Enantioszelektiv szimulált mozgóréteges
folyadékkromatográfia gyógyszeripari alkalmazásának vizsgálata
Doktori (PhD) értekezés
Készítette: Gál Gábor
Témavezetők: Dr. Szánya Tibor, egyetemi docens Dr. Argyelán János, egyetemi docens
Pannon Egyetem
Vegyészmérnöki Tudományok és Anyagtudományok Doktori Iskola Vegyipari Műveleti Intézeti Tanszék
Veszprém, 2010
Enantioszelektiv szimulált mozgóréteges folyadékkromatográfia gyógyszeripari alkalmazásának vizsgálata
Értekezés doktori (PhD) fokozat elnyerése érdekében
Írta:
Gál Gábor
Készült a Pannon Egyetem Vegyészmérnöki Tudományok és Anyagtudományok Doktori iskolája keretében
Témavezető: Dr. Szánya Tibor, egyetemi docens Dr. Argyelán János, egyetemi docens
Elfogadásra javaslom (igen / nem) …………...……….
(aláírás)
Elfogadásra javaslom (igen / nem) …………...……….
(aláírás)
A jelölt a doktori szigorlaton ...%-ot ért el,
Veszprém, …………...……….
a Szigorlati Bizottság Elnöke
Az értekezést bírálóként elfogadásra javaslom:
Bíráló neve: …...…... igen /nem
……….
(aláírás) Bíráló neve: …...…... igen /nem
……….
(aláírás)
A jelölt az értekezés nyilvános vitáján …...%-ot ért el.
Veszprém, ……….
a Bíráló Bizottság elnöke A doktori (PhD) oklevél minősítése…...
………
Az EDHT elnöke
Tartalomjegyzék
Kivonat...6
Abstract ...7
Auszug ...8
I. Bevezetés ...9
II. SZAKIRODALMI ÖSSZEFOGLALÓ ...12
1. Enantiomerek ...13
2. Enantiomerek elválasztási műveletei...14
2.1. Enantiomerek elválasztása kristályosítással ...17
2.1.1. Kristályosító berendezések ...17
2.1.2. Frakcionált kristályosítással kombinált szimulált mozgóréteges folyadékkromatográfia ...17
2.1.3. Kromatográfia és kristályosítás összekapcsolása...20
2.1.4. Kinetikus rezolválás oltásos kristályosítással konglomerátum típusú racém rendszer esetén ...21
2.2. Kromatográfiás enantiomer elválasztás ...24
2.2.1. Nagyhatékonyságú folyadékkromatográfia ...24
2.2.2. Kromatográfiás töltetek ...26
2.2.3. Adszorpciós izotermák ...29
2.2.4. Preparatív folyadékkromatográfia ...32
2.2.5. A frontális adszorpció matematikai leírása...32
2.2.6. Az enantiomerek elválasztása kromatográfiás módszerrel ...38
3. Mozgóréteges kromatográfia ...40
3.1. Valódi mozgóréteges kromatográfia...40
3.2. A szimulált mozgóréteges (Simulated Moving Bed, SMB) folyadék- kromatográfia...42
3.3. Az SMB elméleti ciklusa ...44
3.4. Elméleti analízis lineáris adszorpciós izotermák esetén ...47
3.5. Morbidelli-féle paraméterek meghatározása...48
3.6. Elméleti analízis nem lineáris adszorpciós izotermák esetén ...50
4. Enantiomerek SMB berendezéssel történő elválasztása ...53
4.1. A tervezési paraméterek rögzítése ...53
4.2. A királis állófázis és az eluensrendszer kiválasztása, az oldódási tulajdonságok és a szelektivitás vizsgálata...53
4.3. Az SMB berendezés állandósult állapotának meghatározására szolgáló szimulációs szoftver kiválasztása, a műveleti feltételek optimalizálása...55
III. KÍSÉRLETI RÉSZ ...59
5. Célkitűzés...59
6. Az SS és RS izomerek analitikai HPLC elválasztásának vizsgálata BÉ elegyből ...61
7. Az 4S6S és 4R6S izomerek analitikai HPLC elválasztásának vizsgálata AC elegyből ...63
8. Az S és R izomerek elválasztása az EÉ racém elegyből...64
8.1. A királis kromatográfiás töltetek és eluensek kiválasztása analitikai HPLC-vel ...64
8.2. Töltetvizsgálatok...65
8.3. Adszorpciós egyensúly, elméleti tányérszám (NTP), elméleti tányér-magasság (HETP), szelektivitás, nyomásesés és a műveleti paraméterek meghatározása 66 8.4. Adszorpciós egyensúlyi izoterma adatok meghatározása k’ alapján ...69
8.5. Adszorpciós egyensúlyi izoterma adatok meghatározása frontális adszorpció- deszorpció alapján...70
8.5.1. Mérési körülmények ...70
8.5.2. RG OD FR EÉ 11 mérés...73
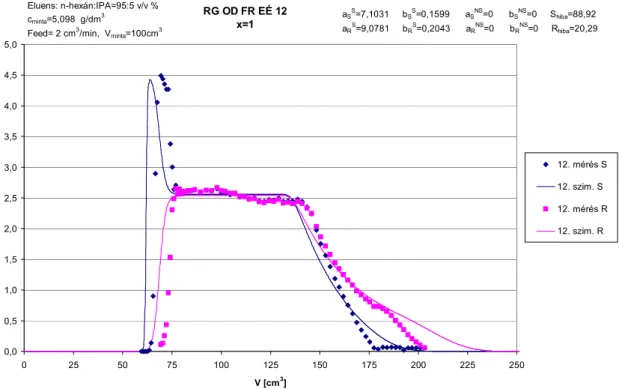
8.5.3. RG OD FR EÉ 12 mérés...74
8.5.4. RG OD FR EÉ 13 mérés...75
8.5.5. Mérési eredmények...76
8.6. Adszorpciós egyensúlyi izoterma adatok meghatározása „hodográf” módszerrel (hullámelmélet) ...78
8.6.1. A számítások eredményeinek összefoglalása ...80
8.7. Adszorpciós egyensúlyi adatok meghatározása SOLVER-EXCEL programmal (bi-Langmuir izoterma, lineáris nem szelektív taggal) ...81
8.7.1. A SOLVER-EXCEL optimalizáló program alkalmazása abszolút hiba négyzetes célfüggvény minimalizáló programmal...82
8.7.2. A SOLVER-EXCEL optimalizáló program alkalmazása relatív hiba négyzetes célfüggvény minimalizáló programmal...83
8.7.3. A SOLVER-EXCEL optimalizáló programmal nyert eredmények összefoglalása...83
8.8. Adszorpciós egyensúlyi izoterma adatok meghatározása tömegmérleg hiba módszerrel...84
8.8.1. A számítógépes programok kibővítése ...84
8.8.2. χ2 tömegmérleg hiba módszer (bi-Langmuir izoterma, lineáris nem szelektív taggal ...87
8.8.3. Optimális szelektív, nem szelektív borítottság meghatározása ...89
8.9. Adszorpciós egyensúlyi izotermák adatainak meghatározása inverz módszerrel ...91
9. SMB művelethez csatolt kristályosítás részletesebb vizsgálata ...95
9.1. Fázisdiagramok ...97
9.2. Oldhatósági adatok meghatározása...99
9.3. Az oldhatósági kísérletek eredményeinek ábrázolása háromszög diagramon 101 9.3.1. Oldhatósági modell számítási eredmények...102
9.4. A metastabil tartományok meghatározása n-hexánban hűtéses kristályosítással S enantiomerben dús SR racém esetén ...103
9.4.1. A kísérleti berendezés ...103
9.4.2. A KR4 mérés eredményei ...104
9.4.3. A KR3 mérés eredményei ...106
9.4.4. A KR1 mérés eredményei ...107
9.4.5. A KR2 mérés eredményei ...108
9.4.6. A kísérleti eredmények összefoglalása ...109
10. SMB művelet és csatolt kristályosítás vizsgálata ...112
10.1. A számításokhoz felhasznált adszorpciós egyensúlyi izoterma adatok összefoglalása ...112
10.1.1. Adszorpciós izoterma adatok hatása a Morbidelli háromszögre ...113
10.2. Az SMB művelet és a kristályosítás összekapcsolása ...114
10.2.1. Az SMB szimuláció rögzített paraméterei...115
10.2.2. A szimulációk bemenő adatai ...116
10.2.3. A szimulációs eredmények kiértékelése ...118
10.2.4. A raffinátum áramok kristályosításos tisztítása ...121
10.2.5. A kristályosítás optimalizálása ...123
11. SMB mérések...125
11.1. Az SMB készülék ...125
11.2. A RG OD SMB EÉ 01-04 mérések eredményei...125
11.3. A RG OD SMB EÉ 06 mérés eredményei...126
11.4. A RG OD SMB EÉ 07 mérés eredményei...128
11.5. A RG OD SMB EÉ 08 mérés eredményei...130
ÖSSZEFOGLALÁS ...135
Irodalomjegyzék ...139
Tézisek ...144
Theses ...146
MELLÉKLET ...148
Kivonat
Napjainkban a regisztrált gyógyszeripari termékek majdnem fele rendelkezik királis struktúrával, ezért a nagy tisztaságban való előállítás követelménye miatt a figyelem középpontjába kerültek. Gyógyszerészeti szempontból csak az egyik optikai izomer éri el a kívánt hatást, míg a másik gyógyászati szempontból inaktív, esetleg toxikus. Ebből adódik az enantiomerek optikai tisztaságának követelménye. Mivel az enantiomerek hasonló fizikai-kémiai tulajdonságokkal rendelkeznek és csak optikailag aktív környezetben mutatnak eltérő viselkedést, az elválasztásuk lehetetlen királis interakció nélkül.
