PE P EP PT TI ID DE EK K A AN N AL A LI IT TI IK KA AI I E EL LV VÁ ÁL LA AS SZ ZT TÁ ÁS S ÁN Á NA A K K V VI IZ ZS SG G ÁL Á LA AT TA A KA K AP P IL I LL LÁ ÁR RI I S S E EL LE EK KT TR RO OF FO O RE R ET TI IK K US U S M M ÓD Ó DS S ZE Z ER RE EK KK KE EL L
Doktori (PhD) értekezés
Készítette
Olajos Marcell
okleveles környezetmérnök
Témavezető
Dr. Hajós Péter
egyetemi docens
Készült a Pannon Egyetem
Kémiai és Környezettudományok Doktori Iskolája keretében
Pannon Egyetem Mérnöki Kar
Analitikai Kémia Intézeti Tanszék 2010
III
Fehérjék glikozilációjának nyomon követése és peptidek analitikai elválasztásának vizsgálata kapilláris elektroforetikus módszerekkel
Értekezés doktori (PhD) fokozat elnyerése érdekében Írta:
Olajos Marcell okleveles környezetmérnök
Készült a Pannon Egyetem Kémiai és Környezettudományok Doktori Iskolája keretében Témavezető: Dr. Hajós Péter
Elfogadásra javaslom (igen / nem)
(aláírás) A jelölt a doktori szigorlaton …... %-ot ért el,
Az értekezést bírálóként elfogadásra javaslom:
Bíráló neve: …... …... igen /nem
……….
(aláírás) Bíráló neve: …... …...) igen /nem
……….
(aláírás) ***Bíráló neve: …... …...) igen /nem
……….
(aláírás) A jelölt az értekezés nyilvános vitáján …...%-ot ért el.
Veszprém/Keszthely, ……….
a Bíráló Bizottság elnöke A doktori (PhD) oklevél minősítése…...
………
Az EDT elnöke
IV
Kivonat
A szerző a dolgozat első részében a glikoproteinek szénhidrát alkotóinak vizsgálatára kidolgozott módszert ismerteti, mely érzékeny fluoreszcenciás detektálású kapilláris gél elektroforetikus analitikai elválasztáson alapszik, mely nagy áteresztőképességű multi- kapilláris formátumban is kivitelezhető. Értékelte a különböző minta-előkészítési módszereket, és hatékony eljárást dolgozott ki a fluoreszcens jelölőanyag nagy moláris feleslegének eltávolítására, mely elektrokinetikus injektálással történő mintabevitel során az elemzést jelentősen megnehezíti. A kidolgozott normál fázisú kromatográfiás módszer teljesen automatizálható. A komplex biológiai minták – mint a humán szérum – szabad cukortartalmának eltávolítását – mely a minták analízisét zavarja – ultraszűrés segítségével sikerült megoldani. Továbbá a jelölt glikánok szerkezetvizsgálatára illékony puffer rendszert dolgozott ki exoglikozidáz enzimemésztések kivitelezésére elektrokinetikus injektálású rendszerekhez.
A dolgozat második részében a glikoproteinek szelektív izolálására és dúsítására kidolgozott, teljesen automatizált boronsav - lektin affinitás kromatográfiás módszert mutatja be a szerző. Részletesen ismerteti a módszer hatékonyságának vizsgálati eredményeit a hőmérséklet, mint meghatározó paraméter, változatásával. Majd az optimalizált módszer humán szérum glikoproteinek izolációjára került alkalmazásra, melynek eredményeit szintén bemutatja. Az affinitás kromatográfiával kombinált minta-előkészítési módszer alkalmasnak bizonyult kevert normál és prosztata karcinómás humán szérum glikozilációs profilja között különbségek kimutatására.
A dolgozat harmadik részében a peptidek kapilláris zóna elektroforézises elválasztásainak modellezése területén elért eredményeit mutatja be. Ismerteti a modellezési megközelítéseket, újszerű paraméter vezérelt módszerrel, kiterjedt kísérleti adatbázisra épülve vizsgálja a különböző modellek predikciós teljesítményét. Ismerteti a szemi-empirikus és gépi intelligenciára épülő (mesterséges neurális hálózat) modellekkel elért eredményeket az elektroforetikus mobilitás szimulációjára, ahol utóbbiak kiváló predikciós hatékonyságát igazolta, rávilágítva az elektroforetikus mobilitás és a peptidek jellemzőinek és az elválasztás paramétereinek nem-lineáris összefüggéseire.
V
Abstract
Glycosylation profiling of proteins and modelling of analytical peptide separations by capillary electrophoretic methods
In the first part of the work the method developed to study carbohydrate moieties of glycoproteins is discussed. The method is based on sensitive fluorescent detection and capillary gel electrophoretic analytical separation, also implemented in high-throughput multi- capillary format. Various sample preparation methods for multi-capillary gel electrophoresis based glycane analysis to support electrokinetic injection are evaluated. Fully automated, effective normal phase chromatography based method was developed for the removal of the excess fluorescent labelling agent that biases the analysis. Problems associated with the high glucose content of such biological samples as normal human plasma were solved by applying ultra filtration. Finally, a volatile buffer system was developed for exoglycosidase based carbohydrate analysis.
The second part of the treatise discusses the fully automated boronic acid - lectin affinity chromatography for the selective isolation and enrichment of glycoproteins. Temperature dependence of the method was investigated; the obtained results are discussed in detail. The sample preparation methodology combined with the optimized glycoaffinity chromatography was utilized for comparative human serum glycosylation profile analysis. By applying the work flow it was possible to comparatively profile glycane patterns of pooled prostate cancer patient serum to differentiate from the healthy serum.
Finally, the third part of the thesis discusses new endeavours in modelling capillary zone electrophoretic peptide separations. Different models are critically evaluated based on their predictive performances according to a novel parameter driven approach. The results for simulation of the electrophoretic peptide mobilities obtained with the two variable semi- empirical Offord-model and machine learning (artificial neural network) techniques are presented. The latter provided outstanding predictive performance, consolidating the conjectures on nonlinear relationships between peptide structural descriptors and their electrophoretic mobilities.
VI
Kurzfassung
Glykoprofilierung von Proteinen und Modellierung von analytischen Peptid Trennungen durch kapillarelektrophoretische Methoden
Der erste Abschnitt dieser Arbeit beschreibt die Entwicklung einer Methode zur Untersuchung von Kohlenhydratresten auf Glykoproteinen. Die Vorgehensweise basiert auf hochsensibler Fluoreszenzdetektion und kapillargelelektrophoretischer Trennung, mitunter im Hochdurchsatzformat der Multikapillarelektrophorese. Verschiedene Methoden der Probenaufbereitung von Glykanen, welche eine elektrokinetische Injektion und die Trennung im Multikapillargelelektrophoreseformat erlauben, werden systematisch evaluiert. Ein vollautomatisiertes Verfahren basierend auf Normalphasen-Chromatographie wurde zur effektiven Entfernung des Überschusses an Fluoreszenzmarkierungreagenz entwickelt und somit die systematische Verzerrung der Analyse minimiert. Mit Hilfe von Ultra-Filtration konnte dem Problem von Glukoseüberschüssen in natürlichen Proben wie menschlichem Serum erfolgreich begegnet werden. Schließlich wurde ein flüchtiges Puffersystem entwickelt, welches die Analyse von Kohlenhydraten in Zusammenhang mit Exoglykosidasen ermöglicht.
Im zweiten Teil der Arbeit wird die selektive Isolation und Anreicherung von Glykoproteinen mittels vollautomatisierter Boronsäure – Lektinaffinitätschromatographie diskutiert. Die Temperaturabhängigkeit der Methode wurde analysiert und die erzielten Ergebnisse detailliert vorgestellt. Probenaufbereitungsverfahren und optimierte Glykoaffinitätschromatographie wurden gemeinsam zur komparativen Analyse der Glykanprofile von menschlichem Blutserum eingesetzt. Unter Verwendung dieses Arbeitsablaufes konnten die Glykosilierungsprofile aus Mischsera von gesunden mit denen von prostatakarzinomerkrankten Patienten verglichen und Unterschiede hervorgehoben werden.
