MTA DOKTORI ÉRTEKEZÉS
SEJTFELSZÍNI RECEPTOROK ÉS INTRACELLULÁRIS JELÁTVITELI RENDSZEREK JELENTŐSÉGE A SZÍV
PUMPAFUNKCIÓJÁNAK SZABÁLYOZÁSÁBAN
SZOKODI ISTVÁN
PhD értekezés tézisei
A DAAM formin hatása az aktin dinamikájára
Barkó Szilvia
Interdiszciplináris Orvostudományok Doktori Iskola D93 Iskolavezető: Dr. Sümegi Balázs
Program: B- 130: Funkcionális fehérjedinamika vizsgálata biofizikai módszerekkel Programvezető: Dr. Nyitrai Miklós
Témavezető: Dr. Nyitrai Miklós
Pécsi Tudományegyetem
Általános Orvostudományi Kar Biofizikai Intézet
2012
PÉCSI TUDOMÁNYEGYETEM KLINIKAI KÖZPONT
ÁLTALÁNOS ORVOSTUDOMÁNYI KAR SZÍVGYÓGYÁSZATI KLINIKA
PÉCS, 2013
dc_119_10
1. Tudományterületi háttér ...1
1.1. A szívizom-kontraktilitás regulációja a téma jelentősége és újszerűsége ...1
1.2. A szívizom-kontraktilitás alapjai ...2
1.3. A szívizom-kontraktilitás klasszikus szabályozó mechanizmusai ...4
1.3.1. A Frank-Starling-mechanizmus ...5
1.3.2. A Gregg-effektus ...6
1.3.3. A frekvencia-erőkifejtés összefüggése ...7
1.3.4. Az adrenerg receptorok szerepe a szívizom-kontraktilitás szabályozásában ...8
1.3.4.1. A β-adrenoceptorok szerepe ...8
1.3.4.2. Az α1-adrenoceptorok szerepe ...9
1.4. A kardiokinek szerepe a szívizom-kontraktilitás szabályozásában ...10
1.4.1. Adrenomedullin ...11
1.4.1.1. Az AM szerkezete, szintézise és szöveti eloszlása ...11
1.4.1.2. Az AM receptorai ...11
1.4.1.3. Az AM élettani hatásai ...12
1.4.1.4. Az AM hatása a szívizom-kontraktilitásra ...12
1.4.1.5. Az AM potenciális jelátviteli mechanizmusai ...12
1.4.1.6. A kamrai AM expresszió patológiás viszonyok mellett ...13
1.4.2. Apelin ...13
1.4.2.1. Az apelin felfedezése, szintézise és szöveti eloszlása ...13
1.4.2.2. Az apelin élettani hatásai ...14
1.4.3. Prolaktin-releasing peptid ...14
1.4.4. Endothelin-1 ...15
1.4.4.1. Az ET-1 felfedezése, szerkezete és szintézise ...15
1.4.4.2. Az ET-1 receptorai ...16
1.4.4.3. Az ET-1 hatása a szívizom-kontraktilitásra ...16
1.4.4.4. Az ET-1 potenciális jelátviteli mechanizmusai ...16
1.4.4.5. Az endogén ET-1 szerepe a szívizom-kontraktilitás szabályozásában ...19
2. Célkitűzések ...20
3. Módszerek ...21
3.1. Kísérleti modellek ...21
3.1.1. Kísérleti állatok ...21
3.1.2. Direkt miokardiális géntranszfer in vivo ...21
3.1.3. Bal kamrai nyomásterhelés modelljei ...21
3.1.4. Felnőtt kamrai szívizomsejtek izolációja ...22
3.1.5. Szívizomsejtek ciklikus mechanikai feszítése ...22
3.2. Vizsgálati módszerek ...23
3.2.1. Hemodinamikai mérések in vivo ...23
3.2.1.1. Vérnyomás monitorozása ...23
3.2.1.2. Balkamra-funkció vizsgálata in vivo ...23
3.2.2. Balkamra-funkció vizsgálata ex vivo ...23
3.2.2.1. Balkamra-funkció vizsgálata izolált patkányszív-preparátumon ...23
3.2.2.2. Balkamra-funkció vizsgálata izolált egérszív-preparátumon ...25
3.2.3. Kontraktilitás vizsgálata izolált szívizomsejteken in vitro ...25
3.2.4.1. Elektrofiziológiai vizsgálatok izolált pitvari preparátumon ...26
3.2.4.2. Patch-clamp mérések ...27
3.2.4.3. Multielektród-array ...27
3.2.5. Fluoreszcens vizsgálati módszerek ...28
3.2.5.1. Intracelluláris Ca2+-koncentráció mérése ...28
3.2.5.2. Intracelluláris pH és NHE aktivitás mérése ...28
3.2.5.3. Intracelluláris ROS termelődés mérése ...28
3.2.6. Western-blot analízis ...29
3.2.7. p38-MAPK assay ...29
3.2.8. RNS meghatározási módszerek ...30
3.2.8.1. Northern-blot analízis ...30
3.2.8.2. Kvantitatív RT-PCR analízis ...30
3.2.9. Radioimmunoassay ...30
3.2.10. Immuncitokémiai és immunhisztokémiai vizsgálatok ...30
3.2.11. Statisztikai elemzés ...31
4. Eredmények és megbeszélés ...32
4.1. Az adrenomedullin szerepe szívizom-kontraktilitás szabályozásában ...32
4.1.1. Az AM pozitív inotrop hatása ex vivo ...32
4.1.2. AM overexpresszió hatása a balkamra-funkcióra ...34
4.1.2.1. Adenovírus-mediálta AM géntranszfer ...34
4.1.2.2. AM overexpresszió hatása a pumpafunkcióra in vivo ...34
4.1.2.3. AM overexpresszió hatása a bal kamrai kontraktilitásra ex vivo ...36
4.1.3. Az AM pozitív inotrop hatását közvetítő jelátviteli folyamatok ...37
4.1.3.1. A cAMP−PKA jelátviteli út szerepe ...37
4.1.3.2. Az AM és az intracelluláris Ca2+ homeosztázis ...38
4.1.3.3. AM overexpresszió hatása egyes jelátviteli utak aktivációjára ...41
4.1.4. Nyomásterhelés hatása az AM bal kamrai génexpressziójára in vivo ...43
4.1.5. AM: a szívizom-kontraktilitás regulátora ...48
4.2. Apelin szerepe szívizom-kontraktilitás szabályozásában ...53
4.2.1. Az APJ-receptor lokalizációja ...53
4.2.2. Az apelin pozitív inotrop hatása izolált patkányszív-preparátumon ...53
4.2.3. Az apelin hatása a Frank-Starling-válaszra ...55
4.2.4. Az apelin hatása a kontraktilitásra izolált szívizomsejteken ...55
4.2.5. Az apelin pozitív inotrop hatásának specificitása ...57
4.2.6. Az apelin pozitív inotrop hatását közvetítő jelátviteli folyamatok ...58
4.2.6.1. A PLC−PKC jelátviteli út szerepe ...58
4.2.6.2. A NHE és a NCX szerepe ...59
4.2.6.3. Az apelin hatása a Ca2+- és K+-áramokra ...60
4.2.6.4. Az apelin hatása az intracelluláris Ca2+-tranziensekre ...61
4.2.7. Az apelin hatása a sejtek közötti kommunikációra ...62
4.2.8. A mechanikai túlterhelés hatása az apelin és az APJ-receptor génexpressziójára ... in vitro és in vivo körülmények között 63 4.2.9. Apelin: a szívizom-kontraktilitás regulátora ...65
4.3. A prolaktin-releasing peptid szerepe a szívizom-kontraktilitás szabályozásában 69.. 4.3.1. A PrRP pozitív inotrop hatása izolált patkányszív-preparátumon ...69
4.3.2. A PrRP pozitív inotrop hatásának jelátviteli folyamatai ...69
4.3.2.2. A PP1 és a PP2A proteinfoszfatázok szerepe ...70
4.3.2.3. A PKCα szerepe ...71
4.3.3. PrRP: a szívizom-kontraktilitás potenciális új szabályozója ...72
4.4. Az endothelin-1 szerepe a szívizom-kontraktilitás szabályozásában ...75
4.4.1. Az ETA- és az ETB-receptorok szerepe az ET-1 pozitív inotrop hatásának ... közvetítésében 75 4.4.2. A nitrogén-monoxid szerepe az ET-1 pozitív inotrop hatásának ... szabályozásában 76 4.4.3. A MAPK-ok szerepe az ET-1 pozitív inotrop hatásának regulációjában ...77
4.4.3.1. Az ERK1/2 szerepe ...77
4.4.3.2. A PLC−PKC jelátviteli út szerepe ...78
4.4.3.3. Az EGFR-transzaktiváció szerepe ...80
4.4.3.4. Az ERK1/2 potenciális célpontjai ...81
4.4.3.5. A p38-MAPK szerepe ...82
4.4.3.6. Az ERK1/2 és a p38-MAPK szignalizáció egymásra hatása ...83
4.4.3.7. A p38-MAPK potenciális célpontjai ...83
4.4.3.8. Az ET-1 hatása a vaszkuláris tónusra ...84
4.4.3.9. ERK1/2 és p38-MAPK: a kontraktilitás prominens modulátorai ...84
4.4.4. A ROS szerepe az ET-1 pozitív inotrop hatásának szabályozásában ...89
4.4.4.1. Az ET-1 hatása az intracelluláris ROS termelődésre ...89
4.4.4.2. A ROS szerepe az ET-1 pozitív inotrop hatásának közvetítésében ...90
4.4.4.3. A NAD(P)H-oxidáz eredetű ROS szerepe az ET-1 pozitív inotrop hatásának ... közvetítésében 90 4.4.4.4. A mitoKATP-csatornák szerepe az ET-1 pozitív inotrop hatásában ...91
4.4.4.5. A ROS produkció és az ERK1/2 aktiváció kapcsolata ...92
4.4.4.6. Az ET-1 hatása a vaszkuláris tónusra ...93
4.4.4.7. A ROS jelátviteli szerepe a kontraktilitás regulációjában ...93
4.4.5. Az endogén ET-1 szerepe a Gregg-effektus szabályozásában ...95
4.4.6. Az endogén ET-1 szerepe a Frank-Starling-válasz szabályozásában ...97
5. Az új tudományos eredmények összefoglalása ...104
6. A kutatási eredmények gyakorlati jelentősége ...106
7. Irodalomjegyzék ...108
8. Saját közlemények ...125
8.1. Az Értekezés alapjául szolgáló közlemények ...125
8.1.1. Eredeti közlemények ...125
8.1.2. Összefoglaló közlemények ...126
8.1.3. Könyvfejezet ...127