Az SMB elválasztás előnye a többi enantiomer-szétválasztási technikával szemben, hogy folyamatos a művelet, az oszlopok kihasználtsága teljes, a termelékenység és a kihozatal magasabb, az eluens felhasználás alacsonyabb, mint a többi kromatográfiás módszeré.
A doktori értekezés a Richter Gedeon Vegyészeti Gyár Nyrt. által előállított kétkomponensű optikai izomer keverékek elválasztását vizsgálja. A szerző a királis racém keverékek elválasztásának megvalósíthatóságát vizsgálta laboratóriumi méretű SMB készülékkel. A cél az értékes optikai izomer 99 m/m %-nál nagyobb tisztaságban történő kinyerése 90 % feletti kihozatallal.
Az SMB méréseket előkészítő szimulációk pontosságát és hatékonyságát a rendelkezésre álló szoftverek továbbfejlesztésével sikerült növelni. Az eredeti szoftvert sikerült kiterjeszteni királisan szelektív és nem-szelektív adszorbensekre is.
Az SMB berendezéshez kapcsolt kristályosítóval tovább sikerült növelni a művelet hatékonyságát.
Kulcsszavak: gyógyszeripari enantiomerek, királis kromatográfiás töltet, enantioszeparáció, preparatív folyadékkromatográfia, szimulált mozgóréteges folyadékkromatográfia (SMB-LC), kristályosítás
Study of enantioselective simulated moving bed liquid chromatographic process application for the pharmaceutical industry
Abstract
Nowadays almost half of the registered pharmaceutical products have chiral structures, accordingly they are of importance in pharmaceutical industry.
Pharmacologically, most often only one optical isomer has proper activity, while the other one is inactive, and possibly toxic. As a consequence, the optical purity of enantiomers has significant importance. As the enantiomers have the same physicochemical properties and they show different characters only in optically active surroundings, the separation of them is unachievable without chiral interactions.
The advantage of the SMB method is that the procedure can be made continuously, the columns are completely utilized, the productivity and yield is higher, and the consumption of the eluent is lower compared to the batch chromatographic process.
The author investigated the separation of chiral racemic mixture - pharmaceutical enantiomers - on a laboratory scale SMB equipment.
In the Ph.D. thesis binary optical isomer mixtures produced by Gedeon Richter Plc.
were studied. The author investigated the feasibility of the separation of a chiral racemic mixture on a laboratory scale SMB equipment. The object was to produce the valuable optical isomer in higher than 99 % m/m purity besides higher than 90 % yield.
The simulation programs were improved, thereby the precision and the efficiency of the calculations were raised. Additionally, new isotherm equations were implemented the computer simulation programs.
The efficiency of the enantiomer separation can be increased by coupled crystallization of the enriched extract and raffinate fractions.
Keywords: pharmaceutical enantiomers, chiral chromatographic packing, enantioseparation, preparative liquid chromatography, simulated moving bed chromatography, crystallization
Die Prüfung der Verwendung der enantioselektiven simulierten Flüssigkeitschromatographie mit bewegender Schicht in der
Pharmaindustrie
Auszug
Heutzutage verfügt fast die Hälfte der registrierten pharmazeutischen Produkte über die chiralische Struktur, deswegen gerieten sie in den Mittelpunkt der Beachtung wegen der Anforderung der Herstellung in großer Reinheit. Aus dem pharmazeutischen Gesichtspunkt erreicht nur eines von den optischen Isomeren die gewünschte Wirkung, während das anderes aus medizinischem Gesichtspunkte inaktiv, eventuell toxisch ist. Die Anforderung der optischen Reinheit der Enantiomeren ergibt sich daraus. Da die Enantiomeren ähnliche physikalische-chemische Eigenschaften haben und sie zeigen nur in optischer aktiver Umwelt ein verschiedenes Verhalten, ihre Trennung ist ohne chiralische Interaktion unmöglich.
Der Vorteil der Trennung durch SMB ist gegenüber der anderen Trennungstechnik der Enantiomeren, dass der Vorgang kontinuierlich ist, die Auslastung der Säulen günstig ist, die Produktivität und das Herausholen höher sind, die Verwendung des Eluens niedriger als die andere chromatographische Technik ist.
Die Doktorarbeit prüft die Trennung der von Gedeon Richter AG hergestellten Mischungen des optischen Isomers mit zwei Komponenten. Der Autor prüfte die Durchführbarkeit der Trennung der chiralischen Racemat-Mischungen mit einem SMB Apparat in Laborgröße. Das Ziel war, für uns wertvolle optisches Isomer in größere als 99 % m/m Sauberkeit zu gewinnen, mit höher als 90 % Ausbeute.
Ich erhöhte mit dem Fortentwickeln der zur Verfügung stehenden Software die Genauigkeit und die Effektivität der Simulationen, die die SMB-Messungen vorbereiten.
Ich erweiterte die ursprüngliche Software auf chiralisch selektiver und nicht-selektiver Adsorbent.
Ich habe den SMB Apparat mit Kristallisation kombiniert, so konnte ich die Effektivität des Verfahrens weiter erhöhen.
Schlüsselwörter: pharmazeutische Enantiomeren, chiralische chromatographische Packung, Enantioseparationen, Preparative Flüssigkeitschromatographie, simulierte Flüssigkeits-chromatographie mit bewegender Schicht, Kristallisation
I. Bevezetés
A kiralitás mindenütt jelen van a természetben, a mikroszkopikus méretű molekuláktól a makroszkópikus szinten élő szervezetekig. Molekuláris szinten, a királis vegyületeknek, mint például a tetraéderes szénatomhoz kötött négy különböző funkciós csoportnak van két egymással nem tükörképi formája. Ezek az úgynevezett enantiomer formák. Több az élő szervezetekben jelenlévő aminosav királis, kölcsönhatásuk más királis molekulákkal sztereospecifikus. Ez különösen fontos a racemátként számon tartott szintetikus gyógyszerek esetén (50:50 %-os keverék a két enantiomer formából), mivel az enantiomerek kölcsönhatása a biológiai receptorokkal különböző hatásokat tud előidézni.
A legtöbb esetben, az egyik enantiomer adja a kívánt gyógyászati hatást, míg a másik a semleges, ártalmatlan. Például azonban a talidomid racém esetén az egyik enantiomer a gyógyszeripari céltermék, míg a másik toxikus méreg [1]. Bár a gyógyszerek sztereospecifikus hatása régóta ismert volt, hosszú ideig nem tanulmányozták, egészen az 1990-es évek elejéig, amíg nem írták elő, hogy tiszta enantiomereket gyártsanak [2]. Ezek az új szabályok már jelentős hatást gyakoroltak a gyógyszeriparra. Az enantiomer gyógyszerek piacának százalékos aránya az összes gyógyszer kereskedelmében az 1990 évi 10 %-ról mintegy 37 %-ra nőtt 2005-ben, amikor az enantiomer gyógyszeripari termékek értékesítése 225 milliárd USD volt [3, 4].
A Richter Gedeon Vegyészeti Gyár Nyrt. a Pannon Egyetem Vegyészmérnöki Intézet Vegyipari Műveleti Intézeti Tanszékét illetve a Kooperációs Kutatási Központot kérte fel a ”Királis vegyületek elválasztására alkalmas folyadékkromatográf (SMB-LC) fejlesztése” nevezetű kutatási téma kidolgozására 2004 és 2007 között. Ezen Ph.D.
dolgozat a fenti K+F feladathoz kapcsolódik.