Der dritte Teil dieser Arbeit beschäftigt sich mit neuen Modellierungsansätzen von kapillar- zonenelektrophoretischer Trennung von Peptiden. Unterschiedliche Modelle werden anhand ihrer Präzision und Vorhersagekraft kritisch evaluiert. Simulationsresultate der elektrophoretischen Mobilität von Modellpeptiden generiert durch den semi-empirischen 2- Variablen Ansatz von Offord sowie künstliche neuronale Netze werden vorgestellt. Letztere erreichten eine außerordentliche Präzision und bestärkten somit die Annahmen über nicht-
VII
lineare Zusammenhänge zwischen Strukturdeskriptoren und elektrophoretischen Mobilitäten von Peptiden.
VIII
Tartalomjegyzék
Kivonat IV
Abstract V
Kurzfassung VI
Rövidítések és jelölésjegyzék 1
Bevezetés 4
1 Irodalmi összefoglaló 6
1.1 Analitikai elválasztásokon alapuló glikán vizsgálatok fluoreszcens jelöléssel 6
1.1.1 A glikán analízis jelentősége 7
1.1.2 Elemzési módok 8
1.1.2.1 HPLC-fluoreszcencia 10
1.1.2.2 Kapilláris gél elektroforézis lézer indukált fluoreszcenciás detektálással (CGE-LIF) 11
1.1.3 Exoglikozidázok 12
1.2 Affinitás kromatográfiás állófázisok glikoproteinek tisztítására és elválasztására 15
1.2.1 Lektin immobilizálási technikák 18
1.2.1.1 Nem-kovalens immobilizálás 18
1.2.1.2 Kovalens immobilizálási technikák 18
1.2.2 Kromatográfiás alkalmazások 20
1.2.2.1 Sorozatos LAC 21
1.2.2.2 Multi-LAC 22
1.2.2.3 Monolitikus lektin affinitás oszlopok 23
1.2.2.4 Lektin affinitás alapú glikán profilozás 23
1.2.2.5 Elektroforetikus alkalmazások 24
1.2.3 Boronsav és más pszeudo-lektinek 24
1.3 Kapilláris zóna elektroforetikus peptid elválasztások modellezése 26
1.3.1 Szemi-empirikus modellek 27
1.3.1.1 A szemi-empirikus modellek hiányosságai 33
1.3.2 Többváltozós modellek 34
1.3.3 Gépi intelligencia módszerek 39
1.3.4 Következtetések 42
2 Kísérleti rész 44
2.1 Anyagok és módszerek komplex szénhidrátok gél elektroforézises elemzéséhez 44
2.1.1 Vegyszerek 44
2.1.2 Oligoszacharid mintaelőkészítés 44
2.1.3 Jelölési reakció 44
2.1.4 Deszialilációs eljárás 45
2.1.5 Mintatisztítás 45
2.1.6 Multikapilláris elektroforézis 46
2.2 Boronsav – lektin affinitás kromatográfia hőmérséklet függésének vizsgálata 48
2.2.1 Felhasznált vegyszerek 48
2.2.2 Boronsav, WGA és BLAC/WGA mikropreparatív glikoaffinitás kromatográfia 48
2.2.3 A minták RP-HPLC és MALDI-TOF-MS analízise 49
2.3 Mikroaffinitás kromatográfiás glikoziláció profilozáshoz felhasznált anyagok és
módszerek 50
IX
2.3.1 Felhasznált vegyszerek és minták 50
2.3.2 Kismolekulájú zavaró mátrix eltávolítása ultraszűréssel 50
2.3.3 BLAC/ConA glikoaffinitás eljárás 51
2.3.4 Az N-glikánok enzimatikus felszabadítása 51
2.3.5 Fluoroeszcens jelölés 52
2.3.6 Poszt-derivatizációs tisztítás 52
2.3.7 Komparatív glikán profilozás kapilláris gél elektroforézissel 52 2.4 CZE peptid mobilitások modellezéséhez felhasznált anyagok és módszerek 53
2.4.1 Vegyszerek 54
2.4.2 Készülékek, műszerek 54
2.4.3 Elválasztási körülmények 55
2.4.3.1 T-CORG elválasztási körülményei 55
2.4.3.2 CBGE-CORG elválasztási körülmények 56
2.4.4 Kísérleti értékelés 57
2.4.4.1 A minták szerkezeti jellemzőinek meghatározása 58
2.4.5 Modellezési módszerek 59
2.4.5.1 T-CORG modellezési fázis 60
2.4.5.2 CBGE-CORG modellezési fázis 62
2.4.5.3 A T-CORG és CBGE-CORG kísérleti adatbázisok egyesítése 62
3 Eredmények 64
3.1 Mintaelőkészítés komplex szénhidrátok multi-kapilláris gél elektroforézises
elemzéséhez 64
3.1.1 A jelölőanyag feleslegének eltávolítása az analízist megelőzően 64 3.1.2 Humán szérum minták monoszacharid tartalmának eltávolítása 66 3.1.3 Illékony puffer rendszer exoglikozidáz emésztéshez 68
3.1.4 Következtetések 69
3.2 A boronsav - lektin affinitás kromatográfia (BLAC) hőmérsékletfüggése 71
3.2.1 Eredmények és diszkusszió 71
3.2.2 Következtetések 76
3.3 Mikropreparatív affinitás kromatográfiás glikoziláció profilozás 78 3.3.1.1 BLAC/ConA mikropreparatív glikoprotein dúsítás 79
3.3.1.2 Komparatív glikozilációs profil vizsgálat 81
3.3.2 Következtetések 83
3.4 Peptidek mobilitásának modellezése kapilláris zóna elektroforetikus elválasztásoknál 84
3.4.1 Kísérleti eredmények 84
3.4.2 Statisztikai értékelés 85
3.4.3 Mintapeptidek töltésszámítása 87
3.4.4 Modellezési eredmények 91
3.4.4.1 T-CORG kísérleti adatbázis modellezési eredményei 91 3.4.4.2 CBGE-CORG kísérleti adatbázis modellezési eredményei 94 3.4.4.3 A CBGE-CORG és T-CORG adatbázisok egyesítése 95
3.4.5 Következtetések 96
4 Összefoglalás 98
Irodalomjegyzék 101
Tézispontok i
Theses iv
A szerző tudományos munkássága viii
Publikációk viii
X
Konferencia előadások ix
Szóbeli előadások ix
Poszterek x
Elismerések xi
Köszönetnyilvánítás xii
Rövidítések és jelölésjegyzék
2-AA 2-amino-benzoesav AAL Aleutra aurantia lektin 2-AB 2-amino-benzamid (2-AA)
ABS Arthrobacter ureafaciens szialidáz 2-AP 2-aminopiridin
%RSD Százalékban kifejezett relatív standard deviáció AAA Alanin aminosavakból álló tripeptid
AGP Alfa-1-savanyú glikoprotein AMF Mandula alfa-fukozidáz
ANN Artificial neural network: mesterséges neurális hálózat ANTS 8-aminonaftalin-1,3,6-triszulfonsav
APTS 8-aminopirén-1,3,6-triszulfonsav
BA Boronic acid: boronsav (m-aminofenil-boronsav) BGE Background electrolyte: háttér elektrolit
BKF Szarvasmarha vese alfa-fukozidáz BLAC Boronsav - lektin affinitás kromatográfia BP-ANN Back propagation artificial neural network BTG Szarvasmarha ivari béta-galaktozidáz CACN Acetonitril koncentráció
CBG Kávébab alfa-galaktozidáz CBGE Háttér elektrolit koncentráció
CBGE-CORG Kísérleti fázis: Puffer koncentráció – szerves oldószer koncentráció CC Charge center: töltésközéppont
CE Capillary electrophoresis: kapilláris elektroforézis CE-MS Tömegspektrometriával kapcsolt kapilláris elektroforézis CGE Kapilláris gél elektroforézis
CMeOH Metanol koncentráció
ConA Concanavalin A: konkanavalin A (lektin) CORG Szerves oldószer koncentráció
CTM Co-translational modification: ko-transzlációs módosulás CV Coefficient of variance: variációs együttható
CZE Capillary zone electrophoresis: kapilláris zóna elektroforézis DDD Aszpargin sav aminosavakból álló tripeptid
DHB 2,5-dihidroxi-benzoesav
DMSO Dimetil-szulfoxid (EOF marker) DSA Datura stratmonium agglutinin DV Doppler sebesség mérés
E4-PHA α-fetoprotein erythroagglutinating phytohemagglutinin EOF Electroosmotic flow: elektroozmotikus áramlás
Err (W) Error function: hiba függvény
ES,C Corrected steric substituent constant: korrigált sztérikus szubsztituens állandó ESI-MS Elektronspray ionizációjú tömegspektrometria
FET Szarvasmarha magzati fetuin
FSCE Free solution capillary electrophoresis: szabad oldat CE
Gal Galaktóz
GDX-ANN Gradient descent ANN using momentum and adaptive learning rate: momentumokat és adaptív tanulási arányú algoritmust használó ANN
GE Gél elektroforézis GlcNAc N-acetil-glükózamin