8.1.4. Hozzászólás ...127
8.2. A PhD Értekezésben nem szereplő további közlemények ...127
8.2.1. Eredeti közlemények ...127
8.2.2. Proceedings folyóiratban ill. könyvben ...129
8.3. A PhD Értekezésben szereplő közlemények ...129
8.3.1. Eredeti közlemények ...129
8.4. Összesített tudománymetriai adatok (lezárva: 2012. december 15.) ...130
9. Köszönetnyilvánítás ...132
AM adrenomedullin
Ang II angiotenzin II
ANP pitvari nátriuretikus peptid
AR adrenerg receptor
AT1-receptor angiotenzin II 1-es típusú receptor
AVP arginin-vazopresszin
BKCa mitokondriális Ca2+ által aktivált K+-csatorna BNP B-típusú nátriuretikus peptid
cAMP adenozin-3',5'-monofoszfát
CGRP kalcitonin gén eredetű peptid (“calcitonin gene-related peptide”) CRLR kalcitoninreceptor-szerű receptor (“calcitonin receptor-like
receptor”)
cTnC szívizom-specifikus troponin-C cTnI szívizom-specifikus troponin-I cTnT szívizom-specifikus troponin-T
DAG diacilglicerol
DP bal kamrai pulzusnyomás
dP/dtmax bal kamrai nyomásgörbe idő szerinti első deriváltjának a maximális pozitív értéke
dP/dtmin bal kamrai nyomásgörbe idő szerinti első deriváltjának a maximális negatív értéke
dTG dupla transzgénikus patkánytörzs
EC50 félmaximális hatás
EF ejekciós frakció
EGFR epidermális növekedési faktor-receptor ERK1/2 extracelluláris szignál által regulált kináz 1/2 ESPVR végszisztolés nyomás-térfogat összefüggés (“end-systolic pressure-volume relationship”)
ET-1 endothelin-1
ETA-receptor endothelin A-típusú receptor ETB-receptor endothelin B-típusú receptor
FS frakcionális rövidülés
GAPDH gliceraldehid-3-foszfát-dehidrogenáz GPCR G-proteinhez kapcsolt receptor
HB-EGF heparin-kötő EGF (“heparin-binding EGF”)
H2O2 hidrogén-peroxid
I1 inhibitor-1 fehérje
ICa befelé irányuló Ca2+-áram IK,sus tartós kifelé irányuló K+-áram
INa feszültségfüggő Na+-áram
Ito tranziens kifelé irányuló K+-áram IP3 inozitol-1,4,5-triszfoszfát
ir- immunoreaktív
JNK c-Jun N-terminális kináz
LacZ β-galaktozidáz
LVEDD bal kamrai végdiasztolés átmérő LVEDP bal kamrai végdiasztolés nyomás LVESD bal kamrai végszisztolés átmérő
MAPK mitogén által aktivált proteinkináz
MEA multielektród-array
MEK1/2 MAPK-kináz 1/2
MIA 5-(N-metil-N-izobutil)-amilorid
mitoKATP mitokondriális ATP-függő K+-csatorna
MKK3/6 MAPK-kináz 3/6
NCX Na+/Ca2+-cseremechanizmus
NHE Na+/H+-cseremechanizmus
NO nitrogén-monoxid
NOS NO-szintetáz
NOS1, nNOS neuronális típusú NO-szintetáz NOS2, iNOS indukálható NO-szintetáz
NOS3, eNOS endotheliális típusú NO-szintetáz
O2−• szuperoxid
p90RSK p90 riboszómális S6-kináz
PAMP proAM N-terminális 20 peptid
PBS foszfát puffer oldat
PCR polimeráz láncreakció
PIP2 foszfatidilinozitol-4,5-biszfoszfát
PKA proteinkináz-A
PKC proteinkináz-C
PLC foszfolipáz-C
PLN foszfolamban
PP1 foszfoprotein-foszfatáz-1
PP2A foszfoprotein-foszfatáz-2A
PrRP prolaktin-releasing peptid
RAMP receptor aktivitást módosító fehérje (“receptor activity modifying protein”)
RIA radioimmunoassay
ROS reaktív oxigén származékok
SD Sprague-Dawley patkánytörzs
S.E.M. mintaközép hibája (“standard error of mean”)
SERCA SR Ca2+-ATPáz
SHR spontán hipertenzív patkánytörzs
SL szarkomerhossz
SOD szuperoxid-dizmutáz
SR szarkoplazmatikus retikulum
τ bal kamrai nyomásgörbe exponenciális csökkenésének időállandója
τF580/640 SNARF-1 F580/640 fluoreszcencia hányados csökkenésének időállandója
TGR(mREN-2)27 egér REN-2 gént expresszáló transzgénikus patkánytörzs
Vmax maximális ballon volumen
WKY Wistar-Kyoto patkánytörzs
1. Tudományterületi háttér
1.1. A szívizom-kontraktilitás regulációja a téma jelentősége és újszerűsége A szívizomsejtek összehúzódó képessége, ún. kontraktilitása alapvető szerepet játszik a szív pumpafunkciójának meghatározásában és ezáltal a vérkeringés optimális szinten tartásában. Az egészséges szív tág határok között képes megfelelni a szervezet metabolikus igényeinek, edzett egyének esetén a szív perctérfogata akár 6-szorosára is emelkedhet. Másrészről a pumpafunkció progresszív romlása keringési elégtelenséghez, kis- és nagyvérköri pangáshoz vezethet (Opie, 2004). A szívelégtelenség a vezető halálokok közé tartozik a fejlett világban. A tünetekkel járó szívelégtelenség prevalenciája 2%-os Európában, mely így legkevesebb 15 millió embert érint (Mosterd és Hoes, 2007).
A szívelégtelenség kimenetele összevethető számos daganatos megbetegedéssel, a hospitalizált betegek 4 éves túlélése csupán 50%-ra tehető (Stewart és mtsai, 2001). A krónikus szisztolés szívelégtelenség kezelése megoldatlan. A széles körben elérhető szívtámogató szerek rövidtávon ugyan drámai módon javítják a balkamra-funkciót és csökkentik a klinikai tüneteket, azonban hosszú távú alkalmazásuk rontja a betegek túlélési esélyeit (Curfman, 1991; Landmesser és Drexler, 2007).
Eddigi ismereteink szerint a szívizom-összehúzódások erejét alapvetően a következő tényezők szabják meg: a Frank-Starling-mechanizmus, az erőkifejtés- frekvencia összefüggés, a szimpatikus idegrendszer aktivitása, valamint egyes keringő hormonok. Egyre több bizonyíték szól azonban a mellett, hogy egy további tényező is fontos szerepet tölthet be a szív pumpafunkciójának regulációjában. Az elmúlt másfél évtizedben számos endogén peptiderg rendszert írtak le a szívizomban, felvetve annak a lehetőségét, hogy ezen peptidek fontos szerepet játszhatnak a szív parakrin/autokrin szintű szabályozási folyamataiban. Fontos kiemelni, hogy az emberi szervezetben fellelhető 367 G-proteinhez kapcsolt receptor (GPCR) alig több mint felénél ismert jelenleg a receptort aktiválni képes endogén transzmitter (Vassilatis és mtsai, 2003; Levoye és Jockers, 2008). Továbbá, a GPCR-ok jelátviteli folyamataiban alapvető szerepet játszó proteinkinázok családjának 518 tagjából, tört részük esetében ismert csupán a fiziológiás szerepük (Manning és mtsai, 2002). Feltételezhető, hogy számos, ez idáig ismeretlen ligand-receptor rendszer vár felismerésre a szívben. A szívizom-kontraktilitást szabályozó fiziológiás mechanizmusok feltárása új molekuláris célpontok azonosításához vezethet, melyek potenciálisan új lehetőségeket teremthetnek a szívelégtelenség kezelésében.
1.2. A szívizom-kontraktilitás alapjai
A szívizomsejtek összehúzódását a plazmamembrán szabályos ütemben bekövetkező potenciálváltozása, az akciós potenciál indítja el. Az extracelluláris térből a depolarizáció során Ca2+ áramlik a szívizomsejtekbe, mely az intracelluláris Ca2+-raktárakból jelentős mennyiségű Ca2+ felszabadulását váltja ki. A nyugalmi, diasztolés szabad intracelluláris Ca2+-koncentráció ([Ca2+]i) 10-7 mol/L-ről közelítőleg 10-6 mol/L-re emelkedik, mely megindítja a kontrakció folyamatát. Majd a [Ca2+]i gyors csökkenését követően a szívizomsejtek relaxálnak (Fabiato, 1983; Bers, 2002a; Opie, 2004). A szívizomban lezajló elektromos változások, az intracelluláris Ca2+-tranziens alakulása és a mechanikai kontrakció/relaxáció között fennálló szoros időbeni összefüggéseket az 1. ábra mutatja be. Végső soron a szívizomsejtek kontrakciós erejét a Ca2+-tranziensek jellemzői és a kontraktilis fehérjék Ca2+ iránti érzékenysége szabja meg.
A plazmamembrán depolarizációja során a sejtbe irányuló Ca2+-áramért (ICa) a feszültségfüggő Ca2+-csatornák, döntően az L-típusú Ca2+-csatornák, nyitása felelős.
Ezen csatornák a plazmamembrán transzverzális tubulusain (T-tubulus) találhatóak. A miofilamentumokat körülölelő szarkoplazmatikus retikulum (SR) a sejtek fő Ca2+-raktára.
A SR terminális ciszternáin helyezkednek el a SR rianodin-szenzitív Ca2+-csatornái. Az L-
1. ábra: A kamrai szívizomsejtek Ca2+-homeosztázisa.
