C H A P T E R 10
Bacterial Photosynthesis*
D A V I D M . G E L L E R
I. Introduction 461 II. The Chromatophore: Center of Bacterial Photosynthesis 462
A. Size, Structure, and Constitution 462 B. Photochemical Oxidation-Reductions 463 C. Photophosphorylation by Chromatophores 469 D . Chromatophore Role in Carbon Dioxide Fixation 471 E. Chromatophore Role in the Photoproduction of Hydrogen 471
III. A General Formulation of Bacterial Photosynthesis 472
IV. Concluding Remarks 476
References 476
I. Introduction
T h e problem of the mechanism of bacterial photosynthesis and its relationship to green plant photosynthesis stands t o d a y essentially as stated by v a n Niel in 1941.1 I n green plants and photosynthetic bacteria light energy is presumed to be utilized by chlorophyllous pigment systems by the simultaneous production of reducing and oxidizing powers, so- called " H " and " O H . " This p r i m a r y light reaction is followed by a series of d a r k reactions. T h e Η is consumed in the reduction of carbon dioxide;
O H is converted to oxygen in the green plant or consumed by reducing power supplied by substrate in the bacteria. As stated by v a n N i e l :1
Green plant: 2 H20 + C 02 -> ( C H20 ) + H20 + 02
Bacteria: 2 H2A + C 02 -> ( C H20 ) + H20 + 2A
T h e similarity between the green plant and bacterial systems appears to be so great t h a t the present reviewer cannot avoid comparing one with the other. This is of particular importance a t the present time when available information is fragmentary.
* This article is not intended as a complete review of the photosynthetic bacteria.
It will deal primarily with the relationship of photochemical reactions of extracts to the photosynthetic activity of the intact bacterial cells. A more complete discussion, including the culture and metabolism of photosynthetic bacteria, may be found in H. Gest and M. D . Kamen, Chapter IV, Volume 5 of "Handbuch der Pflanzenphysio- logie" (1958). The reader is also referred to S. R. Elsden, Chapter 1 of Volume III of "The Bacteria," for a discussion of carbon dioxide fixation, and to M. D . Kamen and J. W. Newton, Chapter 8 of Volume II of "The Bacteria" for a detailed discus
sion of the cytochromes in electron transport systems of photosynthetic bacteria.
461
462 D A V I D Μ . G E L L E R
II. The Chromatophore: Center of Bacterial Photosynthesis
T h e chromatophore was implicated as the photochemical center of bacterial cells by the finding t h a t the carotenoid and chlorophyll pig
ments associated with photosynthesis are present only in the chromato
phores. Evidence of the photochemical function of the chromatophores emerged from experiments showing photochemical oxidation-reductions involving (a) the oxidation of substances by oxygen, (b) the oxidation of substances by compounds other t h a n oxygen, and (c) the coupled oxi
dation and reduction of components of the chromatophores themselves.
Finally, the chromatophores were demonstrated to carry out the conver
sion of light energy to "energy-rich" phosphate bonds, a process termed photophosphorylation. An a t t e m p t will be made to compare the bacterial chromatophore to the green plant chloroplast, and to formulate a working scheme of bacterial photosynthesis.
A. S I Z E , S T R U C T U R E , A N D C O N S T I T U T I O N
The macromolecular organization of the photosynthetic bacterial cell of Rhodospirillum rubrum was found by Schachman et al.2 and Pardee et al.,3 to compartmentalize the pigment system in particles having an estimated diameter of 600 A. and a molecular weight of 30,000,000. I t was estimated t h a t each cell contained 5000 to 6000 of these chromato
phores. Particles of this size were not found in dark-grown R. rubrum cells (which did not contain chlorophyll or carotenoid pigments).
Good evidence for the existence of these chromatophores as such in the intact cells has come from electron microscope studies of sectioned cells.4 - 6 Similar particles have been found in Chromatium.7-9 T h e esti
mated mean diameter of isolated Chromatium chromatophores is 320 A .9 This corresponds to the size of vesicles (200 to 400 A. in diameter) ob
served in electron micrographs of sectioned Chromatium cells by V a t t e r and Wolfe.5 I n this study similar vesicles were found in Rhodopseudo- monas spheroides (400 to 800 A. in diameter) and Chlorobium limicola
(150 to 200 Α.).