GUH Streptococcus pneumoniae hexózaminidáz, rekombináns E. coli-ban HILIC Hidrofil interakciós kromatográfia
HM Heurisztikus módszer
Rövidítések és jelölésjegyzék
HPAEC-PAD Pulzáló amperometriás detektálású magas pH-jú anioncserés kromatográfia
HPLC High performance liquid chromatography: nagyhatékonyságú folyadék kromatográfia i.d. Inner diameter: belső átmérő
IgG Immunglobulin G JBM Kardbab mannozidáz
KKK Lizin aminosavakból álló tripeptid LAC Lektin affinitás kromatográfia LAE Lektin affinitás elektroforézis
LD Kapilláris effektív hossza (injektálástól a detektálás helyéig) LED Light emitting diode: fényemittáló dióda
LIF Lézer indukált fluoreszcenciás detektálás
LM Levenberg-Marquardt training algorithm ANN: Levenberg-Marquardt tanulási algoritmust használó ANN
LT Kapilláris teljes hossza
m Levenberg-Marquardt együttható M Moláris tömeg, molekulatömeg MAH Maackia amurensis hemagglutinin
MALDI-TOF-MS Matrix assisted laser desorption/ionization time-of-flight mass spectrometry:
mátrixszal segített lézer deszorpció ionizációs repülési idő tömegspektrometria
Man Mannóz
MEKC Micelláris elektrokinetikus kromatográfia
MLR Multiple linear regression: többszörös lineáris regresszió MR Molar refractivity: moláris refrakció
MS Tömegspektrometria n Aminosav alkotók száma N Elméleti tányérszám NAN1 Rekombináns szialidáz Neu5Ac N-acetil-neuraminsav NeuNAc N-acetil-neuraminsav NeuNGc N-glikolil-neuraminsav NP-HPLC Normál fázisú HPLC
NMR Mag mágneses rezonancia spektroszkópia P90 90. percentilis
PE Papír elektroforézis pk Protonálódási állandó pK Disszociációs állandó PNGaseF Peptid-N-glikozidáz F enzim
PTM Post-translational modification: poszt-transzlációs módosulás Q Effektív töltés
QSPR Quantitative structure-property relationship: mennyiségi szerkezet-tulajdonság összefüggés
R Stokes-féle hidrodinamikai sugár r² Korrelációs együttható
RBF-ANN Radial basis function artificial neural network RNaseB Ribonukleáz B
RP-HPLC Fordított fázisú HPLC SD Standard deviáció (rövidítés) SE Standard hiba
SNA Sambucus nigra agglutinin
SPG Streptococcus pneumoniae béta-galaktozidáz SSS Szerin aminosavakból álló tripeptid
SVR Support vector regression
t Migrációs idő
T Hőmérséklet
T-CORG Kísérleti fázis: Hőmérséklet – szerves oldószer koncentráció
tEOF EOF marker migrációs ideje
TVSE Two variable semi-empirical model: kétváltozós szemi-empirikus modell U Alkalmazott feszültség
UEA-I Ulex europeaus I agglutinin
v Elektroforetikus migrációs sebesség w Súlytényező (ANN csomópontjainál) WAX-HPLC Gyenge anion-cserélő HPLC
WGA Wheat germ agglutinin: búzacsíra agglutinin (lektin) YYY Tirozin aminosavakból álló tripeptid
ZE Zóna elektroforézis η Dinamikai viszkozitás
µef Effektív elektroforetikus mobilitás σ Standard deviáció (jelölés)
Bevezetés
Bevezetés
Napjainkban a biológiai minták analízise érdekes kihívásokat és lehetőségeket kínál a tudomány és a technológia számára. A biológiai rendszerek komplex folyamatainak mélyreható megismeréséhez új módszerek és anyagok – melyek igen kis mintamennyiségek gyors, hatékony és megbízható analízisét biztosítani tudják – fejlesztése iránt folyamatos az igény. A XX. század második felében sikerült a teljes humán genomot feltérképezni. A nukleotid polimorfizmusok (mint SNP: egypontos nukleotid polimorfizmusok, single- nucleotide polymorphism) vizsgálatára hatékony automatizált RFLP (restrikciós fragmenthossz-polimorfizmus, restriction fragment length polymorphism) vagy PCR (polimeráz-láncreakció, polymerase chain reaction) alapú elektroforetikus (lap gél elektroforézis vagy kapilláris gél elektroforézis) analitikai módszerek állnak rendelkezésre. A fehérje szintézis során a nukleotidok sorrendje egyértelműen meghatározza a képződő polipeptid lánc aminosav sorrendjét, ugyanakkor még mindig távol állunk attól, hogy a biológiai folyamatokat, azok összefüggéseit, rendszerüket teljes körűen ismerjük. A biológiai aktivitást számos tényező befolyásolja, ilyenek például a ko- és poszt-transzlációs módosulások, egyéb a szabályozásban részt vevő molekulák jelenléte.
Az új, korszerű, nagyhatékonyságú bioanalitikai módszerek segítséget nyújthatnak az élő szervezeteket felépítő molekulák és azok komplex összefüggéseinek megismerésében, elvezetve a genomikai érából a proteomika és metabolomika korszakába, új perspektívákat nyitva a modern rendszerbiológia, élettudományok, biotechnológia és biofarmakológia előtt.
Ugyanakkor a bioinformatika eszköztára nélkülözhetetlen az adatok feldolgozásához és összefüggéseik megértéséhez, interpretálásához.
Munkám célja analitikai módszer kidolgozása volt fehérjék glikozilációjának vizsgálatára, elsősorban a komplexebb szerkezetű N-glikoziláció szénhidrát szerkezeteinek analízisére. A kidolgozott metodológia hatékony eszköz lehet a biotechnológia, biofarmakológia és a biomarker kutatások számára egyaránt. Megvizsgáltam továbbá a kapilláris zóna elektroforézises peptid elválasztások előrejelzésére szolgáló modelleket, abból a célból, hogy a nagyhatékonyságú elválasztások egyszerűen optimalizálhatóak, megbízhatóan tervezhetőek legyenek. Lehetővé téve fehérjék emésztményekből való bottom-up azonosítását on-line tömegspektrométerrel kapcsolva (peptide mass fingerprinting: a fehérjék azonosítása a peptidek tömegei alapján, ismert fehérje szekvenciákon vagy akár a genom nukleotid
„Mily boldog, ki a dolgok okát ismerni tanulta…”
Vergilius: Georgica
sorrendjén alapuló adatbázisokkal korreláltatva). A módszer a hatékonyság (elméleti tányérszám) jelentős növelésével jó alternatívát kínálhat a széles körben alkalmazott fordított fázisú kromatográfiás (RP-HPLC-ESI-MS) módszerekkel szemben.
A dolgozat első része a téma irodalmát ismerteti. Bemutatom a fehérjék glikozilációjának vizsgálatára alkalmazott ismert analitikai módszereket, a fontosabb eljárások kritikai értékelésével. Az érzékeny fluoreszcens detektálású módszerek értékelésére különös figyelmet szentelek. Részletesen ismertetem a glikoproteinek szelektív izolációjára és dúsítására szolgáló affinitás kromatográfia alapú módszereket is. Az irodalmi összefoglalás második felében a kapilláris zóna elektroforézisen alapuló peptid elválasztások területén kidolgozott modellezési metodológiákat tekintem át, melyeket a peptidek elektroforetikus mobilitásának becslésére alkalmaznak. Értékelem az egyes modelleket azok pontossága és megbízhatósága alapján.
A dolgozat második felében bemutatom az alkalmazott módszertant, a glikoproteinek szénhidrát alkotóinak vizsgálataihoz felhasznált eszközöket és anyagokat, valamint a peptidek kapilláris elektroforézises elválasztásainak modellezésére kidolgozott újszerű paraméter vezérelt megközelítést. Majd az eredmények részletes bemutatása zárja a dolgozatot.