A gyógyszeriparban, ezen belül a hazai gyógyszer gyártóknál az elmúlt években előtérbe került a közeli fizikai, illetve fizikai-kémiai tulajdonságú anyagok (izomerek, optikai izomerek, biomolekulák) nagytisztaságú előállítása, melyek egyik ígéretes vegyipari műveleti megoldása a preparatív folyadékkromatográfia. A hazai gyógyszeripar tervei között szerepel a preparatív folyadékkromatográfia egyik legkorszerűbb módszerének, a szimulált mozgóréteges (Simulated Moving Bed, SMB) folyadékromatográfiás műveletnek laboratóriumi illetve ipari méretű megvalósítása.
A Ph.D. téma kidolgozása során a Richter Gedeon Vegyészeti Gyár Nyrt. egyik komponensszétválasztási feladatának megoldása kapcsán egy gyógyszer hatóanyag intermedierjeinek - királis észter optikai izomerek (S és R, illetve SS és RS, valamint 4R6S és 4S6S) - elválasztásával foglalkoztam enantioszelektív kromatográfiás töltet és szimulált mozgóréteges preparatív folyadékkromatográfiás művelet illetve készülék alkalmazásával.
Áttekintettem a téma szakirodalmát (Science Direct, Web of Science, EISZ, Chem.
Abstr. Quick Search), mely az általam szétválasztandó molekulák tekintetében rendkívül hiányosnak mutatkozott. Megjegyezném, hogy egyébként az enantiomer elválasztás szakirodalma igen gazdag.
Így a fentieknek megfelelően kísérletekkel kellet meghatároznom az optikai izomer keverékek és a tiszta komponensek oldhatósági adatait különböző, a preparatív kromatográfiában alkalmazott oldószerekben, majd meg kellett határozni különböző enantioszelektív kromatográfiás tölteteken [5] az adszorpciós egyensúlyi adatokat és egyéb vegyipari műveleti jellemzőket (k’, Langmuir állandók, bi-Langmuir állandók, izoterma adatok, NTP, HETP, porozitás (ε), halmazsűrűség (ρH), BET felület adatok, szemcseméret (dp), stb.).
Ezt követően elúciós, frontális adszorpciós-deszorpciós mérésekkel készítettem elő az SMB művelet paramétereinek meghatározását.
A Vegyipari Műveleti Intézeti Tanszéken rendelkezésre álló négy oszlopos, 1:1:1:1 oszlop konfigurációjú, nyitott eluens körű SMB készüléket jelentősen átalakítottam (DAICEL enantioszelektív kromatográfiás töltet, polarimetriás- és UV-detektor, digitális mérlegek, számítógépes adatgyűjtés megvalósítása), így lehetőségem nyílt a laboratóriumi kísérletek során a fontosabb műveletjellemzők enantiomer elválasztásra gyakorolt hatásainak részletes vizsgálatára. Az analitikai kémiai vizsgálatokat GILSON illetve MERCK Hitachi La Chrom HPLC készülékekkel illetve IBZ Chiralyser polariméterrel, ATLAS Betograph, illetve JEOL műszerekkel végeztem.
Az SMB művelet számítására alkalmas számítógépi programot átírtam a művelet matematikai modelljének módosításával (az adszorbens töltet felületén királisan szelektív és nem szelektív felületi csoportokat vettem figyelembe), kompetitív bi-Langmuir adszorpciós izotermát alkalmazva, mely jelentősen javította az SMB kísérletek tervezését, a kísérleti paraméterek beállítását. A fenti matematikai modell alapján készítettem a KROM-Chir, SMB-Chir programokat DELPHI számítógépi nyelven illetve az Excel táblázatkezelő program SOLVER bővítményének alkalmazásával.
1. kép A kislaboratóriumi méretű SMB készülék
A számítógépes szimulációkkal előzetesen meghatározott kísérleti paraméterek felhasználásával laboratóriumi SMB kísérleteket végeztem az 1. képen látható készülékkel, mellyel sikerült mindhárom optikai izomer keverék esetében az előírt termék tisztaságot és kihozatalt elérnem.
Az előírt termék tisztaságok (>99 m/m %) és kihozatalok (>90 %) mellett számítógépi szimulációkkal meghatároztam a termelékenység és oldószer fajlagos maximális illetve minimális értékeit.
Ennek alapján javaslatot tettem a Richter Gedeon Vegyészeti Gyár Nyrt. részére a gyógyszeripari hatóanyag kémiai szintézisen belül az enantioszelektív kromatográfiás művelet helyének kijelölésére.
A Ph.D. téma kidolgozásának befejező részében az SMB művelethez csatolt bepárlásos-hűtéses kristályosítással foglalkoztam. Ehhez kapcsolódva először meghatároztam az S és R valamint a racém SR optikai izomerek oldhatósági adatait n-hexánban és megállapítottam, hogy az oldhatósági diagram kristályosítási szempontból konglomerátum típusú. A kristályosítási anyalúg recirkulációjával jelentősen növeltem a szétválasztási művelet gazdaságosságát, a termelékenységet, miközben az eluens felhasználást csökkentettem.
II. SZAKIRODALMI ÖSSZEFOGLALÓ
A Ph.D. dolgozat kidolgozása során három optikai izomer keverék (S és R, valamint SS és RS illetve 4S6R és 4R6S) elválasztását vizsgáltam királis töltetet alkalmazó kislaboratóriumi méretű SMB-LC készülékkel. A Richter Gedeon Vegyészeti Gyár Nyrt.
által átadott vegyületek (királis észterek) és az előállítandó gyógyszer hatóanyag kémiai összetételét, szerkezeti képletét ismerem, de a titkossági szerződésben leírtak miatt nem hozhatom nyilvánosságra.
A Chemical Abstract illetve a Science Direct szakirodalom kereső programjaival végzett keresés során a fenti vegyületek kromatográfiás elválasztására vonatkozóan szakirodalmi hivatkozást nem találtam. Egyébként általában a királis vegyületekre vonatkozó szakirodalmi publikációk száma rendkívül nagy.
A szakirodalmi összefoglalás elkészítése során áttekintettem a témához kapcsolódó fontosabb könyveket illetve publikációkat a Vegyipari Műveleti Intézeti Tanszék könyvei és publikációi mellett [6-20].
1. Enantiomerek
Az olyan aszimmetriacentrumot tartalmazó királis vegyületeket, melyeknek két egymással fedésbe nem hozható és optikai aktivitást mutató tükörképi szerkezete lehetséges, enantiomer vegyületpárnak nevezzük.
Egy aszimmetriacentrum esetén mindig csak két enantiomer térszerkezet lehetséges, melyekre jellemző, hogy kémiai tulajdonságaik akirális reagensekkel szemben azonosak, a fizikai és termodinamikai állandóik azonosak illetve a fajlagos forgatóképességük nagysága is azonos, csak irányuk ellentétes.
A két aktív módosulat képződésének valószínűsége teljesen azonos. Így egy inaktív, úgynevezett „racém” módosulat alakul ki. Az egy aszimmetriacentrumot tartalmazó vegyületeknek tehát három módosulata lehetséges: két aktív enantiomer és az inaktív racém.
Ha egy molekulában két aszimmetriacentrum van, ezek környezete lehet azonos vagy különböző. Ezenkívül mindegyik centrum rendelkezhet kétféle (R vagy S) konfigurációval. A két azonos környezetű aszimmetrikus szénatomot tartalmazó vegyületek klasszikus példája a borkősav.
A két azonos környezetű aszimmetriacentrumot tartalmazó vegyületek esetén összesen négy módosulat lehetséges: két aktív [(R,R)- ill. (S,S)-], a mezo- [(R,S) = (S,R)] és a racém [(RS)-] módosulat, mely utóbbi a két aktív forma 50-50 %-os elegye. Az aktív formák egymásnak enantiomerjei, s a mezo-formával mindegyikük diasztereomer viszonyban van.
Diasztereomer vegyületpárt alkotnak az olyan, több aszimmetriacentrummal rendelkező optikai izomerek, amelyek a teljes molekulát tekintve nincsenek tükörképi viszonyban egymással, csak részlegesen tükörképei egymásnak.
A diasztereomer párok egymással fedésbe nem hozható, de nem is tükörképi szerkezetek. Ebből adódóan a molekulán belüli atomtávolságok sem mind azonosak, ezért nem azonosak a diasztereomerek kémiai tulajdonságai, fizikai állandói, a forgatás iránya és a forgatóképesség mértéke sem. A diasztereomer vegyületpárok egyszerű fizikai módszerekkel is elválaszthatók egymástól [21].
