated at 150 °C for 30 minutes. The ratio of the diastereomers in the crude product was determined by NMR: cis/trans 82:18. Beige crystals (0.16 g, 82% overall yield of the crude product). The two diastereomers were separated by preparative HPLC (Teknokroma Nucleosil 100 C18 10 µm 25 cm×1 mm; mobile phase composition: A/B 50:50 (A = methanol/water 3:7, B = acetonitrile); flow rate: 3 mL/min). Beige crystals (trans isomer). mp 130.7 – 131.7 °C. 1H NMR: (500 MHz, chloroform-d): 7.38 – 7.11 (5H, m, H-2’, 3’, 4’, 5’, 6’), 7.28 – 7.25 (1H, m, H-8), 7.02 (1H, dm, J 7.5 Hz, H-6), 6.74 – 6.72 (1H, m, H-7), 6.73 (1H, dm, J 6.5 Hz, H-9), 4.74 (1H, s, H-5), 3.77 (1H, dd, J 9.0 and 5.5 Hz, H-3a), 3.73 (1H, m, Hx-1), 3.46 (1H, m, Hy-1), 2.37 (1H, m, Hx-3), 2.28 (1H, m, Hx-2), 2.16 (1H, m, Hy-3), 2.08 (1H, m, Hy-2);
13C NMR: (125 MHz, chloroform-d): 142.2 (C-9a), 137.9 (C-1’), 130.3 (C-2’, 6’), 130.2 (C-6), 129.6 (C-8), 128.7 (C-4’), 128.6 (C-3’, 5’), 117.7 (C-7), 116.7 (C-5a), 114.0 (C-2”), 113.9 (C-1”), 112.2 (C-9), 57.1 (C-3a), 51.1 (C-5), 48.7 (C-1), 39.7 (C-4), 30.0 (C-3), 23.1 (C-2). HRMS (ESI+) m/z calcd. for C20H18N3 [M+H]+ 300.1494, found 300.1503.
cis-(±)-6-Phenyl-1H-2,3,4,4a,5,6-hexahydropyrido[1,2-a]quinoline-5,5-dicarbonitrile (6c cis) and trans-(±)-6-Phenyl-1H-2,3,4,4a,5,6-hexahydropyrido[1,2-a]quinoline-5,5-dicarbonitrile (6c trans). Following method C2, the title compound (6c cis) was isolated. The vinyl precursor (0.20 g, 0.64 mmol) was irradiated at 190 °C for 10 minutes. Beige crystals (0.19 g, 95% overall yield of the crude product). The ratio of the diastereomers in the crude product was determined by NMR: cis/trans 71:29 and by HPLC: CH3CN/H2O 65:35, tR: 19.7 and 18.8 min, 70% and 30%.
The two diastereomers were separated by preparative HPLC (Teknokroma Nucleosil 100 C18 10 µm 25 cm×1 mm; mobile phase composition: A/B 40:60 (A = methanol/water 3:7, B = acetonitrile); flow rate: 3 mL/min). Beige powder (cis isomer). mp 199-202 °C. 1H NMR:
(400 MHz, chloroform-d): 7.44 – 7.43 (5H, m, H-2’, 3’, 4’, 5’, 6’), 7.26 – 7.21 (1H, m, H-9), 6.99 (1H, dm, J 8.5 Hz, H-10), 6.74 – 6.72 (2H, m, H-7, 8), 4.68 (1H, s, H-6), 4.03 (1H, dm, J 11.9 Hz, Hx-1), 3.50 (1H, dd, J 11.5 and 3.4 Hz, H-4a), 2.71 (1H, td, J 12.4 and 3.3 Hz, Hy-1), 2.44 – 2.41 (1H, m, Hx-4), 2.01 – 1.90 (1H, m, Hx-3), 1.89 – 1.81 (1H, m, Hy-4), 1.79 – 1.76 (1H, m, Hx-2), 1.46 – 1.36 (2H, m, Hy-2, 3); 13C NMR: (100 MHz, chloroform-d): 145.9 (C-10a), 136.7 (C-1’), 130.3 (C-3’, 5’), 130.0 (C-7), 129.2 (C-4’), 129.1 (C-9), 128.8 (C-2’, 6’), 120.7 (C-6a), 119.4 (C- 8), 114.3 (C-10), 114.0* (C-2”), 112.9* (C-1”), 60.7 (C-4a), 52.7 (C-6), 48.0 (C-1), 47.2 (C-5), 30.0 (C-4), 24.9 (C-2), 22.4 (C-3). Anal. calcd. for C21H19N3 (313.40): C, 80.48%; H, 6.11%; N,
General Papers ARKIVOC 2016 (v) 164-196
Page 183 ©ARKAT-USA, Inc.