Ismertetem a kidolgozott bioanalitikai módszerrel elért eredményeket, melyek a glikoproteinek szénhidrát alkotóinak hatékony elemzését teszik lehetővé, akár olyan komplex mintákból is, mint a humán szérum. Megvizsgálom a preparatív affinitás kromatográfiás glikoprotein izoláció hőmérséklet függését, hogy a komplex biológiai mintákból maximális kihozatallal kinyerhetőek legyenek a vizsgálat szempontjából fontos komponensek.
Demonstrálom, hogy boronsav - lektin affinitás kromatográfiás glikoprotein dúsítással kombinálva a módszer hatékonysága (az azonosítható glikán szerkezetek száma és azok intenzitása) még tovább növelhető. Végül ismertetem a peptid elválasztások modellezésének eredményeit. Jól megválasztott minta peptidek felhasználásával, és az elválasztás körülményeinek szisztematikus változtatásával kiterjedt kísérleti adatbázist hoztunk létre. A kísérleti adatbázis alapján nem-lineáris modellezési technikák, mint a mesterséges neurális hálózatok, alkalmazásával sikerült megbízható modellt alkotni az elektroforetikus mobilitás előrejelzésére még az olyan komplex hatások esetén is, mint a háttér elektrolit szerves adalékanyag tartalma és az elválasztás hőmérséklete.
Analitikai elválasztásokon alapuló glikán vizsgálatok fluoreszcens jelöléssel
1 Irodalmi összefoglaló
1.1 Analitikai elválasztásokon alapuló glikán vizsgálatok fluoreszcens jelöléssel
A glikoproteinek szénhidrát alkotóinak sokoldalú biológiai szerepének megismerésére napjainkban egyre növekvő tudományos érdeklődés tapasztalható. A glikoproteinek mikroheterogenitásainak a biológiai felismerésben, receptor - ligandum kölcsönhatásokban vagy a sejt-sejt interakciókban, az immunogenitás modulálásában és a fehérjék harmadlagos és negyedleges szerkezetének kialakításában, valamint a bioaktivitás szabályozásában játszott szerepének megismerése különös jelentőséggel bír2. A biofarmakológia rekombináns glikoproteinek iránt támasztott igényei megkövetelik a nagy felbontású, reprodukálható analitikai módszerek kidolgozását a glikoziláció vizsgálatára. A biotechnológiai iparban a rekombináns glikoproteinek fermentációs folyamatának csekély megváltozása a glikoziláció jelentős megváltozásához vezethet, ami a termék farmakológiai alkalmazhatóságát is befolyásolja3. A humán vérkészítmények glikoprotein tartalmának vizsgálata, különösen a glikoziláció különböző megbetegedések következtében bekövetkező megváltozásának megértése, szintén nagy jelentőséggel bír, elsősorban a biomarker kutatás területén4. A glikoproteinek és/vagy glikopeptidek profiljának, szekvenciájának és kapcsolódásának részletes leírása a betegségekkel összefüggő elváltozások megismerése szempontjából kulcsfontosságú5. A fehérjék glikozilációjának megváltozása sok esetben közvetlen kapcsolatba hozható valamely betegséggel, sőt akár a betegség valamely stádiumával is. Az O-glikánok csonkolt formái, az N-glikánok elágazásainak számának növekedése, vagy a túlzott szialiláció, szulfatáció és fukoziláció egyaránt példák a szabálytalan glikozilációs mintázatokra, melyek feltehetőleg kóros elváltozásokkal állnak összefüggésben6.
A glikoprotein analízis a biofarmakológiai ipar számára alapvető fontosságú, mivel a különböző szerkezetű glikánok a termék hatékonyságát és biztonságát egyaránt befolyásolhatják. Ugyanakkor a felszabadított glikánok vizsgálata jelenti a legnagyobb kihívást, mivel természetüknél fogva igen komplex molekulák, nem tartalmaznak kromofór csoportokat és számos izomorf módosulatuk létezik (a kötések típusa és a monomer egységek pozícionális elhelyezkedése alapján). Sőt szinte minden glikoprotein különbözően glikozilált variánsok heterogén keverékéből áll, ezért a felszabadított glikánok számos eltérő szénhidrát
szerkezetet tartalmaznak. A vizsgálni kívánt különböző glikozilációk nagy komplexitása számos bioanalitikai eszköz alkalmazását követeli meg a glikoproteinek szénhidrát részleteinek jellemzésére2. E módszerek közül elsősorban a nagyhatékonyságú folyadék kromatográfiát (HPLC)7-8, tömegspektrometriát (MS)9, magmágneses rezonancia spektroszkópiát (NMR)10, poliakrilamid gél elektroforézist (PAGE)11, mátrix asszisztált lézer deszorpciós ionizációjú repülési idő tömegspektrometriát (MALDI-TOF-MS: matrix assisted laser desorption ionization time of flight mass spectrometry)12-13 és kapilláris elektroforézist (CE)14-17 vagy kapilláris izoelektromos fókuszálást (CIEF)18 érdemes kiemelni. A fenti módszerek némelyikének csatolt technikái, mint az LC-MS vagy CE-MS a szénhidrát analízis számára bíztató lehetőségeket kínálnak5,11,19.
A normál fázisú (NP: normal phase) kromatográfia és a kapilláris gél elektroforézis (CGE) egyaránt kiváló szelektivitást kínál a fluoreszcens jelölésű glikánok analízisére. A normál fázisú kromatográfiás módszer érzékeny, megbízható, széles körben elfogadott és alkalmazott. A szerkezetek azonosítására relatív retenciós idejük alapján adatbázisok is rendelkezésre állnak. A lézer indukált fluoreszcenciás kapilláris gél elektroforézis (CGE-LIF) gyorsabb elemzést tesz lehetővé, mint a HPLC, azonban egyelőre megfelelő adatbázis nem áll rendelkezésre, amellyel a mobilitások alapján a szerkezet azonosítható lenne. Ezért az adatokat más analitikai technikák segítségével, mint a normál fázisú kromatográfia vagy tömegspektrometria, szükséges korreláltatni a módszerfejlesztés és validálás során.
1.1.1 A glikán analízis jelentősége
A fehérje szintézis során a polipeptid lánc kialakulásával egyidejűleg (ko-transzlációs modifikáció, CTM: co-translational modification), illetve transzlációt követően (poszt- transzlációs modifikáció, PTM: post-translational modification) a molekulák további szerkezeti átalakuláson mehetnek keresztül különböző módokon. Ezek a módosulások alapvető változásokat eredményeznek a fehérje tulajdonságaiban, ideértve az élettartamukat illetve biológiai receptor specifikusságukat. A biofarmakológiai termékek jelentős része glikoprotein (például az antitestek), ahol az előállítás üzemi körülményei (közeg, pH, hőmérséklet, … stb.) a glikozilációs mintázatot alapvetően befolyásolhatják, és ezáltal az igen fontos effektor funkciót (glikozilációs kötőhelyek borítottsága, elágazottság mértéke, szialiláció mértéke, kötések sorrendje, … stb.). Ennek következtében a végső termék hatékonysága, immunogenitása, lebomlási ideje is módosul. A biofarmakológiai vállalatok számára jogszabályi előírás a szénhidrát alkotók analízise egy-egy új termék bejegyzését
Analitikai elválasztásokon alapuló glikán vizsgálatok fluoreszcens jelöléssel
megelőzően. A Nemzetközi Harmonizációs Konferencia (ICH: International Conference on Harmonization) előírásainak megfelelően a szerkezeti jellemzés részeként a szénhidrát szerkezet megadása is szükséges (ICH Függelék a Fizikai-kémiai Jellemzésről)20, melynek magában kell foglalnia a szénhidrát tartalom meghatározását (semleges cukrok, amino-cukrok és sziálsavak), a szénhidrát láncok szerkezetét, oligoszacharid mintázatot (antennás profilt) és a glikozilációs helyeket a polipeptid láncon, melyet a lehető legrészletesebben szükséges vizsgálni.