T h e most detailed chemical analyses have been m a d e by Newton and N e w t o n7 , 8 on Chromatium chromatophores. Large amounts of polysac
charide and phospholipoprotein were found. T h e phospholipid moiety contained glycerol, ethanolamine, and phosphate in equimolar amounts.
T h e concentration of bacteriochlorophyll was estimated to be 200 mole
cules per chromatophore. T h e molar ratio of bacteriochlorophyll, carote- noids, and cytochromes was 1 0 : 5 : 1 . Also noteworthy was the presence of large amounts of acid-soluble nonheme ferrous iron.
These chromatophores could be cleaved into "small particle subunits"
by sonic oscillation, with subsequent loss of polysaccharide and protein,
1 0 . B A C T E R I A L P H O T O S Y N T H E S I S 463 resulting in a doubling of the chlorophyll, carotenoid, cytochrome, and phospholipid content with respect to protein. Acid-soluble components, including iron, phosphate, pyridine nucleotide, and flavin, were lost during the degradation. I t is suggested t h a t the bacterial chromatophores consist of repeating subunits cemented together by polysaccharide. P a r t i cles of both types carry out photophosphorylation.1 0
T h e possibility t h a t R. rubrum chromatophores m a y also be aggregates of small subunits is suggested by the fact t h a t small pigmented particles, about 3 0 0 A. in diameter, have been obtained by sonic t r e a t m e n t of cells in viscous media. Photochemically these particles are fully as active as the chromatophores isolated from comparable bacteria in less viscous media.9- η·1 2
As noted by Schachman et al.,2 chromatophores are not found in ex
tracts of dark-grown R. rubrum.5 According to H i c k m a n and F r e n k e l1 2 young light-grown cells of R. rubrum do not contain chromatophores;
instead smaller structures 1 0 0 to 1 5 0 A. in diameter are found distributed throughout the cells. This would appear to conflict with the work of Vatter and Wolfe.5 T h e youth of the cultures is not specified in either case, however; thus it is possible t h a t H i c k m a n and Frenkel's cells m a y contain developmental forms or subunits of chromatophores.
I t should be noted here t h a t members of the photosynthetic bacteria contain large quantities of hematin compounds (as much as 0 . 1 % of the dry weight of the cells) ,1 3 Large amounts of a cytochrome c1 4 have been found to be associated with chromatophores of R. rubrum15'16 and Chro
matium;7'8 quantities of the hematin compound R H P (Rhodospirillum, or Rhodopseudomonas, heme p r o t e i n )1 4 , 1 6 , 1 7 and a cytochrome b1 4 , 1 5 have also been found in R. rubrum chromatophores. Approximately equi
molar quantities of R H P and cytochrome c are present.1 6 T h e concentra
tion of heme pigments has been estimated to be of the order of 0 . 0 1 /xmole/
mg. protein.1 5
B . P H O T O C H E M I C A L O X I D A T I O N - R E D U C T I O N S
1. A E R O B I C P H O T O O X I D A T I O N S
T h e photooxidation of ascorbic acid by cell-free preparations of R.
rubrum was first described by F r e n c h .1 8 , 1 9 These experiments were ex
tended by Vernon and K a m e n2 0 who demonstrated t h a t mammalian cyto
chrome c and reduced 2,6-dichlorophenol-indophenol ( D P I P ) were also photooxidized by these preparations. T h e reaction was heat-labile and relatively insensitive to cyanide, azide, and hydroxylamine. T h e rate of ascorbate photooxidation was increased by the addition of D P I P . Two moles of ascorbate were oxidized per mole of oxygen consumed. Upon addition of large amounts of catalase and ethanol to the mixture, the final
464 D A V I D Μ . G E L L E R
ratio of ascorbate to oxygen consumed was halved; t h e oxidation of ethanol to acetaldehyde was coupled to the photooxidation of ascorbic acid. Hydrogen peroxide was ruled out as the oxidizing agent of ascorbic acid. Hydrogen peroxide m a y have been the product of ascorbate photo
oxidation, however, to serve as the oxidant of ethanol. According to the authors, the reaction could be pictured as a bacterial Hill reaction. W a t e r was presumed to be split photochemically to yield an oxidizing agent which reacted with ascorbic acid; the photochemical reductant from water then reacted with molecular oxygen to form the oxidizing agent of ethanol.