13.41%. Found: C, 80.47%; H, 6.16%; N, 13.19%. HRMS (ESI+) m/z calcd. for C21H20N3 [M+H]+ 314.1652, found 314.1645; and,
Beige crystals (trans isomer). mp 54.6 – 61.9 °C. 1H NMR: (500 MHz, chloroform-d): 7.42 – 7.14 (5H, m, H-2’, 3’, 4’, 5’, 6’), 7.25 (1H, m, H-9), 7.04 (1H, dm, J 8.5 Hz, H-10), 6.89 (1H, dm, J 7.5 Hz, H-7), 6.76 (1H, m, H-8), 4.64 (1H, s, H-6), 4.20 (1H, m, Hx-1), 3.29 (1H, dd, J 11.0 and 2.5 Hz, H-4a), 2.86 (1H, m, Hy-1), 2.14 - 1.42 (6H, m, Hx,y-2, 3, 4); 13C NMR: (125 MHz, chloroform-d): 144.3 (C-10a), 138.0 (C-1’), 130.8 (C-7), 130.6 (C-2’, 6’), 129.5 (C-9), 128.8 (C- 4’), 128.6 (C-3’, 5’), 119.4 (C-8), 118.8 (C-6a), 114.3* (C-2”), 114.2 (C-10), 113.7* (C-1”), 55.7 (C-4a), 49.8 (C-6), 48.8 (C-1), 43.5 (C-5), 28.8 (C-4), 24.5 (C-2), 23.5 (C-3). HRMS (ESI+) m/z calcd. for C21H20N3 [M+H]+ 314.1652, found 314.1657.
General procedure for the radical decyanation (method D). The dinitrile compound (pure diastereomer) (1 eq) was dissolved in dry toluene (80 mL) (distilled from LiAlH4 prior to use) a pale yellow solution was formed. After addition of azobisisobutyronitrile (AIBN) (0.2 eq) and tributyltinhydride (2 eq) at 0 °C, the mixture was stirred for the time indicated below at 80 °C (oil bath). After cooling, an aliquot was taken for the analysis of the diastereomers. To the pale yellow or colorless reaction mixture 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.3 eq) was added and stirred for 30 min. The mixture was filtered through a silica gel plug and the solvent evaporated in vacuum. The crude product was purified as described below.
The ratio of the diastereomers in the crude product was determined by HPLC (Gemini NX C18 RP 25 cm×4.6 mm 5 μm column, CH3CN/H2O 80:20) or 1H NMR.
cis-(±)-1,4-Dimethyl-1,2,3,4-tetrahydroquinoline-3-carbonitrile (9a cis). Following method D, the title compound was isolated. To a mixture of the dinitrile derivative (0.77 g, 3.63 mmol) in 20 mL of toluene, AIBN (0.12 g, 0.73 mmol, 0.2 eq) and tributyltinhydride (2.9 mL, 3.17 g, 10.87 mmol, 2 eq) were added at 0 °C, the reaction mixture was stirred at 90-100 °C for 1.5 hours.
