Ethyl 2-Cyclopentanonecarboxylate and Its Importance in Syntheses*
R O L A N D M A Y E R
Institut jur Organische Chemie der Technischen Universitdt Dresden
P r e p a r a t i o n
The Dieckmann reaction (1,2), an intramolecular Claisen conden
sation (3) of the diesters of adipic acid leads to esters of 2 - c y c l o - pentanonecarboxylic acid (4-6). There are considerable difficulties in working with larger amounts especially with respect to the condensing agent. The applications therefore have steadily increased with the dis
covery of more convenient cyclization agents. Diesters of adipic acid in large quantities may now be cyclized with sodium alcoholate (7a) with yields up to 8 0 % . This cyclization is quite superior both to the original Dieckmann procedure, and to the improved procedure of Pink- ney ( 7 ) , although a substitute for the ethylate would be desirable.
In older work Neunhoeffer and Paschke (8) observed the formation of 2-cyclopentanonecarboxylates during the slow distillation of the monoesters of adipic acid which, however, in the cyclization procedures gave only a minimum yield ( < 1 % ) (9). Direct condensation of c y c l o - pentanone with formates (9) gave yields up to 1 0 % ; here, also, a suit
able condensing agent is lacking. Likewise, a synthesis of the ester from cyclopentanone and oxalate ester failed because of the stability of the oxalo intermediate (10). The action of oxalyl bromide on cyclopentanone with simultaneous splitting out of carbon monoxide was a failure (10a).
Thus, in contrast to cyclohexanone, the peculiarities of cyclopentanone hinder the reaction with oxalyl bromide. The cyclization of the esters of α-haloadipic acid with magnesium (11) (yield: 5 0 % ) or of the esters of butanetricarboxylic acid with ethylate (12) (yield: 4 0 % ) , as well as the action of oxalate esters on the monoester of adipic acid (13), were uneconomical. The high yields recently attained in the cyclization reac
tion (13a) through the use of sodamide could not be obtained in our group. On the other hand, a thorough investigation of the direct cycliza
tion of the monoester monochloride of adipic acid or of adipyl chloride in alcoholic solution seems promising, since it has been reported that adipyl chloride in benzene, ethanol, and triethanolamine formed, with a trace of hydrogen chloride, a cyclic ester in 4 0 % yield (13b).
* Dedicated to the 70th birthday of Dr. W . Treibs.
101
102 R O L A N D M A Y E R
The reaction of solid carbon dioxide with cyclopentanone in the presence of sodamide, and the exchange of the sodium salt with methyl sulfate, gave the methyl ester with a maximum 1 0 % yield (86).
Physical Properties
The colorless, quite fluid, oily, ethyl 2-cyclopentanonecarboxylate is distinguished by its deep blue-violet ferric chloride reaction (alcohol).
On shaking with dilute alkali hydroxides, the white alkali salt precipi- tates; on saponification with alkali, adipic acid is formed, while treat- ment with acids gives cyclopentanone. The phenylhydrazone (m.p. 93°) or the semicarbazone (m.p. 143°) are formed only under specific condi- tions and are not suitable for identification. The I R , Raman and U V
spectra of the ethyl ester, which is prepared in better yield than the methyl ester and also enters more readily into condensation reactions, have been recorded several times (14,14a)-
Enol
The ester of 2-cyclopentanonecarboxylic acid is enolized only slightly in polar solvents, much more so in nonpolar (14,15), which is to be considered in the evaluation of the course of the reaction. [The enol content in alcoholic solution at 25° is 4 - 5 % (14, Ha, 15,15a), in the gaseous state, 2 7 . 5 % (15b).] For the reaction of the enol hydroxyl with diazomethane and orthoformic ester see ref. (15c).
C OtR CO.R
&° -
O H
Preparatively important alkali metal salts are formed from the enol:
(1) by drop wise addition of the ester to a hydrocarbon containing
"powdered" sodium or potassium (unreacted metal fosters secondary reactions and the danger of explosion);
(2) by dropwise addition of the ester to sodium or potassium ethox- ide, dry or dissolved in alcohol (the temperature must be low in order to minimize ring cleavage through alcoholysis);
(3) by dropwise addition of the ester to a water-alcohol solution of potassium hydroxide (16), the potassium salt forming spontaneously.