2. Enantiomerek elválasztási műveletei
Közismert a hatvanas évek gyógyszerbotránya, a Contergan (Thalidomine) esete [1]. A nyugtató hatású gyógyszert széles körben alkalmazták terhes nők gyógyítására is, ám használatának következményeként több ezer csecsemő csökevényes végtaggal jött a világra. Ennek oka a hatóanyag két enantiomerjének eltérő biológiai hatásában keresendő:
az egyik enantiomer nyugtató, a másik magzatkárosító hatású.
Egyebek közt a Contergan irányította rá a figyelmet az enantiomerek fontos voltára, arra, hogy tiszta állapotban való gyártásuk mennyire szükségszerű. Gyógyszereink, növényvédő szereink jelentős része királis vegyület, enantiomerjeik biológia hatása között azonban nem mindig ilyen meglepő a különbség. Például több olyan vegyület van, melynek két enantiomerjét szervezetünk más illatként vagy más ízként érzékeli. Gyakori, hogy a kívánatos biológiai hatást kifejtő enantiomer mellett azonos mennyiségben megtalálható másik enantiomernek bizonyíthatóan nincsen semmiféle káros hatása.
Egy statisztika szerint 1993-ban a szintetikus hatóanyagot tartalmazó gyógyszerek száma 1300 körül volt, közülük kb. 500 tartalmazott királis hatóanyag molekulát. De csak 61 hatóanyaga volt optikailag tiszta. A helyzet azóta sokat javult, de még mindig bőven vannak keverékek gyógyszertári forgalomban. A gyógyszeripart és piacot jelentős mértékben befolyásoló amerikai Federal Drug Administration néhány éve csak tiszta enantiomer hatóanyagot tartalmazó készítmények forgalmazását engedélyezi [7].
Tehát a kémiai reakció során, ha királis vegyületek keletkeznek, feltétlenül számolni kell mindkét konfigurációjú termék képződésével. Gyakorlat szempontjából két lehetséges megoldás létezik az optikai izomerek elválasztására az indirekt és a direkt szintézis.
Indirekt szintézisről beszélünk, amikor a reakciót célirányosan végezzük.
Amennyiben a molekula már tartalmazott aszimmetrikus szénatomot, azzal diasztereomert képzünk. Amennyiben egy új asszimetria centrumot akarunk létrehozni, akkor valamilyen módon kiralitást kell vinni a rendszerbe, például királis katalizátor vagy királis reagens segítségével.
Direkt szintézis során racém elegy keletkezik és azt utólagosan kell elválasztani.
A szerves kémiai szintézisek során általában az enantiomerek azonos arányban keletkeznek. Az így keletkezett racém elegy enantiomerekre való szétválasztása a rezolválás. A rezolválás számos ismert módszere közül a mechanikus eljárás kivételével
mindegyikre érvényes az, hogy az elválasztás csak valamilyen másik királis anyag közreműködésével valósítható meg.
Ha az enantiomerek külön-külön kristályosodnak (konglomerátumot alkotnak), a fejlett enantiomorf kristályok kiválogathatók a halmazból. A mechanikus módszert azonban ritkán alkalmazzák, mert kevés olyan enantiomer van, ami külön-külön kristályosodik, illetve megfelelő méretű kristályok ritkán képződnek. Alkalmazzák viszont az átkristályosítást abban az esetben, amikor az egyik enantiomerből csak kevés van az elegyben.
A biológiai módszer lényege, hogy az enantiomerek oldatából az egyik komponenst megfelelő mikroorganizmussal lebontjuk. Hátránya, hogy ez a komponens elvész.
Valamilyen optikailag aktív reagens hozzáadásával diasztereomer sókat, komplexeket vagy vegyület párokat képzünk. Ha egy A aszimmetria centrummal rendelkező vegyület racém formáját (A, A’) feloldás után B centrumú optikailag aktív vegyülettel reagáltatjuk, a keletkező AB és A’B vegyületek diasztereomer viszonyban lesznek egymással, tehát frakcionált művelettel elkülöníthetők.
A diasztereomerek elválasztása történhet oldhatóság különbség alapján. Pasteur [22]
volt az, aki az első fizikailag is megvalósított enantiomer elválasztást elvégezte borkősav kristályos Na és ammónium sóinak a szétválogatásával. A rezolválás egymással nem elegyedő oldószerek segítségével is megoldható.
A diasztereomer mellől a szabad állapotú enantiomer elválasztását megoldhatjuk úgy is, hogy a rezolváló ágenst és a racém vegyületet megfelelő arányban összekeverjük, majd az oldószert eltávolítjuk és a maradék elegyet szuperkritikus állapotú oldószerrel extraháljuk. Így visszamarad a diasztereomer só, amely később frakcionált kristályosítással elválasztható az oldószertől.
Amennyiben a királis kölcsönhatás szilárd fázisok között játszódik le, a rezolváló ágenssel diasztereomer komplex alakul ki, és a kapott keveréket frakcionált szublimációval elválaszthatjuk.
Az enantiomer elválasztást frakcionált desztillációval is elvégezhetjük, ha a diasztereomer, mely az első frakció után visszamarad, magasabb hőfokon elbomlik, és a só bomlásával a lehetséges termék keletkezik.
A rezolváló ágens kiválasztására különböző számítási módszereket dolgoztak ki, ugyanakkor a fázisdiagramok felhasználásával méréseken alapuló módszereket is javasolnak.
A gyógyszer- és a növényvédőszer-ipar egyre inkább a racém vegyület helyett annak csak egyik enantiomerjét használja fel, így szükség van a rezolválásra.
A rezolválás megoldható a racém elegy optikailag aktív adszorbensen végzett kromatografálásával is. Ilyenkor az enantiomerek az adszorbens molekulákhoz eltérő módon illeszkednek, s így az adszorbensen különböző mértékben kötődnek meg. Részletes leírása a 2.2.6. fejezetben található.
A királis vegyületek nagy enantioszelektivitással történő előállítása a tudományos érdekességen túl gyakorlati szempontból is igen fontos. A királis építőelemek szintézisét lehetővé tevő eljárások, köztük kiemelkedő helyen a homogénkatalitikus enantioszelektív szintézisek mind nagyobb jelentőségre tesznek szert [23].
A 2001-es év Kémiai Nobel-díj kitüntetettjei különböző kémiai reakciókban valósították meg a tükörképi párok szelektív szintézisét. Ezekben a reakciókban átmenetifémeket (ródiumot, irídiumot, titánt, ozmiumot) tartalmazó, úgynevezett enantioszelektív homogén katalizátorokat alkalmaztak. Ezek olyan - reakcióközegben oldott (azaz homogén, az oldószerrel egy fázist képező) - vegyületek (fém komplexek), amelyek a lehetséges két enantiomer közül csupán az egyik tükörképi formát képviselik.
Katalitikus tulajdonságot mutatnak, azaz képesek vegyületeket úgy átalakítani, hogy eközben szerkezetüket és tulajdonságaikat változatlanul megőrzik. A katalitikus enantioszelektív szintézisek legnagyobb jelentősége abban áll, hogy kis mennyiségű optikailag aktív katalizátorral nagy mennyiségű anyagra vihető át a királis információ.
Tiszta enantiomereket hosszú időn keresztül csupán biológiai rendszerek segítségével lehetett előállítani, melyek természetes anyagok szintézisére voltak csak alkalmasak. Ezek a rendszerek igen gyakran sérülékenyek. Az aszimmetrikus katalitikus módszerek azzal az előnnyel bírnak, hogy mind a természetben előforduló, mind a mesterséges vegyületek szintézisére alkalmasak.
2.1. Enantiomerek elválasztása kristályosítással
2.1.1. Kristályosító berendezések
A kristályosító berendezés olyan nyitott vagy zárt műveleti egység, amely keletkező gócok vagy beoltott kristályok segítségével kristályok növesztésére képes. Két típusú kristályosító létezik az oldatkristályosító és az olvadékkristályosító.
A kristályosító után mindig kapcsolnak szűrőberendezést vagy centrifugát, ill.
szárítókat és szitákat. A végrehajtott művelet alapján a kristályosítók lehetnek egyszerű elpárologtató kristályosítók, hűtőkristályosítók, bepárló kristályosítók, vákuum- kristályosítók [24].
2.1.2. Frakcionált kristályosítással kombinált szimulált mozgóréteges folyadékkromatográfia
Az enantiomerek tisztítására, elválasztására alkalmazott kristályosítási eljárások a két enantiomer olvadási viselkedését leíró megfelelő fázisdiagramok (biner olvadáspont diagramok) vagy a megfelelő oldószerbeli oldódási tulajdonságuk (terner oldhatósági diagram) alapos ismeretén alapulnak [25-37].