Diastereomeric ratio in the crude product (HPLC): 67:33 cis/trans. Overall yield of the diastereomers is 85%. The diastereomers were separated by preparative HPLC (see Supplementary Material). The determination of the relative configuration in these compounds were not possible, due to overlap of the relevant aliphatic protons. However, the Boc derivative (8a) proved to be cis diastereoisomer, based on NOE interactions and analysis of the vicinal coupling constants. Since under the reaction conditions of Boc formation the configuration of the carbon atoms do not change, the relative configuration in this compound is also cis. White crystals. mp 88.0-89.5 °C.
1H NMR (500 MHz, chloroform-d): 7.15 – 7.14 (1H, m, H-7), 7.06 - 7.04 (1H, m, H-5), 6.73 – 6.71 (1H, m, H-6), 6.64 (1H, dm, J 7.5 Hz, H-8), 3.49 – 3.45 (2H, m, Hx,y-2), 3.22 – 3.20 (2H, m, H-3), 2.95 (3H, s, NCH3), 1.45 (3H, d, J 7.0 Hz, CH3); 13C NMR (125 MHz, chloroform-d):144.9 (C-8a), 128.9 (C-5), 128.8 (C-7), 124.6 (C-4a), 120.1 (CN), 118.0 (C-6), 112.2 (C-8), 49.6 (C-2), 39.6 (NCH3), 33.7 (C-4), 31.7 (C-3), 20.6 (CH3). Anal. calcd (%) for C12H14N2 (186.25): C 77.38;
H 7.58; N 15.04. Found: C 77.33; H 7.67; N 15.14. HRMS (ESI+) m/z calcd. for C12H15N2 [M+H]+ 187.1230, found 187.1225.
cis-cis-(±)-5-Methyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4-carbonitrile
(9b cis-cis) and cis-trans-(±)-5-Methyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4- carbonitrile (9b cis-trans). Following method D, the title compounds were isolated. To a mixture of the dinitrile derivative (cis isomer) (4.04 g, 17.04 mmol) in 70 mL of toluene AIBN (0.56 g, 34.10 mmol, 0.2 eq) and tributyltinhydride (9.18 mL, 34.08 mmol, 2 eq) were added at 0 °C, the reaction mixture was stirred for at 90 °C for 24 hours. Diastereomeric ratio in the crude product (HPLC): 58:42 cis-cis/cis-trans. The two diastereomers were separated by column chromatography (n-hexane/EtOAc 4:1).
(9b cis-cis) Off-white crystals (1.10 g, 31%). mp 136.4-137.4 °C (n-hexane/EtOAc 6.5:1).
1H NMR (500 MHz, chloroform-d): 7.14 – 7.12 (2H, m, H-6, 8), 6.68 – 6.67 (1H, m, H-7), 6.47 (1H, dm, J 8.5 Hz, H-9), 3.73 – 3.70 (1H, m, H-3a), 3.40 – 3.38 (1H, m, Hx-1), 3.34 – 3.33 (1H, m, Hy-1), 3.24 – 3.23 (1H, m, H-5), 3.13 (1H, dd, J 4.5 Hz, H-4), 2.19 - 2.17 (2H, m, Hx-2, 3), 2.00 – 1.97 (2H, m, Hy-2, 3), 1.57 (3H, d, J 7.0 Hz, CH3); 13C NMR (125 MHz, chloroform-d): 143.9 (C-9a), 128.9 (C-8), 127.0 (C-6), 121.9 (C-5a), 118.4 (CN), 116.8 (C-7), 111.4 (C-9), 59.0 (C-3a), 47.9 (C-1), 37.7 (C-4), 34.8 (C-5), 31.3 (C-3), 24.0 (C-2), 18.4 (CH3). Anal. calcd (%) for C14H16N2
(212.29): C 79.21; H 7.60; N 13.20. Found: C 79.35; H 7.64; N 13.24. HRMS (ESI+) m/z calcd.
for C14H17N2 [M+H]+ 213.1386, found 213.1392.