Since this method has been tested in many cases and found satisfactory so far in all requirements (see Example 2 at the end of this chapter), the use of alkali metals or their alkoxides for the conversion of the cyclopentanone carboxylate to its sodium or potassium salt is now
OR
E T H Y L 2- C Y C L O P E N T A N O N E C A R B O X Y L A T E 103 necessary only in special cases. I t should be noted, however, when using the methyl ester in methanol solution that the sodium salt is obtained in better yield than the potassium salt. Side reactions can occur here through further condensation.
C o n d e n s a t i o n w i t h H a l i d e s
Halides react with the enol salt to form a C-substituted derivative and the alkali metal halide:
COaR CO,R
r ( + B r R ^ > r ~ t ~Rl
OK ο The potassium is split off as a cation (5), the remaining anion ( a )
is stabilized ( b ) and serves as the reactive partner for the positive group, R i+ (9):
COsR COsR COtR
- K+ ,—S Stabilization
"x ^ ^ \ "
OK x> o ~
In Table 1 all of the halide condensation reactions which have been published are summarized. I t can be seen that as the molecular weight of group R increases, there is greater need for higher boiling solvents and longer heating periods. Deviating from the customary condensation procedure, Weizmann and co-workers (17) used potassium hydroxide in solvents of the acetal type, while Arnold et al. (18) carried out the direct condensation of itaconic ester with cyclopentanone ester in the presence of Triton B. While the substitution reactions recorded in Table 1 are assumed b y all investigators to have taken place in the C - l posi
tion and in most cases this has been demonstrated directly through cleavage, the condensation with β-diethylaminoethyl chloride (118) in benzene with sodium hydride or with potassium ί-butoxide in i-butyl alcohol furnishes 6 2 % of the O-alkylated product; this is said to occur also in the condensation with 2-chlorocyclohexanone (120). If these par
ticular reactions can be made more general, then quite new possibilities for the synthesis of the oxa-compounds with an attached five-membered ring appear likely. Diethylaminoethyl chloride, however, also was con
densed without any indication of O-alkylation (117). T h e statement (121) that in condensation with bromoacetone in alcohol solution substi
tution occurs not only on the C - l position, but also on the C-3 atom, if no cleavage and subsequent cyclization is assumed, cannot be explained at this time.