A piros nyilak a kontrakciót kiváltó Ca2+-mozgásokat, míg a zöld nyilak a relaxációt megalapozó, a szabad Ca2+ citoszólból történő eltávolításáért felelős mechanizmusokat jelzik. A fiókábra az akciós potenciál (AP), az intracelluláris Ca2+-szint változása és a kontrakció létrejöttének időbeliségét mutatja.
ATP, ATPáz; NCX, Na+/Ca2+-cseremechanizmus; PLN, foszfolamban; RyR, rianodin-szenzitív Ca2+- csatorna; SR, szarkoplazmatikus retikulum. (Bers, 2002a alapján módosítva)
típusú Ca2+-csatornák és a rianodin-szenzitív Ca2+-csatornák szoros funkcionális kapcsolatban állnak, az ICa aktiválja a SR Ca2+-csatornáit, melyeken keresztül jelentős mennyiségű Ca2+ szabadul fel a SR-ből (Fabiato, 1983). A citoplazmában a [Ca2+]i
nagymérvű emelkedése megindítja a vastag és a vékony filamentumok kapcsolódását, kezdetét veszi a kontrakció. Az erőgenerálás mértékét nagyban megszabja a Ca2+- tranziensek amplitúdója és időtartama. A relaxáció alapfeltétele, hogy a [Ca2+]i kellő szintre csökkenjen. Számos mechanizmus szolgálja a szabad Ca2+ eltávolítását a citoszólból. Ezek közül a legjelentősebbek: (i) a SR Ca2+-ATPáz (SERCA), mely a SR-ba pumpálja vissza a Ca2+-ot; (ii) a szarkolemmális Na+/Ca2+-cseremechanizmus (NCX, “Na+/ Ca2+ exchanger”) és (iii) a szarkolemmális Ca2+-ATPáz, melyek a sejten kívüli térbe juttatják a Ca2+-ot; végezetül (iv) a mitokondriális Ca2+-uniporter, mely révén a Ca2+ a mitokondriumokba kerül. Ezen mechanizmusok közül a SERCA játssza a legfontosabb szerepet, a Ca2+ eltávolításáért fajtól függően 70-92%-ban felelős (Bers, 2002a). A SERCA működését a foszfolamban (PLN) fehérje szabályozza. Defoszforilált állapotában a PLN kötődik a SERCA-hoz és gátolja a Ca2+-pumpa aktivitását. A PLN foszforilációja megszünteti a SERCA gátlását és fokozza a Ca2+ visszavételét a SR-ba. Fontos megjegyezni, hogy a relaxáció kifejezetten energiaigényes folyamat, mivel a citoszólból a Ca2+-ot jelentős koncentrációgradienssel szemben kell eltávolítani, az extracelluláris tér Ca2+-koncentrációja 2-3x10-3 mol/L, míg a SR-ban 5x10-4 mol/L az uralkodó koncentráció (MacLennan és Kranias, 2003).
A szívizomsejtek kontraktilis fehérjéi a vastag és vékony miofilamentumok. A vastag filamentumok miozin nehéz láncokból, kisebb részben egyéb fehérjékből (miozin könnyű láncok, miozinkötő C-fehérje) állnak. A vékony filamentumok vázát az aktin fehérjékből szerveződő dupla helikális struktúra adja. A miofilamentumok kötegei alkotják a miofibrillumokat. A miofibrillumok funkcionális egységei a hosszirányban ismétlődő szarkomerek. Az izomkontrakció mechanizmusára felállított un. “csúszó filamentum”
modell (“sliding filament” model) értelmében, a vastag és a vékony filamentumok közötti ciklikus interakció a miofibrilláris erőgenerálás alapja. A folyamat során a miozin, mint a szív molekuláris motorfehérjéje, alakítja át az ATP hidrolíziséből származó kémiai energiát mechanikai munkává minden egyes szívverés alkalmával. A kontrakció szabályozásában alapvető szerepet játszanak a tropomiozin és troponin fehérjék. Az aktinszálak közötti barázdában helyet foglaló tropomiozin nyugalmi állapotban elfedi az aktin miozinkötő helyeit. A szívizom kontraktilis fehérjéi közül a troponin komplex egyik alegysége, a szívizom-specifikus troponin-C (cTnC), képes megkötni a szabad Ca2+-ot, ez a tulajdonsága emeli a miofilamentumok Ca2+-szenzorává. A troponin komplex a vékony filamentumok vázához a troponin-T (cTnT), a tropomiozint kötő egység, révén kapcsolódik. Végül a troponin-I (cTnI) alegység az aktin-miozin interakciót gátló struktúra.
A szabad Ca2+ kötődése a cTnC-hez konformációs változást hoz létre a troponin komplexben, melynek következtében a tropomiozin mélyebbre kerül az aktinspirál
barázdájában, lehetővé téve a miozinfejek és az aktin filamentumok közötti interakciót, a keresztkötések kialakulását. A miozin aktin iránti affinitása a ciklus folyamán erős ingadozást mutat, mely a ciklicitás molekuláris alapja. Míg nyugalmi állapotban a miozin MgADP-t és anorganikus foszfátot (Pi) köt, addig az izomműködés aktivációja során a keresztkötésekről disszociál a Pi, amelyet a miozinfej erőt generáló konformációs változása követ. Ennek eredményeként a vékony és a vastag filamentumok elcsúsznak egymás mellett, kezdetét veszi a rövidülés. Ezt követően az MgATP az MgADP-t leszorítva kötődik a miozinhoz, a miozinfejek disszociálnak az aktinról. Miután a miozin újfent hidrolizálja az MgATP-t, a miozin újabb kötőhelyen kapcsolódik az aktinhoz.
Konstans MgATP-koncentráció mellett mindaddig folytatódik az aktin-miozin ciklus, míg az aktivátor Ca2+ kellően magas koncentrációban van jelen (Solaro és Rarick, 1998;
Maughan, 2005).
1.3. A szívizom-kontraktilitás klasszikus szabályozó mechanizmusai
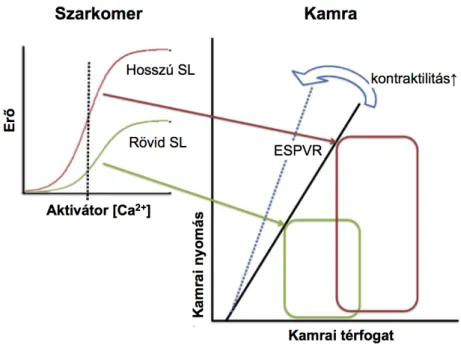
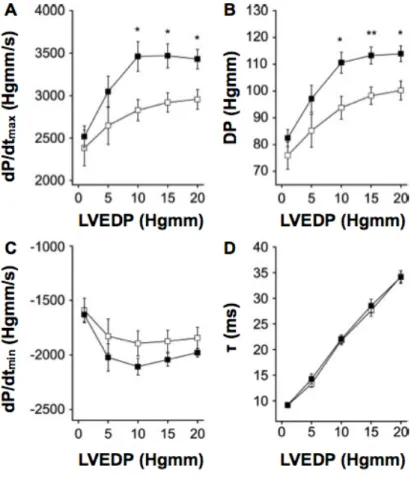
A szívizom-összehúzódások ereje tág határok között változhat. A kontrakciós erő nagyságát számos, párhuzamosan ható tényező bonyolult interakciója szabja meg. E tényezők közé tartozik a nyugalmi rosthosszúság, a szívfrekvencia, a szimpatikus idegrendszer aktivitása, valamint egyes keringő hormonok, illetve a szívben lokálisan termelődő autokrin/parakrin faktorok ingerhatása. A kontraktilitás primer fokozódásán a kontrakciók sebességének növekedését, és ezáltal magasabb csúcsfeszülés illetve csúcsnyomás elérését értjük változatlan előterhelés (“preload”), utóterhelés (“afterload”) illetve szívfrekvencia mellett. A kontraktilitásra, mint a szív inotrop állapotára is szokás hivatkozni (Opie, 2004). A szisztolés és diasztolés funkció számos indexe származtatható a kamrai nyomás és térfogat egyidejű meghatározása révén készített nyomás-térfogat hurkok analízisével. Számos paraméter mellett, a végszisztolés nyomás-térfogat összefüggés (ESPVR, “end-systolic pressure-volume relationship”) vizsgálatával jellemezhető a kontraktilitás, változó elő- és utóterhelés mellett, in vivo viszonyok között. A kontraktilitás fokozódása az ESPVR meredekségének növekedését vonja magával, ily módon azonos végdiasztolés térfogat mellett a végszisztolés nyomást a szív kontraktilis állapota határozza meg (2. ábra) (de Tombe és mtsai, 2010). Molekuláris szinten az emelkedett inotrop állapoton a [Ca2+]i és a kontraktilis fehérjék, elő- és utóterheléstől független, fokozott kölcsönhatását értjük. Ennek hátterében a Ca2+-tranziensek növekedése illetve a kontraktilis fehérjék Ca2+ iránti érzékenységének fokozódása áll.
Fontos kiemelni ugyanakkor, hogy a terhelési viszonyok illetve a szívfrekvencia megváltozásakor bekövetkező celluláris folyamatok nem különíthetőek el élesen a kontraktilitás primer változásakor tapasztaltaktól (Opie, 2004).
1.3.1. A Frank-Starling-mechanizmus
A szívizom rendkívül fontos tulajdonsága, hogy ütésről ütésre képes összehangolni a kamrai telődést a szív pumpafunkciójával változó hemodinamikai viszonyok mellett. A 19.
és 20. század fordulóján Frank békaszíven (Frank, 1895), míg Starling emlős szív-tüdő készítményen (Patterson és Starling, 1914) ismerte fel a fenti összefüggést, mely jelenségre kollektíven Frank-Starling-mechanizmusként hivatkozunk. E mechanizmus révén, adott kontraktilis állapot mellett a kamrai verővolumen (i) arányos a diasztolés telődés fokával (előterhelés), valamint (ii) a verővolumen fenntartható emelkedett artériás nyomással szemben (utóterhelés) az előterhelés fokozása révén (Opie, 2004; de Tombe és mtsai, 2010).