1.1.2 Elemzési módok
Emlős sejtekben a glikoziláció kapcsán két gyakori modifikációt különböztetünk meg: az N-glikánokat (CTM), melyek az aszparagin (Asn) oldalláncok amin-nitrogénjén keresztül kapcsolódnak (Asn-X, ahol X nem prolin, treonin vagy szerin); illetve O-glikánokat (PTM), melyek a szerin (Ser), treonin (Thr), hidroxilizin és hidroxiprolin oldalláncain található hidroxil oxigén atomon keresztül kapcsolódhatnak. A tárgyalt módszerek alapvetően az N- glikánok analízisére szolgálnak, bár az O-glikoziláció vizsgálatára is alkalmasak ezek az analitikai technikák21. A glikoproteinek szénhidrát szerkezeteinek nagy komplexitása okán jellemzésükre általában számos bioanalitikai technika alkalmazása szükséges. E módszerek magukban foglalják az enzimreakciókat, kromatográfiát, tömegspektrometiát (MS), mag mágneses rezonancia spektroszkópiát (NMR), poliakrilamid lap gél elektroforézist (PAGE), nagyhatékonyságú folyadékkromatográfiás technikákat (HPLC), ideértve magas pH-jú anion- cserélő kromatográfiát pulzáló amperometriás detektálással (HPAEC-PAD) és a kapilláris elektroforézist (CE)2,7,10,13,22-23
. A fenti módszerek némelyikének csatolt technikái, mint az LC-MS vagy CE-MS a szénhidrát analízis számára bíztató lehetőségeket kínálnak5,11,19. A glikoproteinek, glikopeptidek és glikánok vizsgálatának jelentősebb analitikai módszereit az 1. ábra foglalja össze.
Napjainkban a biofarmakológiai glikozilációs profilokat legáltalánosabban a glikánok fehérjevázról történő kémiai (például hidrazinolízis által) vagy enzimatikus (pl. peptid-N- glikozidáz F) lehasítását követően határozzák meg. Ezután a szabaddá tett glikánokat megtisztítják a nem-szénhidrát szennyező-komponensektől.
A tömegspektrometria, mint a MALDI (matrix-assisted laser desorption/ionization) /SELDI (surface enhanced laser desorption/ionization)-TOF (time-of-flight) kombinációja ESI-MS (elektron spray ionizációs tömegspektrometria) technikával részletes szerkezeti információkat nyújthat rövid analízisidők mellett24-27. Ugyanakkor az MS adatok igen
összetettek lehetnek és megfelelő feldolgozásuk, interpretációjuk nagy szakértelmet és gyakorlatot igényel. Ráadásul, a tömegspektrometriás mennyiségi meghatározás nem mindig megbízható, és bizonyos minták esetében izobarikus (azonos molekulatömegű) glikánok átlapoló jelei is nehézségeket okozhatnak. E problémák többségét leküzdhetjük, ha a tömegspektrometriás vizsgálatokat elválasztás alapú elemzéssel, és a glikánok exoglikozidáz emésztéses fragmentációjával kombináljuk.
1. ábra: A glikoprotein-analízis leggyakoribb megközelítési módjai
Mivel a szénhidrátok nem tartalmaznak kromofór és/vagy fluorofór csoportokat/részeket, a nagy felbontású bioszeparációs technikáknál, mint a fordított fázisú HPLC (RP-HPLC) vagy CE, széles körben alkalmazott UV és fluoreszcens detektorok nem alkalmazhatóak az elválasztást megelőző derivatizálás nélkül. A HPLC nagyhatékonyságú anion-cserélő kromatográfiás oszlopokkal, pulzáló amperometriás detektálással (HPAEC-PAD: high performance anion exchange chromatography with pulsed amperometric detection) széles körben bevált módszer a biofarmakológiai glikán profilozásra. Általában jelöletlen vagy redukált glikánok vizsgálatára alkalmazzák, és igen jó elválasztást tesz lehetővé töltéssel nem rendelkező és anionos glikánok esetén egyaránt. Azonban az elmúlt évtized során sok glikoprofilozást végző laboratóriumban fluorofór jelölésű glikánok fluoreszcens detektálású
Analitikai elválasztásokon alapuló glikán vizsgálatok fluoreszcens jelöléssel
HPLC analízisével váltották fel. Utóbbi jóval nagyobb érzékenységet biztosít, és különböző állófázisokkal kivitelezhető, ortogonális elválasztást is lehetővé tesz.
A lézer indukált fluoreszcenciás detektálású kapilláris gél elektroforézis (CGE-LIF) jelentős potenciállal rendelkezik a glikán profilozás területén, és az elmúlt években egyre elterjedtebben alkalmazzák. Kapilláris elektroforézises analízishez leggyakrabban alkalmazott fluoreszcens jelölőanyagok a 8-aminonaftalin-1,3,6-triszufonsav (ANTS) és a 8-aminopirén- 1,3,6-triszulfonsav (APTS)15,28-30, melyek kiváló fluoreszcenciás tulajdonságokkal és a hatékony elválasztáshoz szükséges töltéssel egyaránt rendelkeznek. Így a jelölt szénhidrátok elemzése hagyományos kapilláris elektroforézis készülékkel31, kapilláris-sor elektroforézissel (capillary array electrophoresis)32 vagy mikrofluidikai (chip alapú) formában33 egyaránt elvégezhető. Sőt a Beckman Coulter (Fullerton, CA, USA) cég egy igen egyszerűen használható készletet is kifejlesztett a PA800 ProteomeLab™ készülékéhez. A kit 8- aminopirén-1,3,6-triszulfonát (APTS) fluoreszcens jelölőanyagot, bevont (coated) kapillárist, gél elektrolitot és megfelelő standardokat egyaránt tartalmaz. A standardokat és a mintákhoz a módszereket előre elkészítve, az adatok kiértékelésére készen kapják a felhasználók.
1.1.2.1 HPLC-fluoreszcencia
A szerkezetvizsgálat HPLC stratégiája a lehasított, fluoreszcens jelölésű glikánok a 2. ábra által bemutatott három különböző kromatográfiás eljárásra, illetve ezek kombinációira épül. A szerkezetvizsgálatok túlnyomó részében a normál fázisú (NP: normal phase) oszlopokat alkalmazzák hidrofil interakció folyadék kromatográfiás (HILIC: hydrophilic interaction liquid chromatography) módban. Ugyanakkor a módszer, előfrakcionálási lépésként, gyenge anion-cserélő (WAX: weak anion exchanger) kromatográfiával kombinálva (töltés alapján történő frakcionálással), vagy fordított fázisú (RP: reversed phase) kromatográfia segítségével, bizonyos kapcsolódási sorrendek megkülönböztetésére (kétágú N-acetil- glükózamin (GlcNAc) azonosításával) is használható. Tehát, alapvetően a kérdéses minta komplexitása határozza meg a kiválasztott elemzési eljárást34.
2. ábra: A HPLC alapú glikán szekvenálási lehetőségek sematikus összefoglalása
Az ismeretlen minták kiértékeléséhez a relatív skálaként különböző polimerizáció fokú glükóz (NP), arabinóz (RP), vagy ismert töltésű glikánok (WAX) „létráját” futtatják meg. A standardok futtatása ugyanakkor a rendszer stabilitásának ellenőrzését is szolgálja egyben. Az analízis után a skáláról interpolálva minden egyes csúcshoz hozzárendelhetünk egy megfelelő glükóz egységet (GU), arabinóz egységet (AU) vagy töltés értéket. Ezeket az adatokat az adatbázisok35-37 értékeivel összevetve lehetőség nyílik a potenciális szerkezetek meghatározására.
A normál fázisú elválasztásokhoz amid oszlopokat használnak, melynek poláris funkciós csoportjai a cukrok hidroxil csoportjaival kölcsönhatásba lépnek. A mintákat nagy szerves oldószer tartalmú mozgófázissal viszik fel az oszlopra, ahol az állófázison adszorbeálódnak.
Ezt követően vizes fázisú gradienssel kerülnek elúcióra hidrofilitásuk szerinti sorrendben. Ez azt jelenti, hogy általánosságban a nagyobb méretű oligoszacharidok nagyobb hidrofilitásúak, és nagyobb mennyiségű vizes fázist igényelnek az elúcióhoz. Ezt a módszert számos biológiai szempontból jelentős molekula jellemzésére sikerrel alkalmazták21,38-39. A vizsgálatok során leggyakrabban alkalmazott fluoreszcens jelölőanyagok a 2-amino-benzamid (2-AB), 2-amino- benzoesav (2-AA) és a 2-aminopiridin (2-AP), melyeket reduktív aminálással kapcsolnak a mintamolekulákhoz.