TABLE 1 CONDENSATION WITH HALIDES Yield B.p. Halide R Procedure (%) (7mm) Ref. Methyl iodide -CH3 NaOC2H5, cooling 60 105713 19, 20, 21 99 9Ά 9L Methyl bromide —CH3 25 Ethyl iodide —C2H5 NaOC2H5 or Na, 6 hr, toluene 74 10077 20, 23, 26 27, 28 η-Propyl bromide —C3H7 NaOC2H5 85 136.5728 23 t-Propyl iodide —CH(CH3)2 12 hrs, xylene or Na-K alloy in xylene 59 136-7734 20, 29, 30, 31 i-Butyl bromide —CH2CH (CH3)2 KOH in acetaldehyde dipropylacetal 34 138-42715 17 w-Octyl bromide —(CH2)7CH3 K, 7 hr, xylene 85 157-6571 32 n-Decyl bromide —(CH2)9CH3 K, 10 hr, xylene 50 17473 33 4-Phenoxybutyl iodide —(CH2)40C6H5 KOC(CH3)2C2H5, xylene, 45 hr at 110° 67 Me ester 34 under N2 178-8170.08 4-Diethylamino-2-butanone —CH2CH2COCH3 Direct condensation in benzene — 115-2070.1 35 Benzyl bromide —CH2C6H5 K, xylene, cold. If warm, ring cleavage 55 154-571 33 takes place (36) Benzyl chloride —CH2CeH5 KOH in acetaldehyde dipropylacetal at 65 186-90714 17 α-Naphthylethyl bromide (GH^CloH? / 0 Κ in benzene 44 188-970.1 37 3- (m-Methoxyphenyl) w-propyl iodide —(CH2)3C6H4OCH3 K, 8 hr, xylene
—
165-7070.1 38 Allyl bromide —CH2CH=CH2 Na—
125711 39 Cinnamyl bromide —CH2CH=CHCeH5 Na, 10 hr, xylene 60 17973 33 Methyl chloroformate —C02CH3 Na in toluene 95 127-877 40 Methyl bromoacetate —CH2C02CH3— —
14577 40 Ethyl bromoacetate —CH2CO2C2H5 NaOC2H5, or Na or Κ in hydrocarbon 60-85 163-7715 9, 17, 41-47 Ethyl chloroacetate —CH2CO2H5 ——
— 9, 46 104 ROLAND MAYEREthyl 3-bromopropionate Ethyl 2-bromopropionate 7-Bromobutyronitrile Ethyl 5-iodopentanoate Ethyl 11-bromoundecanoate Diethyl a-bromosuccinate Diethyl <*-bromoadipate Diethyl itaconate 7-Chlorocrotyl chloride Ethyl 7-bromo-/3-methyl crotonate Methylene bromide Ethylene bromide 1,3-Dibromopropane l-Bromo-3-chloropropane 1,4-Dibromobutane 1,6-Dibromohexane Methyl bromide Methyl iodide Dimethyl sulfate
—CH2CH2C02C2H5 — 75 187-90/18 47, 48 —CH(CH3)C02C2H5
—
— — 49 —(CH2)3CN—
— 180-274 50 —(CH2)4C02C2H5 Na in toluene—
180-274 51 (—CH2) 10CO2C2H5 Κ in benzene-toluene, sealed tube — Cannot be 52 (—CH2) 10CO2C2H5 distilled —CHCO2C2H5 1 Na in benzene — 211-5710 53 1 CH2C02C2H5 —CHC02C2H5 NaOC2H5; better Κ in toluene 28 216-2076 54 1 (CH2)3C02C2H5 —CH2CHC02C2H5 1 Direct condensation, Triton Β—
— 18 1 CH2C02C2H5 —COCH=CHCH2Cl NaOC2H5 at 0° — 81-770.1 55 —CH2C=CHC02C2H6 - 1 — — 56 1 CH3 Μ —CH2Br K-cpd in excess CH2Br2 6 120-574 16 —CH2CH2Br K-cpd in excess BrCH2CH2Br 21-23 143-474 16 —(CH2)3Br NaOC2H5—
134-572 57 —(CH2)3C1 NaOC2H5—
134.5-5.574 57 —(CH2)4Br K-cpd in excess Br(CH2)4Br 59 144-772 16 —(CH2)6Br K-cpd in excess Br(CH2)6Br 67 16072 16 —CH3 Several hr at 20° in alcohol 50—
102 —CH3 NaOC2H5 at -15°, 6 hr stirring, 67 116-9725 103, 104 temp to 20° Powd. Na in benzene, 48 hr reflux —(CH,), Methanol, NaOCH3 75—
102w (τ1 to 1 d >< ο Ο Ο > W w ο Χ F ί> Η 105