(9b cis-trans) White crystals (0.82 g, 22%). mp 142.8-143.5 °C (n-hexane). 1H NMR (500 MHz, chloroform-d): 7.17 (1H, dm, J 7.5 Hz, H-6), 7.14 - 7.13 (1H, m, H-8), 6.70 - 6.69 (1H, m, H-7), 6.45 (1H, dm, J 9.0 Hz, H-9), 3.63 – 3.62 (1H, m, H-3a), 3.40 -3.39 (1H, m, Hx-1), 3.29 – 3.27 (1H, m, Hy-1), 3.17 – 3.16 (1H, m, H-5), 2.47 – 2.44 (1H, m, Hx-3), 2.35 (1H, dd, J 11.5 and 10.4 Hz, H-4), 2.14 - 2.11 (1H, m, Hx-2), 1.99 -1.97 (1H, m, Hy-2), 1.70 -1.67 (1H, m, Hy-3), 1.58 (3H, d, J 6.5 Hz, CH3); 13C NMR (125 MHz, chloroform-d): 143.9 (C-9a), 128.8 (C-8), 127.1 (C- 6), 123.3 (C-5a), 121.3 (CN), 117.0 (C-7), 111.6 (C-9), 59.8 (C-3a), 48.4 (C-1), 40.3 (C-4), 35.9 (C-5), 33.1 (C-3), 23.8 (C-2), 18.9 (CH3). Anal. calcd (%) for C14H16N2 (212.29): C 79.21; H 7.60;
N 13.20. Found: C 79.45; H 7.64; N 13.25. HRMS (ESI+) m/z calcd. for C14H17N2 [M+H]+ 213.1386, found 213.1390.
cis-cis-(±)-6-Methyl-1H-2,3,4,4a,5,6-hexahydropyrido[1,2-a]quinoline-5-carbonitrile (9c cis-cis) and cis-trans-(±)-6-Methyl-1H-2,3,4,4a,5,6-hexahydropyrido[1,2-a]quinoline-5- carbonitrile (9c cis-trans). Following method D, the title compounds were isolated. To a mixture of the dinitrile derivative (cis isomer) (4.86 g, 19.37 mmol) in 80 mL of toluene AIBN (0.2 eq) and tributyltinhydride (2 eq) were added at 0 °C, the reaction mixture was stirred for 24 hours.
Diastereomeric product ratio in the crude product (HPLC): 72:28 cis-cis/cis-trans. The two diastereomers were separated by column chromatography (n-hexane/EtOAc 4:1).
(9c cis-cis) White crystals (2.50 g, 55%). mp 181.6-183.0 °C (n-hexane/EtOAc 1:1.1). 1H NMR:
(500 MHz, chloroform-d): 7.15 – 7.11 (2H, m, H-7, 9), 6.89-6.87 (1H, dm, J 10.5 Hz, H-10), 6.76 – 6.74 (1H, m, H-8), 3.98 (1H, dm, J 15.5 Hz, Hx-1), 3.30 – 3.25 (1H, m, H-6), 3.13 – 3.09 (1H, m, H-4a), 3.04 - 3.02 (1H, m, H-5), 2.65 – 2.59 (1H, m, Hy-1), 1.91 – 1.89 (1H, m, Hx-3), 1.84 – 1.71 (4H, m, Hx,y-2, 4), 1.52 (3H, d, J 12 Hz, CH3), 1.41 - 1.39 (1H, m, Hy-3); 13C NMR:
(125 MHz, chloroform-d): 146.5 (C-10a), 128.6* (C-9), 127.5* (C-7), 125.4 (C-6a), 119.3 (C-8),
General Papers ARKIVOC 2016 (v) 164-196
Page 185 ©ARKAT-USA, Inc.
119.0 (CN), 114.4 (C-10), 57.9 (C-4a), 48.9 (C-1), 41.7 (C-5), 34.1 (C-6), 32.2 (C-4), 26.1 (C-2), 24.2 (C-3), 18.6 (CH3). Anal. calcd (%) for C15H18N2 (226.32): C 79.61; H 8.02; N 12.38. Found:
C 79.75; H 8.12; N 11.98. HRMS (ESI+) m/z calcd. for C15H19N2 [M+H]+ 227.1543, found 227.1538.