A fázisdiagramok telítési görbéinek jellege szerint az enantiomer rendszerek három alapvető típusát különböztetjük meg. Ezeket először Roozeboom [28] írta le. A konglomerátumok, racém vegyületek és szilárd oldatok által kialakított karakterisztikus biner fázisdiagramok láthatók a 2.1. ábrán.
2.1. ábra A racemát típusainak kételemű fázisdiagramjai: (a) konglomerátum, (b) racém vegyület és (c) pszeudoracemát
A racemátok mindössze 5-10 %-a tartozik a konglomerátumok közé, amely a legkedvezőbb csoport bizonyos, nem-racém elegyekből, frakcionális kristályosítással történő enantiomeres dúsítás megvalósítására. A racemátok 90-95 %-a tartozik a szilárd fázisban jelen levő, úgynevezett racém vegyületek, vagy más néven igazi racemátok közé.
Az utóbbi esetben a fázisegyensúly ismerete még fontosabb, mivel a fázisdiagram tiszta enantiomerek tartománya, amit a biner/terner eutektikus pont helyzete határoz meg, még kisebb. A szilárd oldatok csoportját alkotó racemátok (ún. pszeudoracemátok) viszonylag ritkák.
A terner (oldhatósági) fázis diagram megjelenési formája levezethető a fenti ábrán tárgyalt (2.1. ábra) biner diagramok alakjából. A biner fázisdiagramokra imént tett megállapítás szintén hozzájárul az oldhatósági adatok hozzávetőleges meghatározásához.
Az olvadáspont diagramok meghatározása a terner rendszerek oldhatósági méréseihez képest könnyű.
A 2.2. ábrán két enantiomer és egy oldószer által alkotott rendszer vázlatos oldhatósági diagramja látható a leggyakrabban használt egyenlő oldalú háromszög formájú diagramban.
2.2. ábra (+)- és (-)-enantiomert tartalmazó vegyület vázlatos terner oldhatósági diagramja (T = állandó)
oldószer
A háromszög csúcsai reprezentálják a tiszta komponenseket: az oldószert a felső, míg a (+) és (–) enantiomert a jobb és bal alsók.
A háromszög oldalai (mól vagy tömegtört skálával) a biner rendszereket mutatják: a (+) enantiomer/oldószer és (–) enantiomer/oldószer rendszert a bal illetve a jobb oldalai, míg a háromszög alapja a (+)/(–) enantiomer rendszert, R-rel jelölve azt a pontot, ahol a racém vegyület a mindkét enantiomert 50 %-ban tartalmazza. A háromszög minden egyes belső pontja egy terner elegyet jelöl, ami mind a három komponenst tartalmazza.
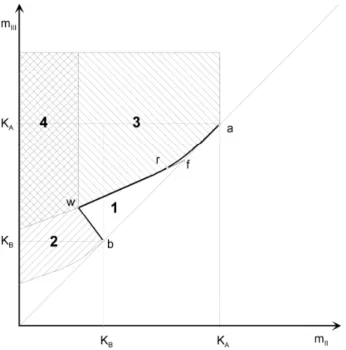
Az A, A’ és C pontok jelentik a tiszta enantiomerek, illetve a racemát oldószerben való oldhatóságát adott hőmérsékleten, következetesen az A-E-C-E’-A’ a terner rendszer oldhatósági görbéje ugyanezen a hőmérsékleten. Ennek következtében az oldhatósági görbe felett telítetlen oldószer (egyfázisú tartomány) van. Az A-E-tiszta (+), A’-E’-tiszta (–) vagy az E-E’-R pontok által határolt területek olyan kétfázisú régiókat jelölnek, ahol az egyik tiszta enantiomerből vagy a racém vegyületből álló szilárd fázis egyensúlyban van az oldhatósági görbén levő összetételű telített oldószerrel. A tiszta enantiomer illetve a racemát tartományában a szakasz osztópontjait a P2’ és P2 pontok mutatják. A megfelelő összetételű folyékony fázist az M illetve az O pontok mutatják. A háromszög tiszta(+)-E- racemát vagy a tiszta(–)-E’-racemát által határolt területei háromfázisú tartományokat jelölnek, ahol két szilárd fázis (tiszta enantiomer és racém keverék) tart egyensúlyt az E vagy E’ pontokkal jelzett eutektikus összetételű folyékony fázissal. Ebbe a tartományba tartozó összetételű, részlegesen dúsított oldatból a tiszta enantiomer kinyerése nem lehetséges. A P3 és P3’ összetételű telített oldatok N vagy K összetételű szilárd fázisra tagolódnak, így jutunk a tiszta enantiomer és a racém elegy keverékéhez. A kapott anyalúg minden esetben eutektikus összetételű.
A tiszta enantiomer kizárólag frakcionális kristályosítással állítható elő abban az esetben, amikor a kiindulási oldat összetétele az A-E-tiszta(+) vagy A’-E’-tiszta(–) pontok által határolt tiszta enantiomer tartományán belül helyezkedett el. Ennek következtében a kromatográfia és a kristályosítás műveletének sorbakapcsolásakor az első, kromatográfiás lépésben egy minimális enantiomeres dúsítást teljesíteni kell, amely meghaladja a terner fázisdiagram eutektikus pontját. Ezt követően az erősen felhígított, telítetlen oldatot (pl.: P1’-es pont) be kell párolni annak érdekében, hogy a tiszta enantiomer tartományát elérjük (pl: P2’-es pont), melynek eredményeképp lehetővé válik a tiszta enantiomer kinyerése a következő, kristályosítási lépés(ek)ben. A betáplált racém elegyet kromatográfiás úton nem lehet megfelelően a P1-es pontba (melynek tisztasága kisebb,
mint az E pontbelié) dúsítani, mivel a bepárlás olyan összetételű elegyet szolgáltat, mint amilyen a P2 vagy P3 pontokban van, melyből a tiszta enantiomerek nem nyerhetők ki kristályosítással. Ha a terner rendszerben az oldhatóság hőmérséklet függése ismert, más módszerek is számításba jöhetnek a kromatográfiásan dúsított oldat túltelítésének megvalósítására.
2.1.3. Kromatográfia és kristályosítás összekapcsolása
A hatékony enantiomer szeparációhoz a folyadékkromatográfia és a frakcionált kristályosítás összetett műveletét javasolják. Első lépésként az egyik enantiomer feldúsítására egy kromatográfiás elválasztó műveletet, főként a szimulált mozgóréteges folyadékkromatográfiát (SMB) ajánlják (Az SMB műveletet lásd részletesebben a 3. fejezetben). Következő lépésként frakcionált kristályosítással végezhető el a cél enantiomer tisztítása.
A (+)/(–)-enantiomerek és az oldószer rendszere egy terner fázisdiagramot alkot.
Látható a 2.2. ábrán, hogy az egyik enantiomert a kívánt tisztaságban akkor lehet kinyerni, ha egy aszimmetrikus összetételű elegyből indítjuk a kristályosítást. Tehát a kromatográfiás szeparációs művelet során nem szükséges magas tisztaságban kinyerni az enantiomert.
A 2.3. ábrán látható az összekapcsolt művelet, amely során mind a két enantiomert tisztítják kristályosítással.
2.3. ábra Kétlépcsős enantiomer elválasztó rendszer
A művelet során az SMB kromatográfiás egységre egy 50:50 m/m % összetételű racém elegyet táplálnak be. A folyamatos szeparáció után a két terméket a raffinátum és az extraktum áramokban vezetik ki. Ebben a két áramban dúsulnak fel a komponensek. Az enantiomerek feldúsulásának mértéke különböző a raffinátum és az extraktum áramokban. A raffinátumban például 80:20 m/m %, míg az extraktumban például 10:90 m/m % összetételűek. Ezt a két áramot betáplálják egy folyamatosan működő kristályosító rendszerbe, ahol a távozó szilárd fázis (kristályok) csak az egyik enantiomert fogja tartalmazni 100 m/m %-os tisztaságban. Az egyensúlyi fázisdiagramnak megfelelően a keletkezett anyalúg tartalmazza mindkét enantiomert. A keletkezett anyalúg összetétele hasonlít a kiindulási racém elegy összetételére, ezért ezt visszavezetik az SMB művelet betáplálási áramába (Az SMB és a csatolt kristályosítás részletezését lásd a 9. fejezetben).
2.1.4. Kinetikus rezolválás oltásos kristályosítással konglomerátum típusú racém rendszer esetén
Konglomerátum típusú racém rendszerek kristályosítással történő elválasztása esetén alkalmazhatjuk az oltásos kinetikus rezolválást (2.4. ábra).