TABLE 1 (Continued) % Yield B.p. Halide R Procedure (%) (7mm) Ref. (with Me ester low yield) η-Propyl iodide (CH2)2CH3 Κ in benzene, 24 hr 75 115-7710 106 η-Butyl iodide —(CH^CHs Κ in xylene, 120 - 130° 82 127-879 107 n-Pentyl bromide (CH2)4CH3 NaOC2H5 44 138-40712 108 n-Heptyl halide — (CH2)eCH3 Na-cpd 65-70 195718 109 n-Octyl halide (CH2)7CH3 Na-cpd 60 205718 109 n-Nonyl halide —(CH2) 8CH3 Na-cpd 60 210718 109 n-Decyl halide —CK^gCHs Na-cpd 55 217718 109 Undecyl halide —(CH2)joCH3 Na-cpd 55 225718 109 Dodecyl halide —(CH2)nCH3 Na-cpd 50 241722 109 2-Iodo-6-methylheptane —CH(CH2)3CH(CH3)2 Κ in xylene, 43 hr at 130-135° ca. 30 142-673 110 1 165-70714 CH3 2-Bromo-6-methylheptane —CH(CH2)3CH(CH3)2 Na-K alloy — — 110 CH3 Benzyl bromide —CH2C6H5 —
— —
111 Phenethyl bromide —(CH2)2C6H5 K-cpd in toluene, 28 hr ca. 60 157-63/2 112 jS-Nephthylethyl bromide —(CH^CioHy Κ in benzene ca. 80 210-1571 112 Cinnamyl bromide —CH2CH=CHC6H5 Na-cpd 48 219719 113 Acrylonitrile —(CH2)2CN Direct condensation with Na-salt 75 176-878 m in dioxane δ-Bromovaleronitrile —(CH2)4CN Na powder in toluene, 18 hr 39 151-270.5 115 o-Cyanobenzyl bromide —CH2C6H4CN Na in toluene, 20 hr 62 m.p. 178-9° 116 o-Cyanophenethyl bromide —(CH2)2C6H4CN Κ in toluene, reflux 3 days 30 180-572 115ROLAND MAYER
Diethjdaminoethyl chloride —(CH2)2N(C2H5)2 Na-cpd in xylene at 0°, then 43 149-5177 117 Diethjdaminoethyl chloride 6 hr at 100° NaH, benzene, 6 hr reflux Mixt. of
—
118 NaH, benzene, 6 hr reflux 0 and C-deriv. KOBu-ί in <-BuOH, 3 hr, then r.t. 50-70 ——
Bromoacetone —CH2COCH3 Na salt, heating in water (28%), alcohol (30%), butyl ether (44%) petroleum ether (55%), toluene (60%)147-879 121 Chloroacetone —CH2COCH3 Na powder in toluene, 6 hr 21 105-10715 122 Chloromethyl methyl —CH2SCH3 K-cpd in benzene Me ester 38 99-10070.01 119 sulfide Et ester 28 155-7710 119 sulfide (also direct ring cleavage) Chloromethyl ethyl sulfide CH2SC2H5 Na in benzene Me 155-6711 119 Chloromethyl ethyl sulfide ester 155-67H 119 Chloromethyl methyl ether —CH2OCH3 K-cpd in benzene (also direct ring cleavage Me ester Et ester
135-6717 12079
119 119 l-Acetoxy-3-iodoacetone —CH2COCH2OCOCH3 *-Butyl ester under N2 to NaNH2 58 137-4770.04 13a l-Acetoxy-3-iodoacetone in benzene; after 8 hr add iodide and heat 5 hr longer 2-Chlorocyclohexanone —CcH90 Na salt in toluene, 2 hr, 80° 56 126-870.02 120 2-Chlorocyclohexanone (also 0.5% O-alkylation m.p. 89-90) 2-Chlorocyclohexanol C-l alkylation +lactone 130° 120 Phenacyl bromide —CH2COC6H5 Na salt in ether, 30 hr 41 m.p. 51° 121 Phenacyl bromide Me ester Na salt in toluene 76 19174 121 (continued)
ETHYL 2-CYCL OPENTAN ONECARB OXYLATE 107
TABLE 1 (Continued) Halide R Procedure Yield B.p. (%) (7mm) Ref. α Note that a few entries are repeated because of recent revision of this table.