(9c cis-trans) White crystals (0.768 g, 17%). mp 144.0 – 144.5 °C. 1H NMR: (500 MHz, chloroform-d): 7.16 (1H, m, H-7), 7.16-7.13 (1H, m, H-9), 6.85 (1H, dm, J 8.5 Hz, H-10), 6.80- 6.77 (1H, m, H-8), 3.89 (1H, dm, J 8.4 Hz, Hx-1), 3.15 (1H, m, H-6), 3.10 (1H, m, H-4a), 2.67 (1H, m, Hy-1), 2.58 (1H, m, H-5), 2.28 (1H, m, Hx-4), 1.90 (1H, m, Hx-3), 1.82 (1H, m, Hx-2), 1.62 (1H, m, Hy-2), 1.54 (3H, d, J 4.8 Hz, CH3), 1.44 (1H, m, Hy-3), 1.38 (1H, m, Hy-4); 13C NMR:
(125 MHz, chloroform-d): 146.4 (C-10a), 128.5 (C-9), 127.4 (C-7), 126.6 (C-6a), 121.1 (CN), 119.3 (C-8), 114.2 (C-10), 58.3 (C-4a), 49.0 (C-1), 42.9 (C-5), 35.2 (C-6), 32.9 (C-4), 25.9 (C-2), 24.2 (C-3), 19.7 (CH3). Anal. calcd (%) for C15H18N2 (226.32): C 79.61; H 8.02; N 12.38. Found:
C 79.29; H 7.99; N 12.00. HRMS (ESI+) m/z calcd. for C15H19N2 [M+H]+ 227.1543, found 227.1532.
cis-(±)-1-Methyl-4-phenyl-1,2,3,4-tetrahydroquinoline-3-carbonitrile (10a cis). Following method D, the title compound was isolated. To a mixture of the dinitrile derivative (1.03 g, 3.73 mmol) in 25 mL of toluene AIBN (0.11 g, 0.70 mmol, 0.2 eq) and tributyltinhydride (3.0 mL, 3.26 g, 11.20 mmol, 3 eq) were added at 0 °C, the reaction mixture was stirred at 90 °C for 2 hours.
Only cis isomer was formed as determined form the crude product by NMR. After the crystallisation white, bright crystals were obtained (0.28 g, 30%). mp 117.3 – 120.0 °C. 1H NMR:
(400 MHz, chloroform-d): 7.39 – 7.08 (6H, m, H-7, 2’, 3’, 4’, 5’, 6’), 6.90 – 6.82 (1H, dm, J 7.3 Hz, H-5), 6.78 – 6.70 (1H, dm, J 8.1 Hz, H-8), 6.69 – 6.61 (1H, m, H-6), 4.45 – 4.37 (1H, m, H-4), 3.51 – 3.31 (3H, m, Hx,y-2, H-3), 3.02 (3H, s, CH3); 13C NMR: (100 MHz, chloroform-d):
145.1 (C-8a), 140.6 (C-1’), 130.0 (C-5), 129.7 (C-2’, 6’), 128.6 (C-7), 128.4 (C-3’, 5’), 127.6 (C- 4’), 121.0 (C-4a), 119.0 (CN), 117.3 (C-6), 111.5 (C-8), 49.0 (C-2), 45.0 (C-4), 39.1 (CH3), 31.9 (C-3). Anal. calcd. for C17H16N2 (248.33): C, 82.22%; H, 6.49%; N, 11.28%. Found: C, 81.89%;
H, 6.50%; N, 11.11%. HRMS (ESI+) m/z calcd. for C17H17N2 [M+H]+ 249.1386, found 249.1382.
cis-cis-(±)-5-Phenyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4-carbonitrile
(10b cis-cis) and cis-trans-(±)-5-Phenyl-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinoline-4- carbonitrile (10b cis-trans). Following method D, the title compound (8b cis-trans) was isolated.