T1 hőmérsékleten legyen az enantiomereket tartalmazó oldat telített, melyet hűtsünk le T2 hőmérsékletre az úgynevezett metastabil zónában. Ha az „a” pontban levő oldatot úgy hűtjük, hogy homokirális (–)enantiomer kristályokat adunk az oldathoz a hűtés során, akkor az oldat összetétele nem jut át közvetlenül a „c” pontba, hanem az „a”, „b”, „c”
trajektóriát követi. Ez az jelenti, hogy (–)enantiomer kristályban dúsabb frakciót kapunk a hűtés elején a „b” pontig, miközben a kristályosítási anyalúg (+)enantiomerre nézve dúsul.
Ezt követően a kristályosítási anyalúg (+)enantiomer koncentrációja lecsökken a racém koncentrációt jelölő „c” pontig. Értelemszerűen a „b”, „c” trajektórián való mozgáskor is (–)enantiomerben dúsabb kristályt kapunk. A teljes „a”, „b”, „c” trajektórián való mozgás után nyert kristály integrálisan racém összetételű, hasonlóan a „c” pontbeli racém anyalúg koncentrációhoz. A fenti elveknek megfelelően létrehozható az úgynevezett “butterfly”
kristályosítás, mely alkalmas konglomerátum típusú racém rendszerekben enantiomerek elválasztására kinetikus rezolválási oltásos kristályosítás alkalmazásával. A módszert a 2.5.
és 2.6. ábrákon mutatom be.
2.4. ábra Oltásos kinetikus rezolválás elve [26]
2.5. ábra A “butterfly” kristályosítás elve háromszög diagrammon [26]
2.6. ábra A “butterfly” kristályosítás megvalósításának elve [26]
Az „a” pontban levő Trac hőmérsékletű telített oldatot TA hőmérsékletig hűtjük és (–)enantiomer kristállyal oltjuk be. Az oldatból kiváló (–)enantiomer kristályokat kiszűrjük és eltávolítjuk. A „b” pontba jutott TA hőmérsékletű anyalúghoz felmelegítés közben (TA- ról Trac-ra növeljük a hőmérsékletet) racém keveréket adunk. Ennek következtében az oldat összetétele a „c” pontba jut. A „c” pontban levő oldathoz (+)enantiomer oltókristályt adunk, miközben hőmérsékletét csökkentjük Trac értékről TB = TA értékig. A rendszer „c”
pontból „d” pontba jut. Ezt követően felmelegítjük TB = TA hőmérséklet értékről az oldatot Trac értékig, miközben racém keveréket adunk az oldathoz. Ennek során „d” pontból visszajutunk az „a” pontba. Ilyen módon a ciklikus művelet első ciklusa befejeződött és a fentieknek megfelelően ismételhető [26].
2.2. Kromatográfiás enantiomer elválasztás 2.2.1. Nagyhatékonyságú folyadékkromatográfia 2.2.1.1. A HPLC elmélete
A HPLC olyan nagynyomású oszlopkromatográfia, amellyel a nem illékony illetve a termikusan nem stabil vegyületek hatékony elválasztása, mennyiségi meghatározása is megoldható.
A nagyhatékonyságú folyadékkromatográfia kifejlődése az 1960-as évek közepén kezdődött. Ekkor még poláris állófázisokat alkalmaztak (alumínium-oxid, szilikagélek). A 70-es évek végétől a technika ugrásszerű fejlődése indult meg. Megjelentek az ún. fordított fázisú töltetek, amelyeket a szilikagél felületének kémiai módosításával hoztak létre. A poláris szilikagél felületén apoláris csoportokat rögzítettek, így a hagyományos polaritásviszonyok (poláris töltet, apoláris mozgófázis – normál fázis) megváltozásával létrejött a fordított fázisú kromatográfia, ahol víz – szerves oldószer elegyeket használnak mozgófázisként.
A klasszikus vagy más néven gravitációs áramlású folyadékkromatográfiával ellentétben a nagyhatékonyságú folyadékkromatográfia esetében a szemcsés töltetet tartalmazó kolonna tetejére feladott elválasztandó mintaelegy öblítő eluens kényszeráramlásban van. Ez a kényszeráramoltatás szivattyúval valósítható meg. A HPLC előnye a klasszikus folyadékkromatográfiával szemben a töltet szemcseméretének jelentős csökkentése, illetve a folyamat felgyorsítása [38-40].
2.2.1.2. A HPLC berendezés fő részei és működése
Az eluens tartályból egy kerámia vagy fém szűrőn keresztül szivattyúzzuk az oldószereket a pumpa segítségével. Az eluenst a mérés megkezdése előtt gázmentesíteni kell.
A nagynyomású szivattyú (pumpa) a HPLC rendszer legfontosabb modulja. A mozgófázis felszívását és áramoltatását a szivattyúval végezzük. Mivel a folyadékkromatográfiában gyakran kell korrozív illetve magas sótartalmú eluensekkel dolgozni, a pumpafejet, dugattyúkat és szelepeket speciális anyagokból készítik amelyek garantálják a tartós hibamentes működést.
Izokratikus üzemmódról beszélünk ha a nagynyomású szivattyú az elválasztás ideje alatt állandó mozgófázis összetételt szállít. Ennél az üzemmódnál megoldható az eluens visszaáramoltatása az eluenstartó palackba, amennyiben nem tartalmaz szennyező komponenst.
Gradiens üzemmódról beszélünk ha időben változtatjuk (növeljük) az eluens erősségét. A modern pumpák 3-4 különféle oldószer keverését tudják elvégezni, a szivattyú szívó és nyomó oldalán egyaránt.
Az injektor (mintaadagoló) a minta bevitelére szolgál. Megkülönböztetünk kézi vagy automata mintaadagolót.
Az analitikai elválsztó kolonnán (oszlop) történik a minta elegy komponenseinek szétválasztása azok kémiai-fizikai tulajdonságai alapján. A kolonnák különféle töltettel (állófázis, szorbens) vannak megtöltve. A kolonna bemeneti részén és végén védő szűrőt alkalmaznak ami egyrészt megakadályozza a töltet kifolyását, másrészt védi a töltetet a mechanikai szennyeződésektől. A kolonnák védelmére ún. előtét vagy védő kolonnákat is alkalmazhatunk, amelyek sokkal rövidebbek mint az analitikai kolonna és ugyanolyan töltetet tartalmaznak (esetleg nagyobb szemcsemérettel).
A detektorok a kolonnáról lejövő, szeparált komponensek detektálására alkalmasak.
A nagyhatékonyságú folyadékkromatográfiában mindig a vizsgálandó minta határozza meg az alkalmazott detektálási módot. A kölönféle detektorok más-más vegyületcsoportok különböző érzékenységgel történő detektálására alkalmasak. A legtöbb módszerleírás tartalmazza a detektálási módot. A méréseim során UV és Chiralyser detektorokat használtam.
Az UV detektorok univerzálisan használhatók azoknál a mérendő komponenseknél amelyek UV fényt nyelnek el. Az UV detektorok a HPLC méréseknél leggyakrabban alkalmazott eszközök, melyek nagy érzékenységgel és alacsony zajszinttel rendelkeznek, így alkalmasak kis minta koncentrációk mérésére. Az UV detektor egy adott hullámhosszon működik, a mérések során mindig 254 nm hullámhosszon detektáltam.
Vannak programozható hullámhosszú ill. spektrum felvételére alkalmas detektorok is.
Az IBZ gyártmányú Chiralyser egy nagy teljesítményű polarimetriás detektor optikailag aktív vegyületek detektálására. A Faraday-kompenzáció elvén működő 426 nm LED fényforással rendelkező műszer lehetővé teszi az optikailag aktív molekulák nem csak az analitikus hanem preparatív detektálását is.
A HPLC technikában használatosak még a diódasoros (Dioda array, DA), a fluoreszcens (FL), az elektrokémiai EC (amperometriás), a tömegszelektív (Mass Spectrometer, MS), a vezetőképesség mérő (ionmérés), törésmutató mérő, radiokémiai és a fényszórásmérő detektorok [40].
A HPLC szoftverek legfőbb feladata a rendszer vezérlése, a detektor adatainak felvétele-tárolása az adatok kiértékelése és dokumentálása. A szoftverek felépítése, megjelenési formája sokféle lehet. A ma használatos modern HPLC szoftverek legtöbbje Windows operációs rendszer felhasználásával működik, ami megfelelő adatbiztonságot és egyszerű kezelhetőséget jelent.