Methyl γ-bromobutyrate —(CH2)3C02CH3 NaOCH3 in methanol, 3 hr 70 135-773 123, m Ethyl bromoisobutyrate Michael addition Κ in xylene, 22 hr reflux — — 125 Ethyl 7-bromocrotonate —CH2CH=CHC02C2H5 Na in toluene 60 156-61°/2 122 Ethyl 4-bromo-2-—CHCH=CHC02C2H5 Na in toluene 60 158-6372 122, 126 pentenoate 1 CH3 77 — 127 Ethyl 4-bromo-3-methyl-—CH2C=CHC02C2H5 — 64 162-872 122 2-butenoate 1 CH3 Ethyl 4-bromo-5-methyl-—CHCH=CHC02C2H5 Na in toluene, 24 hr reflux 26 150-6071.5 122 2-hexenoate 1 CH(CH3)2 1,3-Dibromopropane —(CH2)3Br K-cpd with KOH in excess dibromide 7 hr, 70-80° 50 135-8°/l-2 59a 1,5-Dibromopentane —(CH2)5Br K-cpd with KOH, 7 hr, 80-90° 38 161-1.572 59a 2-Methyl-3-piperidino-2-methylskatyl NaOC2H5 Mannich base 128, 129 methylindole dimethyl exchange sulfate reaction 1, l-Dimethyl-4-oxopiperi-—(CH2)2CO (CH2)2N (CH3)2 Direct condensation with — — 130 dinium iodide 1, l-dimethyl-4-oxopiperidinium iodide Thiacyclohexan-4-one —(CH2)2CO(CH2)2SCH3 — — — 131 methiodide 108 ROLAND MAYER
E T H Y L 2- C Y C L O P E N T A N O N E C A R B O X Y L A T E 109 The condensation involving only one end of a dihalide and in which very reactive five-membered ring compounds are formed, is found to be of particular preparative interest (see Example 3 at the end of this chapter). Under special conditions the halide can condense at both ends to form esters of alkane bis (2-oxocyclopentanecarboxylic acid) (16).
M o r e recently ethylene bromide was condensed at both ends (18a), so that in this series the reaction product with methylene bromide is the only one unknown. These compounds have preparative significance b e - cause with acid cleavage, tetracarboxylic acids are formed, while ketone cleavage forms ( ^ ' - ( d i c y e l o p e n t a n o n y l ) alkane.
Although these condensations are simple preparations, ring opening can occur using improper conditions (use of moist solvents, excess alkali metal, alkoxides, or potassium hydroxide, or overheating), which m a y explain contradictory statements found in the literature. F o r example, numerous investigators have worked with haloacetate condensations in which a slight excess of metal leads to substituted adipic esters (Table 1 ) .
Cyclopentanone is formed b y the hydrolysis of the esters of 2 - c y c l o - pentanonecarboxylic acid with acids or superheated water. Neunhoeffer and Paschke (8) were able to isolate the intermediate, the expected 2-cyclopentanonecarboxylic acid.
Alkali hydroxides hydrolyze the ester to adipic acid. This type of ketone and acid cleavage is only significant in the case of C-substituted derivatives of esters (Table 1 ) , which were subjected to cleavage almost without exception.
In ketone cleavage O-substituted cyclopentanones are formed which, because of the reactive ring carbonyl, can be important starting materials for the ortho ring closure on the five-membered ring (58). Preparatively important ^-substituted adipic acids are formed through the acid cleavage reaction.
The ketone cleavage is usually brought about b y heating with dilute or concentrated acids or hydrochloric acid-acetic acid (109,111) (see Examples 4 and 5 ) and occurs satisfactorily with substituents containing up to 6 carbon atoms. Huckel and Kindler (25) used 2 0 % perchloric acid with success. Also aqueous boric acid is suitable (9). Bromides are
C l e a v a g e t o K e t o n e s a n d A c i d s
C 02H
C 08H
110 ROLAND M A Y E R
carefully treated with 4 0 % hydrobromic acid, in which case the halogen is retained (16):
C 02R (CH2)n-Br
| - ( C H2)n- B r 40 % HBr /
^ 30 min boiling \ / V
η = 1 to 6 Ο Ο
The resulting bromoalkylcyclopentanone cyclizes under alkaline con
ditions with surprising ease to spirocycloalkanecyclopentanones (59a, 59b), which may be rearranged to bicyclo [0.3.x] alkene (59c, 59d) after reduction to the alcohols.
C H .
C H , ( C H2)X
C H2- ( C H8)X- B r °
N ι — r i — S Ha
Ο I y / ( C H ^ x x = 1,2,3,4,5
x/ S
Thiabicycloalkenes (59e) are formed from bromoalkylcyclopenta- nones with alkali metal acid sulfide and hydrogen sulfide.
The acid cleavage reaction in which hydrolysis is carried out with more or less concentrated alkali, goes almost quantitatively. Under cyclization conditions ketones, which are identical with those formed by ketone cleavage are formed from the α-substituted adipic acids.