To a mixture of the dinitrile derivative (cis isomer) (2.28 g, 7.61 mmol) in 50 mL of toluene AIBN (0.25 g, 1.52 mmol, 0.2 eq) and tributyltinhydride (6.1 mL, 6.64 g, 22.82 mmol, 3 eq) were added at 0 °C, the reaction mixture was stirred at 90 °C for 8 hours. Diastereomeric product ratio in the crude product (NMR): cis-cis/cis-trans 50:50. The cis-trans diastereomer was isolated by column chromatography (n-hexane/EtOAc 8:1). White crystals (0.34 g, 16%). mp 205.3-205.6 °C.
1H NMR: (400 MHz, chloroform-d): 7.39 – 7.30 (3H, m, H-3’, 4’, 5’), 7.26 – 7.21 (2H, m, H-2’, 6’), 7.13 – 7.10 (1H, m, H-8), 6.58 – 6.49 (3H, m, H-6, 7, 9), 4.28 (1H, d, J 11.8 Hz, H-5), 3.77 – 3.70 (1H, m, H-3a), 3.47 – 3.35 (2H, m, Hx,y-1), 2.81 (1H, dd, J 11.8 and 10.4 Hz, H-4), 2.50 – 2.44 (1H, m, Hx-3), 2.19 – 2.15 (1H, m, Hx-2), 2.07 – 1.99 (1H, m, Hy-2), 1.79 – 1.73 (1H, m, Hy- 3); 13C NMR: (100 MHz, chloroform-d): 143.7 (C-9a), 141.2 (C-1’), 129.7 (C-6), 129.03 (C-3’,
5’), 128.96 (C-2’, 6’), 128.3 (C-8), 127.8 (C-4’), 122.3 (C-5a), 119.8 (CN), 116.4 (C-7), 111.1 (C- 9), 48.5 (C-5), 47.6 (C-1), 40.7 (C-4), 32.3 (C-3), 23.0 (C-2). HRMS (ESI+) m/z calcd. for C19H19N2 [M+H]+ 275.1543, found 275.1541.
The cis-cis isomer was isolated by HPLC. Beige powder. mp 175.0 – 178.5 °C. 1H NMR:
(500 MHz, chloroform-d): 7.44 – 7.31 (5H, m, H-2’, 3’, 4’, 5’, 6’), 7.17 – 7.14 (1H, m, H-8), 6.72 (1H, dm, J 2.7 Hz, H-6), 6.57 – 6.56 (1H, m, H-7), 6.56 – 6.54 (1H, m, H-9), 4.45 (1H, d, J 5 Hz, H-5), 3.90 – 3.87 (1H, m, H-3a), 3.50 – 3.39 (2H, m, Hx,y-1), 3.29 (1H, dd, J 4.9 and 2.7 Hz, H-4), 2.22 – 2.20 (2H, m, Hx-2, 3), 2.09 – 2.07 (1H, m, Hy-3), 2.05 – 2.00 (1H, m, Hy-2); 13C NMR:
(125 MHz, chloroform-d): 144.0 (C- 9a), 140.2 (C-1’), 129.5 (C-2’, 6’), 129.1 (C-6), 128.7 (C-4’), 128.5 (C-8), 127.9 (C-3’, 5’), 119.9 (C-5a), 118.2 (CN), 116.0 (C-7), 111.2 (C-9), 58.9 (C-3a), 47.6 (C-5), 47.1 (C-1), 38.2 (C-4), 30.5 (C-3), 23.2 (C-2). HRMS (ESI+) m/z calcd. for C19H19N2 [M+H]+ 275.1543, found 275.1545.