1.1.2.2 Kapilláris gél elektroforézis lézer indukált fluoreszcenciás detektálással (CGE-LIF) A lézer indukált fluoreszcenciás detektálású kapilláris gél elektroforetikus (CGE-LIF) oligoszacharid elválasztások gyors, hatékony analízist tesznek lehetővé, mely általában 15 percnél is rövidebb analízisidőt igényel. Leggyakrabban 8-aminopirén-1,3,6-triszulfonát fluoreszcens jelölést (λex: 455 nm) alkalmaznak reduktív aminálással az N-glikánok vizsgálatára. Az APTS jelölést számos publikációban sikeresen alkalmazták különböző oligoszacharidok CGE-LIF analízisére14-15,28,33,40-42
. Az elválasztásokat gyakran semleges bevonatú (PVA coated, poli-vinil-alkohollal bevont) kapillárisokban kivitelezik az elektroozmotikus áramlás és a nem-specifikus fali interakciók visszaszorítása érdekében. A háttér elektrolit (BGE: background electrolyte) enyhén savas kémhatású (pH ~4,75) acetát puffer, mely 0,1% polietilén-oxid gélt is tartalmaz. Az oligoszacharidok a töltés-tömeg arányuknak megfelelően válnak el a rendszerben anódikus detektálással (fordított polaritás).
A több savas csoportot tartalmazó molekulák rövidebb migrációs időket eredményeznek, ezért a migrációs idők változásai alapján a molekulában a sziálsavas csoportok száma könnyen monitorozható. A gél tartalmú futtató puffer szűrő hatásának köszönhetően tovább javítja az elválasztás hatékonyságát. Exoglikozidáz emésztést is alkalmazva a szénhidrát
Analitikai elválasztásokon alapuló glikán vizsgálatok fluoreszcens jelöléssel
szekvenáláshoz, fontos a maradék reagálatlan APTS eltávolítása az emésztés előtt, mivel bizonyos exoglikozidázok aktivitását gátolja, amire Callewaert és munkatársai ismertettek egy módszert41.
1.1.3 Exoglikozidázok
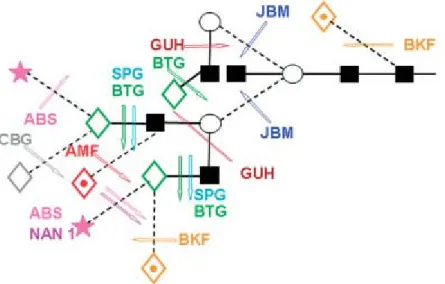
Az exoglikozidáz enzimek alkalmazása az oligoszacharidok szekvenciájának és kapcsolódási sorrendjének megismerésére elválasztás és tömegspektrometria alapú megközelítéssel egyaránt kézenfekvő módszer. Ezek az enzimek az oligoszacharid szerkezet jól meghatározott kötéseit hasítják szisztematikusan, ahogy a 3. ábra is mutatja (újranyomva
43 hivatkozásból). Az emésztetlen minta elemzését követően az egyes glikán szerkezeteket különböző enzimek keverékeinek sorozatával progresszív módon lebontják. Minden egyes emésztési lépés végeztével a mintából az enzimeket eltávolítják, és a terméket újra elemzik.
Az eredeti retenciós illetve migrációs időhöz képest a csúcs eltolódásából a szénhidrát szerkezete visszakövethető. A leggyakrabban alkalmazott exoglikozidázokat (3. ábra) az alábbi felsorolás tartalmazza:
• ABS (Arthrobacter ureafaciens szialidáz): α2-6, α2-3 és α2-8 kapcsolódású nem- redukáló terminális savakat, N-acetil-neuraminsavat (NeuNAc) és N-glikolil- neuraminsavat (NeuNGc) szabadítja fel.
• NAN1 (rekombináns szialidáz): felszabadítja az α2-3 kapcsolódású nem-redukáló terminális sziálsav csoportokat (NeuNAc és NeuNGc).
• AMF (mandula alfa-fukozidáz): α1-3 és α1-4 kapcsolódású nem-redukáló terminális fukóz csoportokat hasítja le. A belső (core) α1-6 kapcsolódású fukózt nem hasítja.
• BKF (szarvasmarha vese alfa-fukozidáz): α1-2 és α1-6 kapcsolódású nem-redukáló terminális fukóz csoportokat hasítja le hatékonyabban, mint az α1-3 és α1-4 kötésű fukózt. A belső (core) α1-6 fukózt is felszabadítja.
• BTG (szarvasmarha ivari béta-galaktozidáz): nem-redukáló terminális galaktóz β1-3 és β1-4 kötéseit hidrolizálja.
• SPG (Streptococcus pneumoniae béta-galaktozidáz): nem-redukáló terminális galaktóz β1-4 kötéseit hidrolizálja.
• CBG (kávébab alfa-galaktozidáz): α1-3 és α1-4 galaktózt hidrolizálja.
• GUH (Streptococcus pneumoniae hexózaminidáz, rekombináns E. coli-ban): β-N- acetil-glükózamint (GlcNAc) szabadítja fel, de a β1-4 kötéssel mannózhoz kapcsolódó GlcNAc-t nem.
• JBM (kardbab mannozidáz): α1-2, α1-3 és α1-6 kapcsolódású mannózt távolítja el.
3. ábra: Az általánosan alkalmazott exoglikozidáz enzimek hasítási helyei
Mind a normál fázisú HILIC, mind a CGE-LIF elválasztások kiváló felbontóképességgel rendelkeznek, ugyanakkor az analízis idő egy nagyságrenddel alacsonyabb a kapilláris elektroforetikus módszerrel, ahol 15 perc alatt kivitelezhetők az elválasztások szemben a 2-3 órás kromatográfiás futásokkal. Azonban a normál fázisú kromatográfiás elválasztás előnye, hogy nagyobb az érzékenysége és a dinamikus működési tartománya a módszernek, így a fő- és mellékkomponensek egyazon mintából jól detektálhatók.
Rudd és munkatársai egy komparatív munka keretében értékelték a módszerek precizitását standard glükóz oligoszacharidok létráját használva mintaként43. Úgy találták, hogy mindkét módszer nagy pontosságú, a retenciós illetve migrációs idők mindkét esetben az elfogadhatónak mondható 2% alatti relatív standard deviáció (RSD) értékeket adtak. A mérések reprodukálhatósága glükóz egységekre normalizált migrációs/retenciós idők esetén 0,2 és 0,3% RSD értékeket eredményezett CGE-LIF illetve NP-HPLC módszerekkel, míg a normalizált csúcsterületek mindkét esetben 3% RSD értéket adtak.
Az analitikai elválasztások érzékenység területén is kiválónak bizonyultak. A normál fázisú kromatográfia kimutathatósági határa (LOD: limit of detection) 7,8 ng/ml értéknek adódott, ahol a fő glikoformok még 3:1 jel-zaj aránnyal detektálhatóak, míg a mennyiségi meghatározás határa (LOQ: limit of quantification) 26 ng/ml értéket adott.
Összehasonlításként, ugyanezek az adatok CGE-LIF analízis esetén 0,45 µg/ml (LOD) és 1,37 µg/ml (LOQ) értékre módosulnak. A CGE-LIF módszer legjelentősebb hátránya a NP-HPLC elválasztásokkal összevetve a némileg kisebb lineáris működési tartomány (1,4-2,5 µg/ml).
Analitikai elválasztásokon alapuló glikán vizsgálatok fluoreszcens jelöléssel
Az elválasztási módszerekkel a glikánok szerkezetének meghatározására is lehetőség nyílik tisztított standardok alkalmazásával. Ugyan az összehasonlításhoz teljes glikán könyvtárra lenne szükség, hogy az összes lehetséges szerkezeti variánst figyelembe vegyük.
NP-HPLC esetében adatbázisok segítséget nyújtanak ebben, így nem szükséges minden esetben a teljes könyvtár beszerzése, illetve megfuttatása a készüléken. Az ilyen adatbázisok összeállítása jelentős munkaigényű, és függ a rendszer stabilitásától. CGE-LIF analízisekhez napjainkban adatbázisok nem állnak rendelkezésre, így a migrációs tulajdonságokat tisztított standardok elektroferogramjai alapján vagy más analitikai módszerekkel (NP-HPLC, MS) korreláltatva van lehetőség kiértékelni. Ugyanakkor mindkét esetben, amennyiben lehetőség van rá, a szerkezetet több megközelítéssel is igazolni szükséges, pl. glükóz egységekből álló oligoszaccharid létrával és exoglikozidáz emésztéssel, valamint tömegspektrometriásan egyaránt.