R ι R
/ \ /
I c o2H ι I
\ \ A
ι ο
C OaH
Thus, whenever a direct ketone cleavage fails, the desired ketone may be obtained through the acid cleavage reaction which is almost always successful. B y the addition of a small amount of alkoxide the acid cleavage reaction goes directly to the esters of adipic acid through alcoholysis (yield: at least 8 0 % , mostly 9 0 - 9 5 % ) . According to D i e c k - mann (60) α,α'-dialkylcyclopentanones (23) may be obtained by cycliza
tion followed by another alkylation:
a * '
.«>.« » . R, R,
•COaR — • I — > Ι I
C 02R C OaR R2
Haworth and co-workers (27) utilized this possibility for syntheses in the steroid field, while Guha and Krishnamurthy (30) synthesized thuj an:
E T H Y L 2- C Y C L O P E N T A N ONECARB O X Y L A T E 111
I Ο I I
/ \ / \ / \
Further extension in the use of the cyclopentanones is merely noted here. Ruzicka and co-workers (37) have described a method in the c y c l o - pentenonephenanthrene series:
Tetrahydro-5-indanone resulted from the work of Prelog and Z i m - mermann (55). While Linstead and Meade (43) prepared bicyclo [0.3.3]
octane from the cyclopentanone ester during the course of their pentalene work, Herz (56) described the syntheses of bicyclo [0.3.5]-decanones and their rearrangement to 1-, 1,5-, 2-, and 2,5-alkylazulenes. Aspinall and Baker (51) synthesized proazulene during their heptalene investigations;
Nunn and Rapson (38) studied the synthesis of 4,5-benzazulene (9).
According to Sorm (45) or Plattner (44) azulenes substituted in the 6-position m a y be obtained through the 1,2-cyclopentanediacetic acid
(43):
C H2- C OtH ( C H2)2- C 02H « \
— • ι —I — • ,1 1
\ / \ \ / \ Ν α ν
C H2- C 02H ( C H2)2- C 02H 3 4 5
4,6-Dimethylazulenes are formed in a corresponding manner from α-bromopropionate esters and the cyclopentanone ester (49), while 5- substituted azulenes are obtained as follows (50):
C OtR ( C H2) , - C 02H ( C H2) , - C OaH . - > r Y - > r Y
Ο Ο C H2- C 02H
5-Alkyl-azulene
Ring-substituted (16) and isomeric (62) cyclopentanone esters need only be mentioned here. I t is self-evident that all of the previously described syntheses are applicable to these. I t is possible, for example, to prepare azulenes or steroids substituted in the five-membered ring.
R e d u c t i o n
According to Mousseron and Jacquier (63) the reduction of the cyclopentanone esters with sodium amalgam gives the cyclopentanol ester
112 R O L A N D M A Y E R
(2,64) and a dimer ( b ) , both of which are readily converted to 1- cyclopentenecarboxylic acid (65).
^ C OaR ^ C OaR C O , R ^ C OtH
kA kA
+v\ ι—ι ~~ * kJ
O H O H O - C O - ^ J I
a b O H
Moreover, sodium in absolute alcohol reduces the carboxyl group (66). According to our experiments, catalytic hydrogenation is the preparative method best suited for cyclopentanol esters ( a ) . The method is advantageous in that the cyclopentanones arising from ketone cleavage m a y also be reduced (66a)} and the splitting out of water (from the cyclic alcohol) is greatly reduced by a work-up using a high vacuum
(see Example 6 at the end of this chapter).
The reduction of the carboxy group to the methylol group (67) m a y be carried out with lithium aluminum hydride, the ring-carbonyl group having been previously protected by acetalization with ethylene glycol
(68):
R R R . | - C O , C2H5 . | - C OaCaH5 LiAlH4 | - C Ha- O H
kA ~* kA-°^
H» ~* kA
ο \ I ο O - C H ,
Otherwise there are formed, among others (68a):
/ C H , C H , - O H yC H , - O H
The reduction to cis- and irans-hydroxycyclopentanecarboxylic esters is said to go rather smoothly with sodium borohydride (68b).