cis-cis-(±)-6-Phenyl-1H-2,3,4,4a,5,6-hexahydropyrido[1,2-a]quinoline-5-carbonitrile
(10c cis-cis). Following method D, the title compound was isolated. To a mixture of the dinitrile derivative (cis isomer) (3.00 g, 9.57 mmol) in 60 mL of toluene AIBN (0.31 g, 1.91 mmol, 0.2 eq) and tributyltinhydride (7.7 mL, 8.36 g, 28.72 mmol, 3 eq) were added at 0 °C, the reaction mixture was stirred at 90 °C for 3 hours. Diastereomeric product ratio in the crude product by NMR: cis- cis/cis-trans 95:5. The crude product was washed with diethyl ether to obtain the pure cis-cis isomer product. The cis-trans isomer was not isolated. White crystals (1.92 g, 71%). mp 162.1- 167.3 °C. 1H NMR: (400 MHz, chloroform-d): 7.40 – 7.29 (5H, m, H-2’, 3’, 4’, 5’, 6’), 7.20 – 7.11 (1H, m, H-9), 6.99 – 6.90 (1H, m, H-10), 6.79 – 6.71 (1H, m, H-7), 6.69 – 6.59 (1H, m, H-8), 4.51 (1H, d, J 5.5 Hz, H-6), 4.05 – 3.99 (1H, m, Hx-1), 3.31 – 3.25 (1H, m, H-4a), 3.17 (1H, dd, J 5.5 and 2.0 Hz, H-5), 2.67 – 2.58 (1H, m, Hy-1), 1.94 – 1.70 (5H, m, Hx,y-2,3, Hx-4), 1.45 – 1.33 (1H, m, Hy-4); 13C NMR: (100 MHz, chloroform-d): 147.0 (C-10a), 140.2 (C-1’), 129.7 (C-7), 129.6 (C-2’, 6’), 128.6 (C-3’, 5’), 128.3 (C-9), 127.8 (C-4’), 123.2 (C-6a), 118.6 (CN), 118.4 (C-8), 113.9 (C-10), 57.7 (C-4a), 48.1 (C-1), 46.9 (C-6), 42.2 (C-5), 31.5 (C-2), 25.5 (C-3), 23.2 (C-4).
Anal. calcd. for C20H20N2 (288.39): C, 83.30%; H, 6.99%; N, 9.71%. Found: C, 82.84%; H, 6.97%;
N, 9.49%. HRMS (ESI+) m/z calcd. for C20H20N2 [M+Na]+ 311.1519, found 311.1527.
General procedure for the reduction of mononitrile derivatives (method E). The precursor mononitrile compound (1 eq) was dissolved in dry MeOH (distilled from Mg/I2 prior use) (50 mL).
At 0 °C di-tert-butyl dicarbonate (Boc2O) (2 eq) and NiCl2×6H2O (0.4 eq) were added under argon atmosphere. Subsequently NaBH4 (10 eq) was added in small portions (within 45-55 min), keeping the temperature below 5 °C (the reaction is strongly effervescent, while adding NaBH4 and the reaction mixture turns black). The mixture was allowed to warm up and was stirred for 1 hour at ambient temperature, followed by adding of 25 % aq. NH3 (100 mL) and the mixture was stirred for a further 1 h. After evaporation to dryness, the residue was taken up in 300 mL of H2O/EtOAc 2:1 and the phases were separated. The inorganic layer was extracted with EtOAc (4x50 mL) and washed with saturated NaHCO3 solution (2×50 mL). The organic phase was dried over MgSO4, filtered, evaporated to dryness and purified by column chromatography.
![Table 5. Various measured SSAO biological activities [inhibition (IC 50 ) and substrate binding constant (K m )] of the prepared compounds 11a-c and 12a-c as well as the references 2-BEA and 4-PBA Compound IC 50 (µM) K m (µM) 2-BEA a 0.56 ± 0.12 4](https://thumb-eu.123doks.com/thumbv2/9dokorg/1385112.114521/9.892.105.478.425.651/various-biological-activities-inhibition-substrate-compounds-references-compound.webp)