A szívben a kamrafunkciós görbe (ún. Starling-görbe) csúcspontja 2.2-2.3 µm szarkomerhossznál figyelhető meg, a Starling-görbe felszálló ága ezen pontig tart. A 2.2-2.3 µm szarkomerhossz esetén éri el a miozin és az aktin közötti kölcsönhatás a maximumát. E felett és ez alatt az aktin-miozin interakció hatásfoka csökken. A vázizomtól eltérően, a szívizom esetén a szarkomerhossz ritkán haladja meg a 2.4 µm-t, ugyanis a
2. ábra: A Frank-Starling-mechanizmus és a miofilamentumok hosszfüggő aktivációja.
Adott kontraktilis állapot mellett szoros összefüggés áll fenn a kamrai végszisztolés nyomás és a végszisztolés térfogat között (ESPVR, “end-systolic pressure-volume relationship”). A kontraktilitás fokozódása az ESPVR meredekségének növekedését vonja magával. A kamrai töltőtérfogat emelkedése (vörössel ábrázolt nyomás-térfogat hurok) a végszisztolés nyomás növekedésével jár, ami lehetővé teszi, hogy (i) adott szisztolés nyomás mellett emelkedjen a verőtérfogat, illetve (ii) megtartott maradjon a verőtérfogat emelkedett szisztolés nyomással szemben. A Frank-Starling-mechanizmus alapja a miofilamentumok Ca2+-érzékenységének módosulása a szarkomerhossz (SL) változása esetén. A kontrakciós erő és az aktivátor Ca2+-koncentráció közötti összefüggés felfelé és balra tolódik a SL növelésekor (zöld: rövid SL; vörös: hosszú SL), a miofilamentumok szintjén jelentkező fokozott erőgenerálás magasabb kamrai nyomást eredményez magasabb végszisztolés térfogat mellett (vörös nyomás-térfogat hurok). (de Tombe és mtsai, 2010 alapján módosítva)
szívizom masszív elasztikus apparátusa (extracelluláris kollagén, intracelluláris titin óriásmolekulák) védő szerepet játszik a miofilamentumok túlnyújtásával szemben.
Fiziológiás körülmények között a szív a Starling-görbe felszálló szakaszán működik.
Szívelégtelenségben a tágult szív a kamrafunkciós görbe csúcsán, ugyanakkor további hosszfüggő tartalékok hiányában kénytelen működni (Opie, 2004).
A Frank-Starling-mechanizmus a miofilamentumok Ca2+-érzékenységének hosszfüggő változásán alapul, a kontrakciós erő és az aktivátor Ca2+-koncentráció közötti összefüggés felfelé és balra tolódik a szarkomerhossz növelésekor (2. ábra). Az izom megnyújtásakor egyrészt fokozódik a cTnC Ca2+ iránti érzékenysége, másrészt nő a miofilamentumok közti kooperatív interakció hatékonysága (Allen és Kentish, 1985;
Campbell, 1997; Fuchs és Martyn, 2005; de Tombe és mtsai, 2010). Számos hipotézis született annak magyarázatára, hogy a szarkomerek szintjén milyen változások közvetítik a miofilamentumok hosszfüggő aktivációját. Felvetették, hogy nyújtás hatására a vékony és vastag filamentumok közti oldalirányú tér (“lattice spacing”) csökkenése váltja ki a Ca2+-érzékenység fokozódását (McDonald és Moss, 1995). Ettől eltérően, mások a vastag filamentumokat lehorgonyzó óriásfehérje, a titin, szerepét vetették fel. Mivel a fehérje rendelkezik mind aktin, mind miozin kötőhelyekkel, elképzelhető, hogy a titin feszülése befolyásolhatja az aktin-miozin interakciót (Linke, 2008). Habár a pontos mechanizmust nem ismerjük, bizonyítást nyert, hogy a jelenség nem az aktin-miozin ciklus egyes átmeneteinek kinetikai változásával, hanem az egyes aktin-miozin keresztkötések számának növekedésével magyarázható (Édes és mtsai, 2007). A szívizom feszítésekor fellépő azonnali választ a kontrakciós erő további, lassú fokozódása követi, melyet Anrep- effektusként tartanak számon (von Anrep, 1912). A gyors komponenstől eltérően, a lassú fázis hátterében a Ca2+-tranziensek növekedése áll (Kentish és Wrzosek, 1998), melyért feltehetőleg a szarkolemmális Na+/H+-cseremechanizmus (NHE, “Na+/H+ exchanger”) és a NCX fordított irányú működése felelős (Cingolani és mtsai, 2011).
1.3.2. A Gregg-effektus
A koronáriák perfúziós nyomásának növekedése esetén fokozódik a szív kontraktilitása és oxigénfogyasztása, melyre Gregg-effektusként hivatkozunk (Gregg, 1963). Kezdeti elképzelések szerint a perfúziós nyomás emelkedése a rosthosszúság növekedésével jár (kerti öntözőcső effektus, “garden hose effect”), mely a Frank-Starling-mechanizmus révén váltja ki a kontrakciós erő fokozódását (Arnold és mtsai, 1968). Kimutatták azonban, hogy papilláris izmon a Gregg-effektus jelentékenyebb, mint a Frank-Starling- válasz (Schouten és mtsai, 1992). Kiemelendő, hogy a Gregg-effektus ineffektív áramlási autoreguláció mellett jut szerephez, az autoreguláció tartományán belül a perfúziós nyomás növelése minimális hatást gyakorol a kontrakciók erejére (Bai és mtsai, 1994). A jelenlegi feltételezések szerint, a Gregg-effektusért döntően a kapillárisok szintjén
jelentkező térfogatváltozás felelős (Dijkman és mtsai, 1998). A perfúziós nyomás emelkedése a szívizomsejtek membránjának deformálódását váltja ki, mely a stretch- aktivált inocsatornák aktivációja révén fokozza a [Ca2+]i-t és a kontrakciós erőt (Lamberts és mtsai, 2002; Westerhof és mtsai, 2006). Nyitott kérdés, hogy a fokozott nyírófeszültség hatására az endotheliumból esetlegesen felszabaduló faktorok szerepet játszanak-e a Gregg-effektus kialakításában (Ramaciotti és mtsai, 1993; Dijkman és mtsai, 1997).
1.3.3. A frekvencia-erőkifejtés összefüggése
Bowditch békaizmon tett eredeti megfigyelése szerint, az ingerlési frekvencia növelése fokozta a kontrakciók erejét (Bowditch, 1878), mely jelenséget frekvencia-erőkifejtés összefüggésként illetve Treppe-hatásként tartanak számon. Az emlősfajok döntő hányadában az egészséges szív pozitív frekvencia-erőkifejtés összefüggést mutat, mely jelenség a nagyobb testű emlősök esetében kifejezettebb. A fizikai terhelés fokozódásakor ezen adaptív mechanizmus is hozzájárul a perctérfogat növekedéséhez (Opie, 2004;
Endoh, 2008). Szívelégtelenségben e mechanizmus kimerül, illetve egyenesen negatív frekvencia-erőkifejtés összefüggés tapasztalható mind in vitro (Pieske és mtsai, 1995), mind in vivo körülmények között (Hasenfuss és mtsai, 1994). Megjegyzendő, hogy a többi emlősfajtól eltérően a patkányok és az egerek szíve minimális pozitív (Georgakopoulos és Kass, 2001) illetve negatív Treppe-effektust mutat (Simor és mtsai, 1997; Tanaka és mtsai, 2005) a kísérleti körülmények függvényében.
A Frank-Starling-effektussal ellentétben, a pozitív frekvencia-erőkifejtés összefüggés hátterében a Ca2+ SR-beli fokozott felhalmozódása és a Ca2+-tranziensek következményes növekedése áll. A frekvencia emelésekor egyrészt fokozódik a Ca2+
sejtekbe történő beáramlása az L-típusú Ca2+-csatornákon keresztül, másrészt az intracelluláris Na+ akkumulációja csökkenti a Ca2+ eltávolítását a NCX előreható működésének (“forward”, Na+ be, Ca2+ ki) gátlása révén (Endoh, 2008). A negatív Treppe- effektus egér és patkányszív esetében feltehetőleg az akciós potenciál speciális karakterisztikájának köszönhető. Más fajokhoz (nyúl, tengerimalac, etc) képest, az akciós potenciál extrémen rövid és tüskeszerű. A frekvencia növelése a NCX előreható működésének és így a Ca2+-kiáramlásnak kedvez (Bers, 2002b; Tanaka és mtsai, 2005).
1.3.4. Az adrenerg receptorok szerepe a szívizom-kontraktilitás szabályozásában A perctérfogat szabályozásában alapvető szerepet játszik a szimpatikus idegrendszer aktivitása vészhelyzetek során (“fight-or-flight” response, “harcolj vagy menekülj” reakció).
A katekolaminok (noradrenalin, adrenalin) a szív adrenerg GPCR-aihoz (AR), döntően a β1-AR-okhoz, kötődve váltják ki komplex szívhatásaikat: fokozzák a szívösszehúzódások erejét, gyorsítják a relaxációt (pozitív lusitrop hatás) és emelik a szívfrekvenciát (pozitív chronotrop hatás). A β-AR-okhoz viszonyítva az α-AR-ok szerepe kevésbé tisztázott (Xiao és mtsai, 2006), feltehetőleg kóros állapotokban jutnak jelentősebb szerephez (Jensen és mtsai, 2011).