The reduction of the ring carbonyl to a methylene group has not yet been carried out with derivatives of the type,
CO,R
since even with careful reduction (69) cleavage products are also formed and consequently the synthesis of spirans is not possible. On the other hand spirans are directly accessible when the carbonyl group is protected
(70). If the ester group is not desired in the final product, then the Clemmensen reduction with hydrochloric acid is used which, in one operation hydrolyzes, decarboxylates, and reduces:
E T H Y L 2- C Y C L O P E N T A N O N E C A R B O X Y L A T E 113
C O , R
R i ι —rR l
ο
After the pharmacological activity of the chaulmoogra acid series became known, Bokil and Nargund (52) synthesized 11-cyclopentyl- undecanoic acid [ R i = ( C H2) i0C O O H ) ] in the above manner. Unfortu
nately some of the ω-halogenoalkanoic acids, necessary for the primary condensation, are difficult to obtain, so that up to now this method has become of preparative value only for certain members. An indirect method is possible using α-halogendicarboxylic acids, but the center carboxyl group can be decarboxylated only with difficulty (53,54).
In the electrolytic reduction of a cyclopentanone ester using a lead cathode, the cyclopentanol ester, cyclohexanol, the cyclopentane ester, and pinacol are formed (71). W e could obtain pinacol readily using zinc and glacial acetic acid and thereby continue successfully in the azulene and tropolone series (9):
ο C OtR Rx Ri
R i _
O H O H
R ^ - C H ^ C O ^ J J
In the electrolysis of the sodium salt of the ethyl ester, diethyl 2,2'-dioxo [bicyclopentyl] -Ι,Ι'-dicarboxylate was obtained along with other products (18a). 1- (/?-Diethylaminoethyl) - l - c a r b e t h o x y - 2 - c y c l o - pentanol was formed in 6 5 % yield by the reduction of the ketoester with aluminum isopropoxide in isopropyl alcohol (117). With respect to methylcyclopentanols, see reference (102).
C y a n o h y d r i n s
The activated carbonyl group of the cyclopentanone ester readily adds hydrocyanic acid (72) to give a distillable cyanohydrin, which serves as starting material for 1,2-dicarboxylic acids (72a). Similar reactions are successful with methyl derivatives (72b).
C O2R C O2R C O2R
O C N C O , R
In contrast to the cycloheptanone derivatives which were investi
gated by Plattner (73), the equilibrium in the C-substituted c y c l o pentanone esters lies on the side of the cyanohydrins, which thus become suitable starting materials for ortho-ring closures in the cyclopentane
114 R O L A N D M A Y E R
system. A disadvantage is that with long chain substituents the c o m pounds do not distill well and so are often difficult to purify. I t is possi
ble, however, in most instances to use the crude product (9). As an example:
T h e G r i g n a r d a n d R e f o r m a t s k y R e a c t i o n s
U p to now the 2-cyclopentanonecarboxylates have been reacted un
equivocally only at the active hydrogen of the enol {74a). A method for lengthening the chain of carboxylic acids by 5 carbon atoms from the M g B r salt of the enolic form of the ester depends on this reaction. On the other hand in carbon-substituted derivatives the carbonyl group reacts readily; thus a 6 0 % yield is obtained in the action of β-phenyl- ethylmagnesium bromide on l-carbethoxy-l-methyl-2-cyclopentanone
(74b). A benzazulene synthesis {74) should be pointed out as a prepara
tive possibility (9):
C Ha- C OaR
Π
C O2R \ y \
/ /• ο
Grignard
Ο \
B r - M g - C Ha- CeH5
C Ha- CeHe
Reformatsky
C Ha- C O , H
B r - C H , - C OaR + Zn
Besides this numerous 1,2-disubstituted 5-membered carbocyclics were obtained with the Grignard reaction (75) as test substances for physical measurements and made possible the further identification of petroleum-naphthene fractions. The combined yield, even with long- chain alkylenes, is mostly over 6 0 % (74c).
a .