2.2.2. Kromatográfiás töltetek
A királis állófázisú kromatográfiát csakúgy, mint a kromatográfiát, általában már a kezdetektől is inkább tiszta komponensek előállítására alkalmazták, mint analitikai célokra.
Ipari felhasználásuknak sokáig magas áruk szabott határt, emiatt csak különleges esetekben alkalmazták őket, mint például biomolekulák elválasztására, ahol más módszer nem volt alkalmazható. Ezzel szemben néhány petrolkémiai és cukoripari folyamat esetében nyilvánvalóvá vált, hogy az adszorpció illetőleg a kromatográfia elvének alkalmazásával még néhány kisebb értékű termék is nagyon gazdaságosan állítható elő.
Kezdetekben a természetes polimereket – pl. cellulóz-triacetátot – és a mesterséges polimereket – pl. a poli-trifenil-metil-metakrilátot vagy a poli-aminosavészter-akrilamidot – hatalmas üvegkolonnákba töltve alkalmazták gyógyszeripari aktív komponensek tiszta enantiomerjeinek izolálására.
Ezt követően Pirkle („brush type” királis állófázisok) és Okamoto (cellulóz- és amilóz-karbamátok és -észterek) úttörő munkája révén, akik a szilikagél felületére kötötték meg a királis elválasztó elemeket, vált lehetővé olyan kolonnák kifejlesztése, amelyek már használhatóak voltak a nagyhatékonyságú kromatográfiában [13]. Ezen állófázisok alkalmazásával a különböző farmako-kinetikai, farmakológiai és farmako-dinamikai tulajdonságokkal rendelkező enantiomerek is vizsgálhatóvá váltak.
Királis állófázisnak nevezünk minden olyan kromatográfiás elválasztásra alkalmas állófázist, amely
• kismolekulájú királis monomerekből felépülő természetes, vagy szintetikus polimer szerkeztű anyag:
-tiszta polimerek
-szervetlen váz polimer bevonattal
A legjelentősebbek az oligo- és poliszaharidok valamint ezek származékai, a poliakrilamidok, poliakril-észterek és a protein-alapú fázisok [14].
• a hordozó fázis (polimer, szilika gél) felületére rögzített kismolekulájú szelektort tartalmaz:
-szervetlen anyag (pl.: szilika gél) felületének módosításával -szerves polimer hálóba beültetett királis molekulákkal létrehozott fázisok
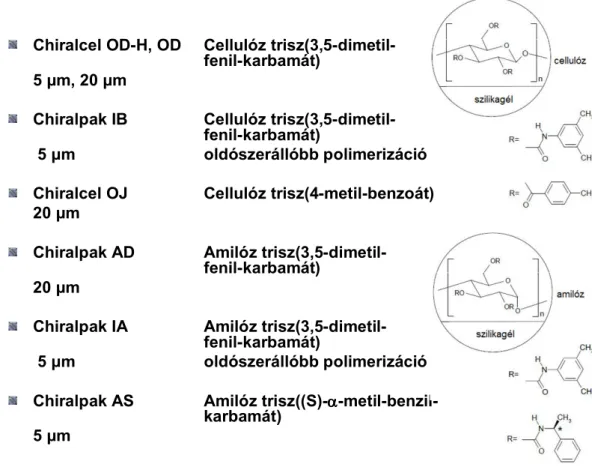
Ilyenek: az aminosav származék, „brush type” CSP (Chiral Stationary Phase), koronaéter, borkősav származék és ciklodextrin alapú fázisok. Az utóbbi években számos cég fejlesztett ki királis állófázisokat (2.1. táblázat). Jelenleg több, mint 100 - HPLC-hez alkalmazható - ipari CSP létezik.
Név Márkanév
Cellulóz tri-acetát Chiralcel OA
Cellulóz trisz(3,5-dimetil-fenil-karbamát) Chiralcel OD
Cellulóz trisz(4-metil-benzoát) Chiralcel OJ
Amilóz trisz(3,5- dimetil-fenil-karbamát), 10 µm Chiralpak AD Amilóz trisz(3,5- dimetil-fenil-karbamát), 5 µm Chiralpak IA * Cellulóz trisz(3,5- dimetil-fenil-karbamát), 5 µm Chiralpak IB *
Cellulóz tri-benzoát Chiralcel OB
Amilóz trisz[(S)-fenil-etil-karbamát] Chiralpak AS
Poli[(S)-N-akril-oilfenil-alanin-etil-észter] Chiraspher
3,5-Dinitro-benzoil-fenil-glicin DNBPG
Keresztkötésű di-(3,5-dimetil-benzoil)-L dialliltartaramid Kromasil CHI-DMB Keresztkötésű di-(4-terc-butil-benzoil)-L dialliltartaramid Kromasil CHI-TTB Tetrahidro-aminofenantrén 3,5-dinitro-benzamid WHELK-O I
* - jó oldószerálló polimer alapú királis töltet
2.1. táblázat Királis állófázisok
Francotte szerint öt fő HPLC-CSP osztály létezik a kialakuló minta-CSP komplex alapján [14]. Az I-es típus (vagy Pirkle típus) vonzó-taszító, főleg Π-elektron donor- akceptor mechanizmuson alapuló minta-állófázis komplexeket tartalmaz. A II-es típust egy
cellulózszármazékkal lehet szemléltetni, melyet a királis üregekben létrejövő vonzó kölcsönhatások jellemeznek. A III-as típusú CSP-k, mint a ciklodextrinek, koronaéterek
„befogadó” komplexeket képeznek. A IV-es típusú CSP-knél a minta a diasztereomer fémkomplex része (királis ligandumcsere kromatográfia). Az V-ös típusú CSP egy protein (bovin szérum albumin), a minta-CSP komplexek létrejötte hidrofób és poláris kölcsönhatásokon alapszik.
A méréseket egy részét a II-es típusú CHIRALCEL OD tölteten (2.7. ábra) végeztem. A töltet 5 vagy 20 µm-es szemcseméretű szilikagélre (fizikai lecsapással) felvitt cellulóz 3,5-dimetil-fenil-karbamátot tartalmaz. Ez egy normál fázisú analitikai oszlop, azonban jó kapacitása miatt preparatív elválasztásokra is alkalmazható. Főként szteroidok, egyszerű funkciós vegyületek elválasztására szolgál. A töltet hátránya, hogy az oldószereknek csak egy meglehetősen szűk csoportjánál alkalmazható. A legtöbb HPLC- ben eluensként gyakran alkalmazott oldószer (pl.: aceton, kloroform, dimetil-szulfoxid, etil-acetát, metilén-klorid, THF), még igen kis mennyiségben is tönkreteheti.
2.7. ábra A CHIRALCEL OD töltet felépítése
Ezt a hátrányt küszöbölték ki egy a közelmúltban forgalomba hozott töltettípusnál, a CHIRALPAK IB-nél. A töltet szinte teljesen megegyezik a CHIRALCEL OD-H töltettel, a különbség abból adódik, hogy ebben az esetben a 3,5-dimetil-fenil-karbamátot fizikai lecsapás helyett kémiai keresztkötéssel rögzítették a felületre, ezáltal növelve annak oldószer ellenállóságát [9, 14, 41, 42].
2.2.3. Adszorpciós izotermák
Adszorpciós egyensúlyról akkor beszélünk, ha egy időegység alatt az aktív felületre érkező és onnan eltávozó molekulák száma megegyezik. Az adszorpciós egyensúly függvénye a hőmérsékletnek, a nyomásnak, valamint a megkötődő részecske folyadékfázisbeli koncentrációjának.
Azt az összefüggést, amely megadja, hogy állandó hőmérsékleten hogyan változik az adszorbeált anyag mennyisége a folyadékfázisbeli koncentrációjának függvényében adszorpciós izotermának nevezzük (2.8. ábra). Jelentősége, hogy széles koncentráció- tartományban leírja az adott komponens megoszlását a folyadék és az adszorpciós fázis között. Emiatt az adszorpciós izotermák átfogó vizsgálata és ismerete információt ad a folyamatban résztvevő anyagokról és az adszorpció során végbemenő kölcsönhatásokról [39, 43].
ck, pk q 8
qk
2.8. ábra Langmuir izoterma
Az adszorpciós izotermákat a hőmérséklettel (T), az adszorbeálandó komponens adszorbensbeli koncentrációjával (qk), és az adszorbeálódó komponens folyadékfázisbeli koncentrációjával (ck) jellemezik.