1.3.4.1. A β-adrenoceptorok szerepe
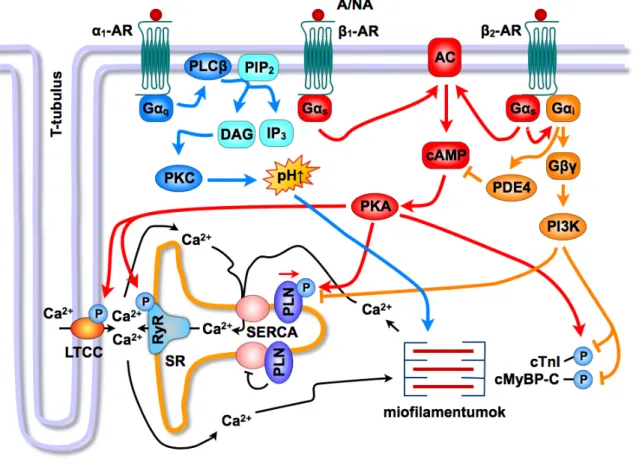
A szívizomsejtek β-AR-ai között a β1-AR altípus a domináns. A β1-AR a Gs-protein révén képes aktiválni az adenilátciklázt, mely az ATP-ből történő ciklikus adenozin-3',5'- monofoszfát (cAMP) átalakítást katalizálja. Az intracelluláris cAMP-szint emelkedése a proteinkináz-A (PKA) aktivációját váltja ki. A PKA szimultán képes fokozni számos fehérje foszforilációját (L-típusú Ca2+-csatornák, PLN, rianodin-szenzitív Ca2+-csatornák, cTnI, miozinkötő C-fehérje) (Dzimiri, 1999; Bers, 2002a; Xiao és mtsai, 2006). A β1-AR aktivációt követő pozitív inotrop hatás létrejöttében alapvető szerepet játszik az intracelluláris Ca2+-tranziensek növekedése, melynek a hátterében döntően a PLN fehérje PKA-függő foszforilációja áll (Luo és mtsai, 1994; Kiss és mtsai, 1997). A PLN Ser-16 helyen történő foszforilációja megszünteti a SERCA gátlását és fokozza a Ca2+
visszavételét a SR-ba. A SR telítődése révén növekszik a felszabadítható Ca2+
mennyisége és másodlagosan fokozódik a szívizom kontraktilis ereje (MacLennan és Kranias, 2003). Ezt a folyamatot támogatja a plazmamembrán L-típusú Ca2+-csatornáinak foszforilációja, az ICaL amplitúdójának növekedése serkenti a SR rianodin-szenzitív Ca2+- csatornáin keresztül történő Ca2+ felszabadulást (Bers, 2007) (3. ábra). Felvetették továbbá, hogy az auxotóniás kontrakciót végző szívizomban a cTnI foszforilációja is hozzájárul az összehúzódások erejének fokozódásához, azonban ennek pontos mechanizmusa még nem ismert (Layland és mtsai, 2004; Takimoto és mtsai, 2004). A pozitív lusitrop hatás hátterében is összetett folyamatok állnak. A PLN foszforilációja következtében, a SERCA fokozott működése az aktivátor Ca2+ SR-ba történő gyorsult visszavételét eredményezi (Luo és mtsai, 1994). Továbbá ismert, hogy a cTnI PKA-függő foszforilációja révén csökken a miofilamentumok Ca2+-érzékenysége (Wolska és mtsai, 2002) (3. ábra). A β1-AR-ok mellett a β2-AR-ok is megtalálhatóak a szívben, melyek a teljes β-AR populáció 20-30%-át teszik ki. Kiemelendő, hogy a β2-AR altípus a serkentő Gs mellett a gátló hatású Gi fehérjét is aktiválja (Xiao és mtsai, 2006). Ezzel összefüggésben, a β2-AR jelátvitelre jellemző, hogy az adenilátcikláz–cAMP–PKA aktiváció térben és időben korlátozott. Az L-típusú Ca2+-csatornák foszforilációján túl
egyéb fehérjék (pl. PLN) foszforilációja nem tapasztalható, a pozitív inotrop hatás mérsékeltebb, ahhoz pozitív lusitrop hatás nem társul (Jo és mtsai, 2002; Xiao és mtsai, 2006) (3. ábra).
1.3.4.2. Az α1-adrenoceptorok szerepe
Az α1-AR-ok stimulációja is képes befolyásolni a szív inotrop állapotát, azonban ez mind minőségileg, mind mennyiségileg eltér a β1-AR hatástól. Az α1-AR-ok által közvetített pozitív inotrop hatás szerényebb mértékű, döntően a miofilamentumok Ca2+ iránti érzékenysége fokozódik, az intracelluláris Ca2+-tranziensek nem vagy csak mérsékelten növekednek (Endoh és Blinks, 1988; Puceat és mtsai, 1990; Terzic és mtsai, 1992; Terzic és mtsai, 1993; Endoh, 2008). Az α1-AR-ok közül az α1A- és az α1B-AR altípusok jelenlétét mutatták ki emlősszívben, az egerektől az emberig bezárólag (Brodde és Michel, 1999;
Jensen és mtsai, 2009). Az α1A- és az α1B-AR altípusok ellentétes hatást gyakorolnak a kontraktilitásra: az α1A-AR-ok izgalma pozitív inotrop hatással jár, melyet az α1B-AR-ok ellensúlyozni igyekszenek (Gambassi és mtsai, 1998). Az α1-AR-ok a Gs–adenilátcikláz–
cAMP–PKA jelátviteli rendszertől független mechanizmusokra hatnak. Tradicionális
3. ábra: A β- és α-AR-ok inotrop hatását közvetítő jelátviteli mechanizmusok.
AC, adenilátcikláz; cMyBP-C, szívizom-specifikus miozinkötő C-fehérje; cTnI, szívizom-specifikus troponin-I; DAG, diacilglicerol; IP3, inozitol-1,4,5-triszfoszfát; LTCC, L-típusú Ca2+-csatorna; PDE4, foszfodiészteráz-4; PI3K, foszfatidilinozitol-3-kináz; PIP2, foszfatidilinozitol-4,5-biszfoszfát; PKA, proteinkináz-A; PKC, proteinkináz-C; PLC, foszfolipáz-C; PLN, foszfolamban; RyR, rianodin-szenzitív Ca2+-csatorna; SERCA, SR Ca2+-ATPáz; SR, szarkoplazmatikus retikulum.
felfogás szerint, az α1A-AR a Gq/11-fehérjén keresztül képes aktiválni a sejtmembránban található foszfolipáz-C (PLC) enzimet, mely a foszfatidilinozitol-4,5-biszfoszfátot (PIP2) inozitol-1,4,5-triszfoszfátra (IP3) és diacilglicerolra (DAG) hasítja. A DAG a proteinkináz-C (PKC) enzimet aktiválja, mely számos intracelluláris fehérje foszforilációs állapotát képes befolyásolni (Terzic és mtsai, 1993). Az α1-AR stimuláció a miofilamentumok Ca2+ iránti érzékenységét részben az intracelluláris pH (pHi) alkalikus irányú eltolása (Gambassi és mtsai, 1992; Terzic és mtsai, 1992), részben a miofilamentumok foszforilációja (Terzic és mtsai, 1993) révén fokozza. Felvetették a PKC szerepét ezen folyamatokban (Endoh és mtsai, 1993) (3. ábra), ugyanakkor mások szerint az α1-AR stimuláció a miofilamentumok Ca2+-érzékenységét a könnyű miozin láncok foszforilációja útján, a PKC-től függetlenül váltja ki (Andersen és mtsai, 2002). Az α1A-AR-tól eltérően, az α1B-AR altípus a Go- fehérjén keresztül fejti ki a hatását (O-Uchi és mtsai, 2008). A pontos mechanizmus, mely révén az α1B-AR-ok ellensúlyozni képesek α1A-AR-ok hatását a szívizom-kontraktilitásra, nem tisztázott.
1.4. A kardiokinek szerepe a szívizom-kontraktilitás szabályozásában
Egyre több adat szól a mellett, hogy a szívizmot felépítő sejtek közötti kommunikáció (szívizomsejt-szívizomsejt, endothelsejt-szívizomsejt, fibroblaszt-szívizomsejt interakció) fontos szerepet játszik a szív pumpafunkciójának szabályozásában. A szívizomzat sejtjei által szekretált fehérjék alapvető jelentőséggel bírnak ezen parakrin/autokrin szintű regulációban (Winegrad, 1997; Brutsaert, 2003; Prabhu, 2004; Westerhof és mtsai, 2006;
Kakkar és Lee, 2010). A vázizomból felszabaduló miokinek (Pedersen és mtsai, 2007) illetve a zsírszövet által szecernált adipokinek (Walsh, 2009) mintájára, a szívizom által termelt és szekretált fehérjéket összefoglalóan kardiokineknek nevezték el (Doroudgar és Glembotski, 2011). A kardiokinek számát megközelítőleg hatvanra teszik jelenleg (Frost és Engelhardt, 2007; Belmont és mtsai, 2008; Stastna és mtsai, 2010). A kardiokinek döntően a GPCR-ok családjához tartozó sejtfelszíni receptorokat aktiválva váltják ki biológiai hatásaikat. Az emberi szervezetben 367 GPCR található (Vassilatis és mtsai, 2003), ezek közül közelítőleg 200 receptor fejeződik ki a szívben (Hakak és mtsai, 2003;
Salazar és mtsai, 2007). Fontos kiemelni, hogy a GPCR-ok alig több mint felénél azonosították a receptort aktiválni képes endogén ligandot (Vassilatis és mtsai, 2003;
Levoye és Jockers, 2008), továbbá az ismert kardiokinek töredéke esetén tisztázott, hogy pontosan milyen biológiai funkcióval bírnak (Doroudgar és Glembotski, 2011). A jelen disszertációban bemutatott vizsgálatok célja az adrenomedullin, az apelin, az endothelin-1 és a prolaktin-releasing peptid szívizom-kontraktilitásra gyakorolt hatásának tisztázása volt.