C OaR
fa Br—Mg—R8
Enol H y d r o x y l E x c h a n g e w i t h H a l o g e n
According to Rapson and Robinson (76) the enol hydroxyl group is replaced by chlorine using PC15, while with P B r5 the reactive bromo
E T H Y L 2 - C Y C L O P E N T A N O N E C A R B O X Y L A T E 115 compound is formed (9). Since the halide is readily reactive, the 2 - substituted cyclopentanecarboxylic acids or their derivatives are con
veniently obtained [for the formation of cycloalkanone derivatives see ref ( 7 7 ) ] :
C 02R C 02R C 02H
— / e . g .
O H Br ( C H2)n- C 02H
C O - C I
. / e . g .
— — • Cycloalkanone derivatives Br
R e p l a c e m e n t of the C a r b o n y l O x y g e n b y S u l f u r
T h e thioesters (77a, 77b), obtained by the following reaction, have been investigated thoroughly in recent times (77b); it was discovered
X 02R / C 02R / C OTR / H2S . _ / _^ , /
i ^ j l- O H - Η , Ο ' \ J ~ S H "~ \J=S
that, in contrast to the condensation of halides with 2-cyclopentanone- carboxylic esters, no carbon-substituted derivatives are obtained, but exclusively S-alkyl or S-acyl derivatives instead. U p to now (77b) methyl iodide, bromoacetate, β-bromopropionate, bromoacetone, and acetyl chloride (Examples 7 and 8) have been condensed. Carbon- substituted thioesters of the above type are not yet possible using the roundabout method of starting with a suitable cyclopentanone carboxylic ester.
The ring system of these thiocyclopentanone esters is surprisingly stable and it is not opened under the conditions for acid cleavage. Instead 2-thiocyclopentanonecarboxyl acids, present completely in the thio-enol form, are obtained; as also in the case of the ester, this m a y be oxidized quantitatively to the disulfide. B y the addition of thioglycolic acid to the carbonyl group of ethyl 2-cyclopentanonecarboxylate and esterification with ethanol, the following compound is formed (77c):
c o2cFH6
r £ s- C He- C OeCeH ,
S - C H J - C O J C J H .
on distillation it is converted to the diethyl ester of 2-(carboxymethyl- thio)-l-cyclopentene-l-carboxylic acid, which m a y also be obtained by the direct condensation of bromoacetate with thiocyclopentanonecar- boxylic ester (77b).
116 R O L A N D M A Y E R
4,5-Cyclop*enteno-l,2-dithiol-3-thione is isolated from the reaction of cyclopentanonecarboxylic esters with phosphorus pentasulfide (77d).
C o n d e n s a t i o n w i t h A m i n e s
Under 80° the amine adds primarily to the ring carbonyl and water splits out, so that both possible condensation products (a) and (b) are obtainable when ammonia is added (2):
C O2R C O2R
= N — R j / ^ - N H - R i
b
Normally the exocyclic double bond is preferred on the basis of bond strain (19,20). These Schiff bases can be converted to heterocyclic compounds through the reactive ester groups which are present on the ring.
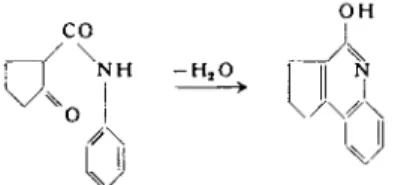
In this manner aniline (78) or substituted anilines (79) reacted below 80° with the ring carbonyl, but cyclized to the 4-hydroxyquinoline derivative on distillation; e.g.,
Schiff bases (see Example 9 at the end of this chapter) were also obtained with α-naphthylamine (79a), 5,6,7,8-tetrahydro-l-naphthyl- amine (79a), l-amino-6-methoxynaphthalene (79a), 8-aminoquinoline
(79b), and 5-aminoquinoline (79b). With dimethylamine (79c), ethyl 2-dimethylamino-l-cyclopentene-l-carboxylate was obtained.
In the reaction with benzylamine, investigated by us, the primary addition product is stable (33), splits out water at 70° and m a y close to a seven-membered ring:
c oAR C O2R
lXY- N H - C H2- C6H5 U N - C H2- CEH5
While practically no amides are formed below 80°, these are obtained in high yield by warming the reaction mixture. The numerous amides which have been prepared (Table 2) m a y be cyclized to 2-hydroxy- quinoline derivatives (80):