A CGE-LIF analízishez elméletileg akár 96 minta párhuzamos előkészítésére (96 well plate formátumban) is lehetőség van, és az elemzés során az injektálás is történhet a plate-ről.
A legjobb eredmény elérése érdekében az APTS jelölési reakcióhoz, a reagenseket egész éjszakán át szükséges sötétben inkubálni reduktív atmoszférában. Ugyan a minták és a megfelelő emésztmények analíziséhez szükséges idő a 15 percet sem éri el általában, a jelölési és enzimemésztési lépések a teljes analízis időt két napra növelik (azonban akár 96 mintát is elemezhetünk ez idő alatt).
Összegzésként elmondható, hogy az ismertetett analitikai elválasztások egyaránt alkalmasak komplex N-glikánok szénhidrát láncainak szerkezetvizsgálatára. A fluoreszcens detektálású HPLC módszer nagy érzékenységű, robosztus és megbízható eszköz a glikánok mélyreható szerkezeti jellemzésére. A rendelkezésre álló adatbázisok megkönnyítik a munkát, mivel nem szükséges nagyszámú standardot minden elemzés során megfuttatni a kiértékeléshez. Ugyanakkor a módszer igen időigényes, mivel egy-egy elválasztás több mint két órát vesz igénybe. Ezzel szemben a CGE-LIF igen hatékony, gyors analízist tesz lehetővé, gyakran 10 percnél is rövidebb elválasztásokkal. Ugyanakkor a fluoreszcens jelölés 10 órás inkubálást igényel a reakció tökéletes lejátszódásához. A módszer rutinszerű használatához szerkezet meghatározó adatbázis létrehozása és validálása is indokolt lenne, azonban egyelőre ilyen még nem áll rendelkezésre, ugyanakkor minden kétséget kizáróan a közeljövőben már ez a feladat is megoldódik. A CGE-LIF módszer reprodukálhatósága némileg jobb, azonban a lineáris működési tartománya szűkebb, mint a kromatográfiás módszeré. Mindkét módszer lehetővé teszi exoglikozidáz enzim emésztmények analízisét is.
1.2 Affinitás kromatográfiás állófázisok glikoproteinek tisztítására és elválasztására
Ismerve a növényi lektinek nagy specifikusságát, nem is lehet meglepő, hogy immobilizált formáik egyre elterjedtebbek a glikoproteinek analízisében. A gyakori lektin immobilizációs technikák a reverzibilis nem-kovalens kötésektől a különböző állófázisokhoz való kovalens kapcsolásokig terjednek. Ebben a fejezetben a lektinek immobilizálási technikáit tekintem át különböző állófázisokon, lektin affinitás kölcsönhatásokon alapuló kromatográfiás és elektroforetikus bioszeparációs módszerekhez.
A glikánok a komplex sejtszerveződések közvetítői, melyek jellemzőek a sejt típusára és életszakaszára. Minden élő szervezet tartalmaz változatos szénhidrát szerkezetekkel borított sejteket44, ezért a glikoprotein-analízis a tudományos érdeklődés középpontjában áll. A lektinek nem-immun eredetű fehérje molekulák, melyek szénhidrátokkal specifikus kölcsönhatásokba lépnek anélkül, hogy módosítanák őket. A legtöbb lektin célzottan kapcsolódik a cukor egységekhez, mint az N-acetil-neuraminsav (NeuNAc), N-acetil- glükózamin (GlcNAc), galaktóz, fukóz vagy mannóz45. A növényi magvakban nagymennyiségű lektin található, ezért leggyakrabban innen izolálják őket. Feltételezhetőleg a csírázás folyamatában és a magok túlélésében egyaránt fontos szerepük van. Ugyancsak feltételezik, hogy a növényi sejtek felületén a glikoproteinek megkötésében is részt vesznek46. Továbbá állati szervezetekben is jelen vannak lektinek, melyek például a glikoproteinek kiválasztásának szabályozásában játszanak szerepet47. Sőt számos egyéb biológiai funkciót ellátnak a sejtadhézió szabályozásától, a glikoprotein szintézisen át, különböző vérfehérjék szintjének szabályozásáig48. Ugyancsak ismert, hogy a lektinek fontos szerepet töltenek be az immunrendszerben, mivel képesek felismerni a patogénekre jellemző szénhidrátokat49.
A szénhidrát felismeréséhez általában megfelelő konformációra van szükség, és gyakran a szomszédos cukoregységek minősége is meghatározó a kölcsönhatás szempontjából. A legtöbb irodalmi forrás feltételezi, hogy a lektinek elsődlegesen monoszacharidokkal alakítanak ki kötéseket, ugyanakkor tudott, hogy összetett szénhidrátokkal egy nagyságrenddel nagyobb affinitást mutatnak50. Gyakran fém-szénhidrát kölcsönhatások is fontos szerepet játszanak a lektinek megkötési folyamatában. Például a mannóz-kötő lektinek krisztallográfiás vizsgálatai kimutatták, hogy a szénhidrát megkötés csak Ca2+ ionok
Affinitás kromatográfiás állófázisok glikoproteinek tisztítására és elválasztására
jelenlétében játszódik le, míg más típusú lektinek a megfelelő szubsztráttal fémionok hiányában is képesek reagálni51-52.
A bioelválasztásoknál leggyakrabban használt lektinek összefoglalását az 1. táblázat tartalmazza53. Ahogy korábban is utaltam rá, lektinek nem kizárólag növényekben fordulnak elő. Néhány állati lektint már a növényi lektinek előtt felfedeztek, azonban ezeket szénhidrátkötő fehérjékként már jó ideje nem alkalmazzák54.
Ahogy az 1. táblázatban is látható különböző glikoproteinek és glikopeptidek izolálásához a szénhidrátszerkezettől függően más-más lektinek használata javasolt. Például, a galektinek az N-acetil-laktózamin tartalmú szénhidrátokra specifikusak, melyek az N- és O-glikánokban egyaránt megtalálhatóak55, a földimogyoró agglutinin az O-glikánokra specifikus56, a konkanavalin A (ConA) az N-glikánok oligomannozil mintázatait ismeri fel57, míg az aleuria- aurantia lektin a fukóz tartalmú oligoszacharidok széles skálájához mutat affinitást58.
A szénhidrátok felé mutatott nagy specifikusságuk miatt a növényi lektineket különböző glikánvegyületek – glikoproteinek, glikopeptidek, glikolipidek vagy szabad glikánok – tisztításánál előszeretettel alkalmazzák45. Nem kizárólag az széleskörűen alkalmazott egyféle lektinnel végzett affinitás kromatográfiát (LAC, single LAC) ismerjük59-60, hanem különböző lektinek kombinációit multi-LAC (MLAC)61 vagy sorozatos LAC1 formájában is kidolgozták, melyek különösen hasznosak hasonló méretű és töltésű, de eltérő szerkezetű glikánok komplex keverékeinek frakcionálására. Az általános eljárások során a megkötött glikánokat a lektin affinitás oszlopról az egyensúly haptén cukrokkal történő megbontásával eluálják. A LAC nagyhatékonyságú folyadékkromatográfiás (HPLC) módszerként alkalmazva, nagytisztaságú glikán-vegyületeket eredményez a további szerkezetvizsgálatokhoz50.