1.4.1. Adrenomedullin
1.4.1.1. Az AM szerkezete, szintézise és szöveti eloszlása
Új vazoaktív peptidek után kutatva, 1993-ban Kitamura és munkatársai egy rendkívül erőteljes hipotenzív hatással bíró peptidet izoláltak humán pheocromocytoma kivonatból (Kitamura és mtsai, 1993a), melyet adrenomedullinnak (AM) neveztek el. A humán AM 52 aminosavból áll és egy hattagú gyűrű struktúrát tartalmaz. A patkány AM 2 aminosavval rövidebb és további 6 aminosavban tér el a humán peptidtől (Samson, 1999; Hinson és mtsai, 2000; Ishimitsu és mtsai, 2006; Yanagawa és Nagaya, 2007). Az AM mérsékelt homológiát mutat a kalcitonin gén eredetű peptiddel (CGRP, “calcitonin gene-related peptide”), ezért a peptidet a kalcitonin/CGRP/amylin családhoz sorolták (Hinson és mtsai, 2000). A humán preproAM 185 aminosavból áll, az N-terminális szakasz 21 aminosavból álló szignál peptidjének lehasításával jön létre a 164 aminosavat tartalmazó prohormon (Kitamura és mtsai, 1993b). A proAM további hasításával keletkezik a 20 aminosavból álló proAM N-terminális 20 peptid (PAMP, “proadrenomedullin N-terminal 20 peptide”), mely önálló biológiai hatásokkal rendelkezik (Kitamura és mtsai, 1994). Végezetül, a 93. és 94.
illetve a 148. és 149. pozíciók közötti hasítással jön létre az érett AM (Kitamura és mtsai, 1993b). Humán és patkány szövetek vizsgálata során jelentős AM expressziót találtak a mellékvese velőállományában, szívben, vesében, tüdőben és a központi idegrendszer számos régiójában (Sakata és mtsai, 1993; Ichiki és mtsai, 1994; Sakata és mtsai, 1994;
Isumi és mtsai, 1998). Az egyes szervek AM expressziója szoros korrelációt mutat azok vaszkularizáltságával, mivel az érendothelium sejtjei termelik legaktívabban ezt a fehérjét (Sugo és mtsai, 1994a; Sugo és mtsai, 1994b; Isumi és mtsai, 1998).
1.4.1.2. Az AM receptorai
Az AM mRNS szintekkel párhuzamosan, autoradiográfiás vizsgálatok specifikus AM kötőhelyek létét igazolták a szervezetben (Eguchi és mtsai, 1994; Owji és mtsai, 1995).
Munkánk kezdetekor nagyfokú bizonytalanság övezte, hogy az AM milyen receptoron keresztül váltja ki a hatását. Számos tanulmány szerint CGRP[8-37]-tel, egy CGRP- receptor antagonistával, az AM egyes biológiai hatásai kivédhetőek voltak (Eguchi és mtsai, 1994; Entzeroth és mtsai, 1995; Ikeda és mtsai, 1996), felvetve annak a lehetőségét, hogy az AM specifikus CGRP-receptorokon keresztül hat. Specifikus AM- receptorok után kutatva, a GPCR-ok működésének új paradigmáját ismerték fel. A CRLR (“calcitonin receptor-like receptor”) árvareceptor képes kötni mind az AM-t, mind a CGRP- t. A kötődés specificitását egy, a receptorhoz kapcsolódó, úgynevezett receptor aktivitást módosító fehérje (RAMP, “receptor activity modifying protein”) határozza meg. A CRLR/
RAMP-1 komplex CGRP receptorként funkcionál, míg a CRLR és a RAMP-2 illetve a RAMP-3 kombinációjához specifikusan az AM képes kötődni (McLatchie és mtsai, 1998;
Gibbons és mtsai, 2007). Továbbá, AM-receptorként működhet az L1 és az RDC-1 árvareceptor is (Kapas és mtsai, 1995a; Kapas és mtsai, 1995b; Hanze és mtsai, 1997;
Autelitano, 1998).
1.4.1.3. Az AM élettani hatásai
Az AM és specifikus kötőhelyeinek szöveti eloszlását tekintetbe véve valószínűsíthető, hogy a peptid autokrin/parakrin módon hatva fontos szerepet játszhat a szisztémás vérnyomás, a regionális véráramlás és a só-víz háztartás szabályozásában (Samson, 1999; Hinson és mtsai, 2000). Az AM intravénás infúziója potens, hosszan tartó hipotenzív hatást vált ki (Kitamura és mtsai, 1993a; Parkes, 1995; Parkes és May, 1997). Mind in vitro, mind in vivo körülmények között, a peptid egyike a legerőteljesebb koronária dilatátoroknak (Entzeroth és mtsai, 1995; Parkes és May, 1997; Yoshimoto és mtsai, 1998; Terata és mtsai, 2000; De Matteo és May, 2003). Az AM intrarenális adminisztrációja során jelentékenyen fokozódik a vese artériák véráramlása, a nátriurézis valamint a diurézis (Ebara és mtsai, 1994; Jougasaki és mtsai, 1995a; Hirata és mtsai, 1995). Mindezen effektusokat kiegészítik az AM központi idegrendszeri hatásai, a peptid csökkenti mind a víz-, mind a sófelvételt (Murphy és Samson, 1995; Samson és Murphy, 1997).
1.4.1.4. Az AM hatása a szívizom-kontraktilitásra
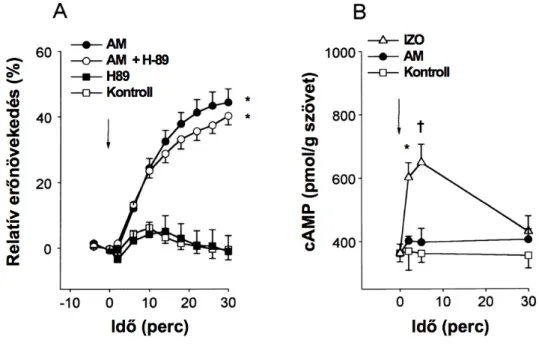
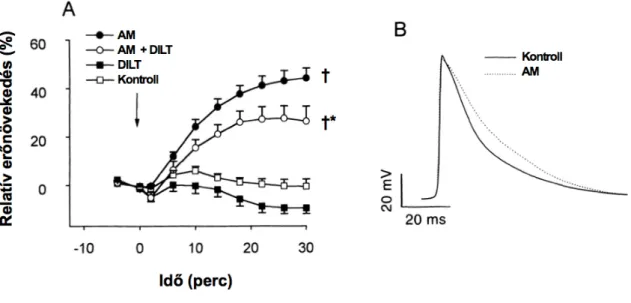
Az AM intravénás adását a perctérfogat jelentős mérvű emelkedése követi (Ishiyama és mtsai, 1993; He és mtsai, 1995; Parkes, 1995; Parkes és May, 1997; Charles és mtsai, 1997), felvetve, hogy a perifériás rezisztencia csökkenéséből adódó bal kamrai utóterhelés redukciója mellett a peptid a kamrai kontraktilitást direkt módon is befolyásolhatja. Az AM igen kifejezett kamrai génexpressziója (Sakata és mtsai, 1993), valamint specifikus kötőhelyeinek jelenléte (Owji és mtsai, 1995) egy autokrin/parakrin szintű szabályozó kör létezésére utal a miokardiumban. Ezzel összhangban, kezdeti adataink szerint az AM dózisfüggő pozitív inotrop hatást váltott ki spontán verő izolált patkányszíven (Szokodi és mtsai, 1996).
1.4.1.5. Az AM potenciális jelátviteli mechanizmusai
Ismert, hogy az AM számos sejttípus esetén, mint vaszkuláris simaizomsejtek (Eguchi és mtsai, 1994; Ishizaka és mtsai, 1994), érendothelsejtek (Shimekake és mtsai, 1995) és mesangialis sejtek (Chini és mtsai, 1995), képes fokozni az intracelluláris cAMP szintet.
Összhangban ezen megfigyelésekkel, az AM szívizomsejtekben is növeli a cAMP szintet (Ikeda és mtsai, 1996; Sato és mtsai, 1997), felvetve a lehetőségét, hogy az AM pozitív inotrop hatásának a közvetítésében szerepet játszhat az adenilátcikláz−cAMP−PKA
jelátvivő rendszer aktivációja, mely a kamrai kontraktilitás egyik kulcs regulátora (Bers, 2002a). Mindazonáltal, a peptid inotrop hatásáért felelős jelátviteli mechanizmusok feltárása várat magára.
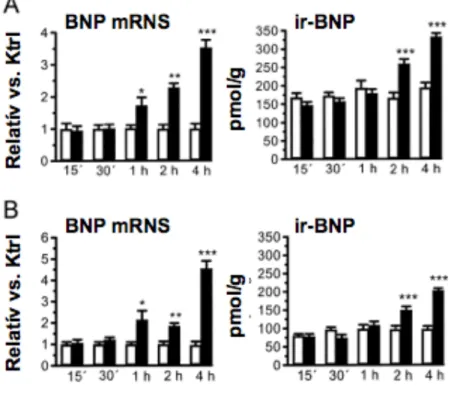
1.4.1.6. A kamrai AM expresszió patológiás viszonyok mellett
Az AM potenciális kórtani szerepére utal, hogy a peptid plazmaszintje magasabb szívelégtelen betegekben (Jougasaki és mtsai, 1995b; Nishikimi és mtsai, 1995; Kato és mtsai, 1996; Jougasaki és mtsai, 1996). Immunhisztokémiai vizsgálatok szerint fokozott AM szintézis jellemzi az elégtelen funkciójú humán kamrai izomzatot (Jougasaki és mtsai, 1995b), valamint a miokardium által szekretált AM feltehetőleg hozzájárul az emelkedett plazmakoncentrációhoz szívelégtelenségben (Jougasaki és mtsai, 1996). Továbbá, a szívhipertrófia egyes állatmodelljeiben is erőteljesebb kamrai AM termelődést figyeltek meg (Shimokubo és mtsai, 1995; Shimokubo és mtsai, 1996; Ishiyama és mtsai, 1997).
Azonban az AM bal kamrai expresszióját szabályozó mechanizmusok, illetve az aktiváció időbelisége nem ismert.
1.4.2. Apelin
1.4.2.1. Az apelin felfedezése, szintézise és szöveti eloszlása
Az APJ-receptort, az egyik legrégebbi árvareceptort, O'Dowd és munkatársai 1993-ban azonosították. A humán APJ-receptort kódoló gén a 11-es kromoszóma hosszú karján (q12.1) helyezkedik el. A receptor az angiotenzin II (Ang II) 1-es típusú receptorának (AT1- receptor) transzmembrán régiójával 54 %-os hasonlóságot mutat, azonban az Ang II nem kötődik hozzá (O'Dowd és mtsai, 1993). Tatemoto és munkatársai 1998-ban izoláltak marha gyomor kivonatból egy endogén peptidet, mely specifikusan kötődik az APJ- receptorhoz (Tatemoto és mtsai, 1998). A peptidet apelinnek nevezték el (APJ endogen ligand).