A továbbiakban a mértékegységek:
qk: [mg adszorbeálódó komponens/cm3 adszorbens]
Kk: [cm3 folyadék/cm3 adszorbens]
ck: [mg komponens/cm3 folyadék]
ak: [cm3 folyadék/cm3 adszorbens]
bk: [cm3 folyadék/mg adszorbeálódó komponens]
N: a komponensek száma
1. Lineáris-izoterma
Lineáris izotermának nevezzük az olyan összefüggést, amelyben az adszorbeálandó komponens adszorbensbeli és folyadékfázisbeli koncentrációja között lineáris a kapcsolat:
k k
k K c
q = ⋅ (2-1)
A lineáris összefüggés általában a valós rendszerekre csak nagy körültekintés mellett használható, mert általában csak kis koncentrációknál érvényes.
2. Langmuir-izoterma
A gázokra és a folyadékokra általánosan érvényes adszorpciós izoterma egyenletet Langmuir vezette le az alábbi egyszerűsítéseket feltételezve:
• minden aktív centrum egy részecskét köt meg, az adszorpció legfeljebb monomolekuláris réteget alkothat a felületen,
• az adszorbeált molekulák között nincs kölcsönhatás,
• az adszorbens felületén adott számú, energetikailag egyenértékű aktív centrum található,
• az adszorpciós réteg és az adszorbeálandó komponens között dinamikus egyensúly van.
A Langmuir - egyenlet matematikai alakja:
k k
k k
k b c
c q a
= +
1 (2-2)
3. bi - Langmuir-izoterma
A kromatográfiás elválasztások esetében az adszorbens felülete nem tekinthető homogénnek. Ilyen eseteknél szokás alkalmazni ezt a fajta izotermatípust, amely két - adszorpciós szempontból eltérő - aktív centrummal rendelkező felületet feltételez. A modell így a két független részizoterma összegéből tevődik össze:
k NS k
k NS k k S k k S k
k b c
c a c b
c q a
+ +
= +
1
1 (2-3)
A bi-Langmuir-izotermák alkalmazása kevésbé elterjedt, de királis töltetek adszorpciós tulajdonságainak modellezésekor kifejezetten előnyösnek bizonyult.
4. kompetitív Langmuir-izoterma
Az egykomponensű rendszerekre vonatkozó Langmuir-izoterma többkomponensű rendszerekre való kiterjesztése:
∑
=+
= N
k k k k k k
c b c q a
1
1
(2-4)
A kompetitív izotermák egyik fontos tulajdonsága, hogy az elválasztási tényező nagysága független lesz alkalmazásukkor az összetételtől.
5. kompetitív bi-Langmuir-izoterma
Néha a kompetitív Langmuir izotermánál összetettebb adszorpciós izotermát kell alkalmazni, ami az izoterma kiterjesztését jelenti két adszorpciós helytípussal rendelkező modellre:
∑
∑
= =+ + +
= N
k
k NS k
k NS k N
k k S k k S k k
c b
c a c
b c q a
1 1
1 1
(2-5)
Ezt az összefüggést gyakran használják enantiomerek adszorptív elválasztása esetén, a változó szelektivitás leírására. Ekkor az izotermát lineáris taggal, vagy újabb Langmuir taggal egészítik ki.
2.2.4. Preparatív folyadékkromatográfia
Az elmúlt évtizedekben új feladatként jelentkező közeli fizikai, fizikai-kémiai tulajdonságú anyagok (izomerek, optikai izomerek) elválasztásának megoldása a hagyományos vegyipari műveletekkel (rektifikáció, folyadék-folyadék extrakció, folyadék- szilárd extrakció, kristályosítás, stb.) nem lehetséges, mivel a szétválasztáshoz szükséges egyensúlyi egységek száma (NTP = Number of Theoretical Plates = elméleti tányérszám) 100-nál is nagyobb lehet. Ekkora elméleti tányérszámot a hagyományos vegyipari műveleti egységekben műszakilag igen nehéz megvalósítani, másrészt megvalósítás esetén a berendezések költségesen üzemeltethetők [38, 39].
Az elméleti tányérszám 102–105 is lehet olyan adszorbenssel töltött nyugvóréteges berendezésekben, ahol az aktív töltet szemcseméretét 5-200 µm közötti. Ezek a berendezések (folyadék adszorpciós berendezés, folyadék kromatográf stb.) alkalmasak a fenti elválasztási fokozatok megoldására.
Az igen szerteágazó adszorpciós-ioncserés műveletek közül a Preparatív folyadék kromatográfia művelet az egyik legfontosabb a vegyészmérnöki gyakorlatban. A
„preparatív” jelzőn – nem az analitikai készülékeket – a „termelő” folyadék kromatográfiás berendezéseket értjük.
2.2.5. A frontális adszorpció matematikai leírása
A kromatográfiás elválasztások, csak úgy mint sok egyéb tisztítási folyamat az oldatban levő komponensek egyensúlyi illetve nem-egyensúlyi adszorpcióján alapulnak.
A frontális adszorpció szükségképpen félfolyamatos üzemvitelű, mely során koncentráció-változtatást alkalmazunk lépcsős függvény szerint a műveleti egység belépő felületén.
Tételezzük fel példaként, hogy az i komponenst folyadékból távolítjuk el adszorpcióval, izoterm körülmények mellett.
Az adszorbenst előzetesen tökéletesen regeneráltuk. Az i komponens végighaladva az adszorbens tölteten megkötődik, majd az adszorbens kapacitásának kimerülése után áttör az adszorpciós oszlopon. Az áttörési görbe a z= H helyen felvett koncentráció-idő görbe
(
ci(
H,t) )
(2.9. ábra).2.9. ábra Egyedi komponens frontális kromatogramja
Ha az adszorbens töltet porozitása ε, az adszorpciós oszlop keresztmetszete A és a feldolgozandó folyadék térfogati sebessége B állandó az idő és hely függvényében, akkor a következő differenciál-egyenletet írhatjuk fel az adszorpciós oszlopra:
(
1)
=0
∂
⋅ ∂
⋅
+
∂
⋅ ∂
⋅
−
+
∂
⋅ ∂
z i z
i t
i
t A c t
A q z
B c ε ε (2-6)
Az axiális keveredést elhanyagoltuk és feltételeztük, hogy a szilárd fázis átlagos i komponens koncentrációja qi az adszorbens szemcsében. Az oszlop keresztmetszete A.
Tételezzük fel, hogy kis térfogati sebességeknél a folyadék és szilárd fázis az i komponensre nézve bármely z helyen és bármely t időpontban egyensúlyban van egymással (lokális, pillanatszerű egyensúly feltétele). A továbbiakban ezt egyensúlyi adszorpciónak nevezzük.
Az egyensúlyi adszorpciót leggyakrabban az adszorpciós izotermákkal ábrázolják.
Az adszorpciós izoterma kapcsolatot teremt a folyadékban oldott anyag koncentrációja és a szilárd fázisban lévő koncentráció között, a teljes koncentrációtartományra állandó hőmérsékleten. Az adszorpciós egyensúlyi izoterma legyen az alábbi összefüggéssel megadva:
( )
ii f c
q = (2-7)
az egyensúlyi adszorpció feltétele alapján:
(
1)
=0
∂
⋅ ∂
⋅
+
∂
⋅ ∂
∂
⋅ ∂
⋅
−
+
∂
⋅ ∂
z i z
i i c
i t
i
t A c t
c c
A q z
B c
i
ε
ε (2-8)
a (2-8)-at átrendezve:
( )
0 1
=
∂ + ∂
∂
∂
∂
− ∂
+ z
i t i
i c
i t
c z
c
c A q
B
i
ε ε
(2-9)
írjuk fel a ci =
( )
z,t függvény teljes deriváltját:−1
=
∂
⋅ ∂
∂
⋅ ∂
∂
∂
i t c z
i
c z z
t t
c
i
(2-10)
A (2-7) egyenlet alapján mondhatjuk, hogy qi legfeljebb ci-n keresztül függ z-től.
a (2-9) és (2-10) egyenletekből:
(
1)
1 '0
k v c
q u v
t z
i i
i
i c i c
c i
= +
∂
− ∂ +
=
=
∂
∂
ε ε
(2-11)
ha ci≈0 akkor
c K k q
ci
i i
ε ε ε
ε = −
∂
− ∂
=1 1
' (2-12)
A megfigyelt ci koncentrációjú folyadékelem haladási sebessége (
ci
u ) az adszorbens töltetben kisebb, mint a v0/ε lineáris sebesség. A haladási sebességet az adszorpciós egyensúlyi izoterma ci helyen vett deriváltja határozza meg.
Tekintsünk először egy olyan esetet, amikor az i komponens adszorpciós egyensúlya kedvezőtlen (2.10. ábra).