1. táblázat: A glikopeptidek és oligoszacharidok affinitás kromatográfiájában leggyakrabban használt lektinek Triviális rövidítés Lektin forrása Monoszacharid specifitás Haptén cukor Megjegyzések a kötésről Konkanavalin A
ConA Canavalia ensiformis α-D-mannóz
α-D-glükóz α-metil-D-mannozid Gyenge kötés biantennás glikopeptidekhez, erős a magas mannóz/hibrid típushoz Borsó lektin Pisum sativum α-D-mannóz α-metil-D-mannozid Bi- és triantennás belső L-fukóz tartalmú glikopeptidek
Lencse lektin Lens culinaris α-D-mannóz α-metil-D-mannozid Bi- és triantennás belső fukóz tartalmú glikopeptidek
RCA-1 Ricinus communis β-D-galaktóz
N-acetil- β-D-galaktózamin Laktóz β(1→4) kötésű terminális galaktozil csoportokat tartalmazó glikopeptidek L4PHA
leukoagglutinin Phaseolous vulgaris β-D-galaktóz N-acetil-D-
galaktózamin Tri- és tetraantennás glikopeptidek E4PHA
erytroagglutinin Phaseolous vulgaris β-D-galaktóz N-acetil-D-
galaktózamin Bi- és triantennás glikopeptidek és oligoszacharidok
SNA Sambucus nigra β-D-galaktóz Laktóz Sziálsav tartalmú glikopeptidek NeuAc-α(2→6)Gal terminális szekvenciával LTA Lótusz lektin Tetragonobulus pupureas α-D-fukóz Fukóz A glikán külső részén fukózt tartalmazó glikopeptidek
GS-I-B4 Griffonia (Bandeiraea) simplicifolia 1-34
α-D-galaktóz
N- α-acetil-D-galaktózamin Raffinóz Terminális α-galaktozil csoportot tartalmazó oligoszacharidok
UEA-I Ulex europeaus I α-D-fukóz Fukóz Külső α-L-fukóz csoportot tartalmazó oligoszacharidok
HPA Helix pomatria N- α-acetil-D-galaktózamin N-acetil-D- galaktózamin
Terminális α-GalNAc tartalmú N- és O-kötésű oligoszacharidok nagy affinitással
Jacalin/Jackfruit
lektin Artocarpus integrioflia α-D-galaktóz α-D-metil-galaktozid O-α-galaktozil kapcsolódású terminálist tartalmazó glikopeptidek és oligoszacharidok
PNA Földimogyoró
lektin Arachis hypogaea α-2-N-acetil-galaktózamin
β-Gal(1→3)GalNAC Galaktóz O-β-galaktozil kapcsolódású terminálist tartalmazó glikopeptidek és oligoszacharidok
AAL Aleuria aurantia α-D-fukóz L-fukóz N-acetil-glükózaminhoz α1-6 vagy N-acetil-laktózaminhoz α1-3 kapcsolódású fukóz
MAH, MAA, MAL Maackia amurensis Sziálsav, meghatározott
triszacharidok Laktóz MAH II csak bizonyos sziálsavat vagy meghatározott triszacharid szerkezeteket tartalmazó szénhidrátokat köt
DSA, DSL Datura stramonium N-acetil-glükózamin Kitin hidrolizát Felismeri a β1-4 kötésű N-acetil-glükózamin oligomereket,
Affinitás kromatográfiás állófázisok glikoproteinek tisztítására és elválasztására 1.2.1 Lektin immobilizálási technikák
A lektinek különböző hordozón történő immobilizálása során olyan felületet alakítanak ki, mely mind a lektin mind a hordozó anyag fizikai-kémiai jellemzőivel rendelkezik. A maximális stabilitás, és a mintafehérjék nem-specifikus kölcsönhatásainak minimalizálása érdekében ideális esetben a hidrofób kötőhelyeket nem tartalmazó, hidrofil jellegű felszínek az optimálisak. Egy másik szempont a lektin és az állófázis felületének összekapcsolását végző, úgy nevezett linker típusa. Jó linkerrel az immobilizált lektinek a természetes állapotukhoz hasonlóan viselkednek, azaz felismerik és megkötik a célmolekulákat, sztérikus gátlás nem lép fel. A különböző mátrixok jellemzéséről több áttekintő tudományos publikáció is készült62-63.
1.2.1.1 Nem-kovalens immobilizálás
A ConA reverzibilis csatolása hidrofób kölcsönhatással polymyxin B (egy dekapeptid) felülethez egy jó példa a nem-kovalens lektin immobilizálásra64. Első lépésben a polymyxin B a primer amin csoportjain keresztül kovalens kötéssel egy felületi plazmon rezonanciás (SPR:
surface plasmon resonance) érzékelő lemezre került rögzítésre. Az így létrejött felületi réteg képes spontán adszorbeálni a ConA molekulákat erős hidrofób kölcsönhatással (KD=1,9×10-10 M). Érdekes módon azt tapasztalták, hogy az így kapott állófázis nagyobb kötőkapacitással rendelkezik, mint a kovalensen immobilizált forma.
A glikozilált lektin indirekt adszorpciója kovalensen kötött lektinekhez egy másik érdekes megközelítés a nem-kovalens immobilizálásra65. Griffonia simplicifolia lektint (GS-I-B4) az aszparaginen keresztül kapcsolódó magasabb rendű mannóz cukorláncaival kötöttek sepharose állófázisra kapcsolt ConA lektinhez, és így oligoszacharidokat választottak el α- kötésű D-galaktóz csoportjaik mennyisége alapján. Az eredmények igazolták, hogy a lektin megőrizte glikán kötő képességét a gyors, kényelmes és kvantitatív immobilizálási módszerrel. Hasonló módszerrel immobilizáltak ConA-t fémes kölcsönhatásokkal réz(II)- iminoecetsav állófázishoz66.
1.2.1.2 Kovalens immobilizálási technikák
Az immobilizált lektinek az affinitás alapú állófázisok kialakítására glikokonjugáltak67, felszabadított glikánok68, glikopeptidek69, poliszacharidok, oldható sejtösszetevők, sőt egész sejtek elválasztására, izolálására egyaránt70 nagy potenciállal rendelkeznek. Ugyanakkor gyakran csak laboratóriumi méretekben sikerült a módszert kidolgozni, aminek az oka
elsősorban az agaróz hordozóban keresendő. Ez a legáltalánosabban használt hordozó a lektinek immobilizálására, azonban mind a kémiai, biológiai és mechanikai stabilitása korlátozott71.
Egyre növekvő tudományos érdeklődés tapasztalható, az újszerű kovalens immobilizálási technikák iránt, melyek stabilabb hordozóra épülnek. A következőkben a különböző kovalens immobilizálási módszereket tekintem át.
A lektin affinitás kromatográfiához, ahogy a publikációk nagy száma is tükrözi, leggyakrabban agarózon immobilizált lektineket használnak1,45,59-61,72-75
. Annak ellenére, hogy az alapötlet – az agaróz mint hordozóanyag használata a lektin immobilizálására – azonos, számos különböző reaktív csoportot és kémiai módszert alkalmaznak a cél elérésére. Ennek eredményeként több megfelelő, megbízható aktivált hordozó és metodológia elérhető napjainkban, melyek nagy része könnyen kivitelezhető.
Az egyik közkedvelt módszer a lektinek immobilizálására a lektinek amin csoportját használja ki. A kötést három módon lehet kialakítani:
• A CNBr hordozó imidokarbonát csoportjának reagáltatása az amin csoporttal haptén cukor jelenlétében, ami a lektin szénhidrát-kötőhelyét megvédi76;
• N-hidroxi-szuccuinimid-észter reagáltatása az amin csoporttal76;
• Schiff-bázis képzés a formil- és amin csoportok közt, amit nátrium-ciano- borohidriddel való redukálás követ, alkil-amidot képezve77.
A legtöbb affinitás kromatográfiában használt lektin agaróz gélhez kötve napjainkban már kereskedelemből beszerezhető, és megfelelő oszlopba töltés után használatra kész.
A szilika tresyl aktiválását már az 1980-as évek elején kidolgozták enzimek és affinitás ligandumok immobilizálására78, és mind a mai napig széles körben alkalmazzák lektinnel bevont szilika affinitás állófázisok kialakítására. Általában a szilikát szilanilálják 3- merkaptopropil-trimetoxiszilánnal szobahőmérsékleten enyhe körülmények között. A tresylált szilikára a lektinek immobilizálását 4 órás óvatos keveréssel foszfát pufferben (pH 8,0) valósítják meg, amit Tris-HCl pufferrel (pH 8,0) a megmaradt szabad tresyl csoportok blokkolása követ, stabil oszlopot eredményezve.
A lektinek felülethez való kapcsolására 1,1’-karbo-diimidazol derivatizált szilikát is alkalmaznak, mely egy ötlépéses protokollal valósítható meg. A glikoproteinek az aktivált szilikához igen jó hatékonysággal kapcsolhatók enyhe körülmények között79.
Az elmúlt években a szilikán való lektin immobilizálási technikákat egyre növekvő a tudományos érdeklődés kísérte. A 3-merkaptopropil-trimetoxiszilánnal derivatizált szilika állófázisokra a fetuin izolálása magzati szarvasmarha szérumból búzacsíra agglutininnel