A humán apelin gén, mely a 77 aminosavból álló preproapelint kódolja, az X kromoszómán (q25–26.1) található. A prepropeptid C-terminális utolsó 23 aminosavból álló szakasza teljes mértékben megegyezik az ezidáig vizsgált emlős fajokban, valamint enyhe homológiát mutat az Ang II-vel. A preproapelin C-terminális szakaszának enzimatikus hidrolízise révén alakulnak ki a rövidebb, biológiailag aktív formák, mint az apelin-36, az apelin-17, az apelin-13, és az apelin-12 (Tatemoto és mtsai, 1998; Lee és mtsai, 2000). A felsorolt változatok mind képesek kötődni az APJ-receptorhoz, a rövidebb fragmentumok erőteljesebb biológiai hatással rendelkeznek. A 12 aminosavnál rövidebb formák azonban már nem képesek biológiai választ kiváltani (Tatemoto és mtsai, 2001).
Az apelin és az APJ-receptor expressziója széleskörűen kimutatható a központi idegrendszerben, a szívben, a tüdőben, a vesékben, a hasnyálmirigyben és a
zsírszövetben (Lee és mtsai, 2000; O’Carroll és mtsai, 2000; Hosoya és mtsai, 2000;
Kawamata és mtsai, 2001; Boucher és mtsai, 2005).
1.4.2.2. Az apelin élettani hatásai
A szöveti eloszlásának megfelelően, az apelin szerteágazó biológiai hatásokkal rendelkezik. A felfedezése óta eltelt időben kimutatták, hogy az apelin fokozza a diurézist (De Mota és mtsai, 2004), csökkenti a táplálékfelvételt, előnyösen befolyásolja a cukor- és zsíranyagcserét (Castan-Laurell és mtsai, 2011), antinociceptív (Xu és mtsai, 2009) illetve immunmodulátor effektussal bír (Horiuchi és mtsai, 2003; Leeper és mtsai, 2009).
Az apelinnel kapcsolatos kutatások korán felvetették, hogy a peptid szerepet játszhat az értónus szabályozásában. Immunhisztológiai módszerekkel igazolták az apelin és az APJ-receptor jelenlétét különböző kaliberű artériákban és vénákban, az erek endothelsejtjei mellett a peptid és a receptora kisebb mértékben a vaszkuláris simaizomsejtekben is megtalálható (Kleinz és Davenport, 2004?; Kleinz és mtsai, 2005).
Az apelin intravénás adása átmeneti vérnyomásesést váltott ki patkányokban (Lee és mtsai, 2000; Tatemoto és mtsai, 2001). A peptid vazodilatátor hatását feltehetőleg a NO közvetíti, mivel az effektus kivédhető volt L-NAME adásával, mely a NO-szintetázt (NOS) gátolja (Tatemoto és mtsai, 2001). Megjegyzendő, hogy az apelin vazokonstriktor hatást indukált humán vena saphenán illetve arteria mammarián, amennyiben az ereket megfosztották az endotheliumuktól (Katugampola és mtsai, 2001; Maguire és mtsai, 2009).
Az apelin prekurzora (Lee és mtsai, 2000; O’Carroll és mtsai, 2000;
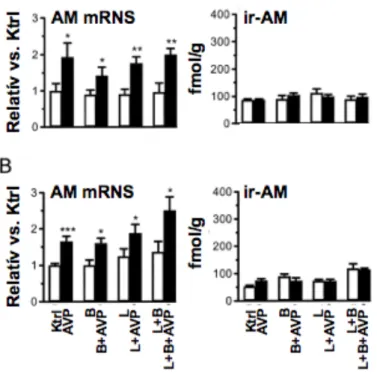
Kawamata és mtsai, 2001), továbbá maga az APJ-receptor is jelentős mennyiségben expresszálódik a szívben (O’Carroll és mtsai, 2000; Hosoya és mtsai, 2000), ami arra utal, hogy a peptid autokrin/parakrin módon befolyásolhatja a szívműködést, azonban munkánk kezdetekor ismeretlen volt az apelin-APJ rendszer funkcionális jelentősége a miokardiumban. Továbbá, feltárásra várt, hogy az APJ-receptor illetve az apelin expressziója miként változik a szívizomban patofiziológiás viszonyok között.
1.4.3. Prolaktin-releasing peptid
A prolaktin-releasing peptidet (PrRP) 1998-ban izolálták Hinuma és munkatársai marha hipotalamusz extraktumból, mint a hGR3/GPR10 árvareceptor potenciális ligandját (Hinuma és mtsai, 1998). A marha prepropeptid 98 aminosavból áll, melyből a 31 illetve 20 aminosavból álló érett peptidek (PrRP-31 és PrRP-20) képződnek. A marha PrRP-31 a humán, patkány illetve egér eredetű peptiddel 90, 83 illetve 80%-os azonosságot mutat (Hinuma és mtsai, 1998). Habár a peptidet eredetileg, mint a hipofízis elülső lebenyéből történő prolaktin szekréció lehetséges regulátorát azonosították, további vizsgálatok nem
támasztották alá azt az elképzelést, hogy a PrRP klasszikus hipofiziotrop hormonként hatna (Samson és mtsai, 2000; Sun és mtsai, 2005). Mára bizonyossá vált, hogy a PrRP sokrétű biológiai hatással bír (Sun és mtsai, 2005; Onaka és mtsai, 2010), többek közt befolyásolja a hipotalamusz-hipofízis-mellékvese tengely működését (Maruyama és mtsai, 2001), a táplálékfelvételt (Takayanagi és mtsai, 2008), valamint a fájdalomérzést (Laurent és mtsai, 2005). Felvetették továbbá, hogy a PrRP részt vehet a vérnyomás centrális szabályozásában. Éber patkányokban a PrRP intracerebro-ventrikuláris adagolása mellett szignifikánsan emelkedett az artériás középnyomás (Samson és mtsai, 2000). A PrRP kaudális ventrolaterális nyúltvelői területeken alkalmazott mikroinjekciója dózisfüggő módon növelte az artériás középnyomást, a szívfrekvenciát, valamint a renális szimpatikus idegek aktivitását (Horiuchi és mtsai, 2002). A peptid centrális hatásaitól eltérően, nem ismert, hogy a PrRP perifériás kardiovaszkuláris hatásokkal is rendelkezik- e. Számos perifériás patkány szövetben sikerült specifikus PrRP kötőhelyek létét kimutatni, melyek közül a szívben észlelték a legmagasabb fokú kötődést (Satoh és mtsai, 2000). Ezen megfigyelések felvetik, hogy a peptid direkt módon is befolyásolhatja a szívműködést.
1.4.4. Endothelin-1
1.4.4.1. Az ET-1 felfedezése, szerkezete és szintézise
Az 1980-as évek közepén ismerték fel, hogy az endothelium eredetű relaxáló faktor (EDRF, “endothelium-derived relaxing factor”) (Moncada és mtsai, 1976; Furchgott és Zawadzki, 1980) mellett az érendothelsejtek vazokonstriktor természetű faktorokat is termelnek (EDCF, “endothelium-derived constricting factor”) (Rubányi és Vanhoutte, 1985;
Hickey és mtsai, 1985; O’Brien és mtsai, 1987), melyek révén az endothelium parakrin módon képes befolyásolni az értónust (Rubanyi, 2011). Az ismeretlen vazoaktív faktort Yanagisawa és munkatársai 1988-ban azonosították. Sertés eredetű endothelsejt-kultúra felülúszójából izoláltak egy rendkívül erőteljes érszűkítő hatású peptidet, melyet endothelinnek neveztek el (Yanagisawa és mtsai, 1988). A peptid 21 aminosavból áll és 2 intramolekuláris diszulfid hidat tartalmaz. Az eredetileg izolált endothelin-1 (ET-1) mellett további két izoformát azonosítottak, az ET-2 és az ET-3 az ET-1-hez képest 2 illetve 6 aminosavban tér el (Inoue és mtsai, 1989). A humán ET-1 génje a 6-os kromoszómán helyezkedik el. A 203 aminosavból álló prepro-ET-1-ről furin-szerű endopeptidáz hasítja le a big-endothelint (Kedzierski és Yanagisawa, 2001). A biológiailag inaktív, 37-41 aminosavat tartalmazó intermediereket az endothelin konvertáló enzim alakítja át érett ET-1 molekulává (Xu és mtsai, 1994).
1.4.4.2. Az ET-1 receptorai
A szívben számos sejttípus, mint az endothelsejtek, vaszkuláris simaizomsejtek, szívizomsejtek és fibroblasztok, szekretálják az ET-1-et. A peptid hatásait két GPCR, az ETA- és az ETB-receptorok közvetítik (Kedzierski és Yanagisawa, 2001). Szívizomsejtek mindkét receptor altípust expresszálják, az ETA-receptorok jelentős dominanciája mellett (85−90%) (Molenaar és mtsai, 1993).
1.4.4.3. Az ET-1 hatása a szívizom-kontraktilitásra
A peptid sokrétű biológiai effektussal rendelkezik a szívben. Az értónus szabályozásán túl, az ET-1 hatással van a szívizomsejtek növekedési folyamataira, a fibroblasztok proliferációjára, illetve a sejtek túlélésére (Kedzierski és Yanagisawa, 2001; Kohan és mtsai, 2011). Továbbá, az ET-1 a szívizom-kontraktilitás erőteljes stimulátora (Sugden, 2003; Brunner és mtsai, 2006; Endoh, 2008). A vizsgált emlősfajok zömében az ET-1 pozitív inotrop hatást váltott ki, így patkány (Kelly és mtsai, 1990; Krämer és mtsai, 1991), nyúl (Li és mtsai, 1991; Takanashi és Endoh, 1991; Wang és mtsai, 2000; Chu és Endoh, 2005), tengerimalac (Ishikawa és mtsai, 1988), macska (Cingolani és mtsai, 2006; De Giusti és mtsai, 2008), kutya (Chu és mtsai, 2003; Czóbel és mtsai, 2009) és emberi miokardiumban illetve izolált szívizomsejtekben (Schomisch-Moravec és mtsai, 1989;
Pieske és mtsai, 1999; MacCarthy és mtsai, 2000; Goldberg és mtsai, 2000). Egér szívizomsejteken ugyanakkor ellentmondásos a peptid hatása, egyes szerzők szerint az ET-1 pozitív (Pi és mtsai, 2002), mások szerint negatív inotrop effektust hozott létre (Sakurai és mtsai, 2002; Nishimaru és mtsai, 2007; Nishimaru és mtsai, 2008). Felnőtt egérszíven azonban nem ismert, hogy az ET-1 miként befolyásolja a kontraktilitást.
Számos adat utal arra, hogy az ET-1 inotrop hatását az ETA-receptorok közvetítik (Goldberg és mtsai, 2000; Takeuchi és mtsai, 2001; De Giusti és mtsai, 2008), ugyanakkor az ETB-receptorok szerepe nem tisztázott.
1.4.4.4. Az ET-1 potenciális jelátviteli mechanizmusai 1.4.4.4.1. A PLC-PKC jelátviteli út
Az ET-1 pozitív inotrop hatása hasonlóságot mutat az α1-AR-ok effektusával: elsősorban a miofilamentumok Ca2+-érzékenysége nő, az intracelluláris Ca2+-tranziensek mérsékelten fokozódnak csupán (Yang és mtsai, 1999; Talukder és mtsai, 2001). Az ET-1 inotrop effektusát közvetítő pontos szubcelluláris mechanizmusok feltárásra várnak. Az ET-1 jelátvitelére magas fokú komplexitás jellemző. Klasszikus módon, az ETA-receptor a Gq/11- fehérjén keresztül képes aktiválni a PLC–PKC kaszkádot szívizomsejtekben (Sugden, 2003; Brunner és mtsai, 2006). Számos vizsgálat szerint az ET-1 pozitív inotrop hatásáért döntően a NHE PKC-függő aktivációja felelős (Krämer és mtsai, 1991; Chu és mtsai,
2003; Zolk és mtsai, 2004) A fokozott NHE aktivitás intracelluláris alkalizációhoz vezethet, mely növeli a miofilamentumok Ca2+-mal szembeni érzékenységét (Krämer és mtsai, 1991; Goldberg és mtsai, 2000). Másrészt, a NHE aktivációja révén intracelluláris Na+ akkumuláció jöhet létre, mely indirekt módon fokozhatja a NCX fordított irányú működését (Na+ ki, Ca2+ be), és így hozzájárulhat a sejtekbe történő megnövekedett Ca2+
beáramláshoz (Yang és mtsai, 1999; Perez és mtsai, 2001). A NCX mellett, az ET-1 az L- típusú Ca2+-csatornák nyitása révén is elősegítheti az intracelluláris Ca2+-tranziensek növekedését (Watanabe és Endoh, 1999).
1.4.4.4.2. Az ERK1/2 és a p38-MAPK jelátviteli út
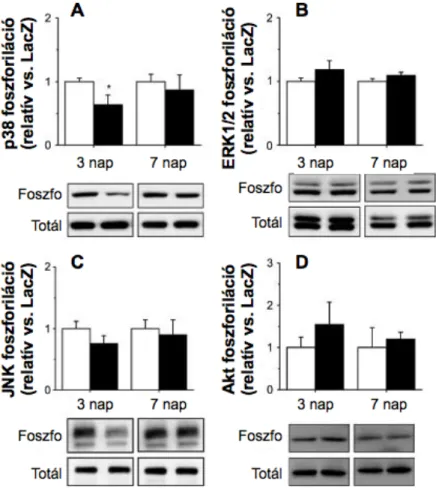
Szívizomsejtekben az ET-1, a PLC–PKC kaszkád mellett, potens aktivátora a mitogén által aktivált proteinkinázoknak (MAPK-ok) (Sugden, 2003; Schorlemmer és mtsai, 2008).
Felvetették, hogy a MAPK-ok családjába tartozó extracelluláris szignál által regulált kinázok (ERK1 és ERK2, továbbiakban ERK1/2) és a p38-MAPK szerepet játszik a szívhipertrófia és a szívelégtelenség kialakulásában (Bueno és Molkentin, 2002; Fuller és mtsai, 2008; Rose és mtsai, 2010). Kezdetben, mind az ERK1/2-ről, mind a p38-MAPK-ról úgy tartották, hogy serkentik a szívizomsejtek növekedését GPCR stimuláció (Ang II, ET-1) hatására (Clerk és mstai, 1998). Azonban transzgénikus állatmodelleken nyert adatok szerint a MAPK-ok eltérő feladatot töltenek be a hipertrófiás válaszban. Az ERK1/2-t foszforiláló MAPK-kináz 1 (MEK1) aktivált formájának szívizom-specifikus overexpressziója mérsékelt fokú koncentrikus bal kamrai hipertrófiát eredményez, az állatok hosszú távon sem mutatnak hajlamot a szívelégtelenség kialakulására (Bueno és mtsai, 2000). Ezzel szemben, a p38-MAPK-t aktiváló MAPK-kinázok, az MKK3 illetve az MKK6, domináns negatív mutáns változatát expresszáló egerekben a miokardiális p38- MAPK jelátvitel krónikus gátlása progresszív szívhipertrófiához, majd szívelégtelenséghez vezetett. Az eredmények arra utalnak, hogy a p38-MAPK gátolja a szívizomsejtek növekedési folyamatait in vivo körülmények között (Braz és mtsai, 2003). A patológiás viszonyoktól eltérően, keveset tudunk a MAPK-ok fiziológiás szerepéről a szívben. Az MKK3bE overexpressziója esetén, a tartósan fokozott p38-MAPK aktivitás a kontrakciós erő csökkenéséhez vezetett patkány eredetű izolált szívizomsejteken (Liao és mtsai, 2002). Ugyanakkor a p38-MAPK akut aktivációjának jelentősége a szívműködés szabályozásában tisztázásra vár. Továbbá, nem ismert, hogy az ERK1/2 direkt képes-e befolyásolni a szívizom kontraktilis állapotát intakt szíven.
1.4.4.4.3. A nitrogén-monoxid szerepe
A nitrogén-monoxid molekula a kardiovaszkuláris rendszer fontos regulátora mind élettani, mind patofiziológiás állapotokban (Pacher és mtsai, 2007). Az NO központi szerepet tölt be az értónus szabályozásában, mivel az NO közvetíti az endotheliumfüggő vazodilatációt
(Furchgott és Zawadzki, 1980; Palmer és mtsai, 1987). ET-1 stimuláció során az érsimaizomsejtek ETA-receptorai vazokonstriktor hatást közvetítenek (Cannan és mtsai, 1995), míg az érendothelsejtek felszínén található ETB-receptorok izgalma NO felszabadulást vált ki, mely az érszűkítő effektus ellen hat (Lerman és mtsai, 1992; Wang és mtsai, 1994).
A miokardiumban termelődő NO fontos szerepet játszik a szívizom-kontraktilitás szabályozásában (Kelly és mtsai, 1996; Massion és mtsai, 2003; Ziolo és mtsai, 2008). Az NO szintézisét a NOS enzimek végzik, miközben L-argininből L-citrullint hoznak létre. A NOS két izoformája, a neuronális típusú NOS (nNOS, NOS1) (Xu és mtsai, 1999) és az endothelialis típusú NOS (eNOS, NOS3) (Seki és mtsai, 1996), folyamatosan, alacsony szinten termeli az NO-t Ca2+/kalmodulin-dependens módon a szívben (Ziolo és mtsai, 2008; Zhang és Casadei, 2012). A harmadik izoforma, az indukálható NOS (iNOS, NOS2), kizárólag patofiziológiás viszonyok között (iszkémiás-reperfúziós károsodás, szívelégtelenség) expresszálódik. Az iNOS nagy mennyiségű NO-t képes termelni Ca2+- independens módon (De Belder és mtsai, 1993).
Fiziológiás körülmények mellett az NO termelődés jelentősen gyengítette a β-AR inotrop választ in vitro (Balligand és mtsai, 1993; Ebihara és Karmazyn, 1996; Gyurko és mtsai, 2000; Godecke és mtsai, 2001) és in vivo (Keaney és mtsai, 1996). Ugyanakkor más vizsgálatok szerint a NOS farmakológiai illetve genetikai inaktivációja nem fokozta a β-AR inotrop effektust (Vandecasteele és mtsai, 1999; Martin és mtsai, 2006). Habár a fent ismertetett módon, az NO képes ellensúlyozni az ET-1 vazokonstriktor hatását (Lerman és mtsai, 1992; Wang és mtsai, 1994), tisztázásra vár, hogy a peptid inotrop effektusát az NO képes-e módosítani.
1.4.4.4.4. A reaktív oxigén származékok szerepe
A normál aerob anyagcsere folyamán a szívben reaktív oxigén származékok (ROS,
“reactive oxygen species”) képződnek, mint a szuperoxid (O2−•) és a hidrogén-peroxid (H2O2). Fiziológiás viszonyok között a ROS termelődést az antioxidáns molekulák hatékonyan képesek ellensúlyozni (Giordano, 2005). Patofiziológiás állapotokban, mint krónikus szívelégtelenségben, azonban a ROS termelődése jelentékenyen fokozódik.
Ismert, hogy az oxidatív stressz számos kóros folyamatot indít el, hozzájárul a szívhipertrófia, az apoptotikus és nekrotikus sejtelhalás, valamint az intersticiális fibrózis kialakulásához, melyek összességében a szív pumpafunkciójának zavarához vezetnek (Sawyer és mtsai, 2002; Giordano, 2005; Zimmet és Hare, 2006; Murdoch és mtsai, 2006). A fokozott ROS produkció közvetlenül is ronthatja a kontraktilitást, a szabadgyök molekulák csökkenthetik a SERCA (Xu és mtsa, 1997; Li és mtsai, 2006; Tang és mtsai, 2010) és az L-típusú Ca2+-csatornák működését, a cisztein molekulák tiolcsoportjainak (-SH) oxidációja révén (Gill és mtsai, 1995; Fearon és mtsai, 1999; Zima és Blatter, 2006).