Vernon and K a m e n2 0 also have shown t h a t extracts of R. rubrum cata
lyze the photooxidation of reduced m a m m a l i a n cytochrome c. One mole of oxygen was consumed for every four moles of reduced cytochrome, in a cyanide-insensitive reaction which was associated with the chromato
phores.2 1 A cyanide-sensitive " d a r k " cytochrome oxidase was also found;
this was associated with particles smaller t h a n the chromatophores.
Furthermore, the photooxidase was found to be more heat-stable (63°C.
for 10 minutes) t h a n the " d a r k " oxidase; a temperature of 80°C. for 10 m i n u t e s2 0 completely inactivated the photooxidase. T h e photooxidase of R. rubrum catalyzed the oxidation of either R. rubrum cytochrome c
(cytochrome c2) or mammalian cytochrome c, whereas R. rubrum D P N H - cytochrome c reductase reduced only cytochrome c2 .2 1 T h e mechanism proposed for cytochrome c or D P I P photooxidation was similar [except for equation (3)] to t h a t for ascorbate:
2 H20 h v > 2 (H) -f 2 (OH) (1)
2 (H) + 02 > " H202" (2)
" H202" + 2 H + + 2 Fe++cyt > 2 Fe+^cyt. + 2 H20 (3) 2 (OH) + 2 H + + 2 F e ^ c y t > 2 Fe+^cyt. + 2 H20 (4) 4 Fe++cyt. + 4 H+ -f 02 > 4 Fe+^cyt. + 2 H20
2. A N A E R O B I C L I G H T - I N D U C E D O X I D A T I O N - R E D U C T I O N S
T h e Hill reaction of the · chloroplast2 2 has not been found in extracts of photosynthetic bacteria; this has been attributed to the absence of the oxygen evolution system in the bacteria.1 Recently it has been possible to demonstrate the equivalent of the Hill reaction in bacterial systems.
Frenkel1 1* 2 3 has described the enzymic photooxidation of reduced flavin mononucleotide ( F M N ) coupled to photoreduction of oxidized diphos- phopyridine nucleotide ( D P N ) by R. rubrum chromatophores under a n a erobic conditions. Oxidized triphosphopyridine nucleotide ( T P N ) would not substitute for D P N in this system. Succinate could be used instead of reduced F M N ; the rate of reduced diphosphopyridine nucleotide
10. B A C T E R I A L P H O T O S Y N T H E S I S 465 ( D P N H ) formation with succinate was several fold greater t h a n t h a t with F M N as hydrogen donor. This reaction could be visualized as a reaction of the photochemical Η with D P N , coupled with oxidation of the hydrogen donor, reduced F M N or succinate, by photochemically generated O H . T h e general significance of this reaction will be taken up in a later section.
V e r n o n2 4 has described the photoreduction of T P N by R. rubrum chro
matophores. Unlike Frenkel's system, D P N was not active; reduced tri- phosphopyridine nucleotide ( T P N H ) formation [as indicated by reduced glutathione (GSH) formation by glutathione reductase and oxidized glutathione (GSSG)] was inhibited by the addition of various hydrogen donors. T h e puzzling point concerned the source of reducing power for T P N . According to present concepts, utilization of Η alone should result in excess O H and hence inhibition of the system. Therefore, either the present scheme will have to be modified, or the experiments re-examined.
T h e necessary reducing power for T P N reduction (for consumption of OH) m a y be derived from reducing agents present in the chromatophore preparations. I n this case, the effect observed is analogous to FrenkePs observations.
More recently Vernon and A s h2 5 have confirmed Frenkel's findings;
magnesium ions, cyanide, and a preparation of photosynthetic pyridine nucleotide r e d u c t a s e2 6 were required for optimum photoreduction of D P N by R. rubrum chromatophores supplemented with succinate. Furthermore, V e r n o n2 7 has noted t h a t these chromatophore preparations carry out the anaerobic photooxidation of a variety of reduced dyes and other sub
stances [reduced 2,6-dichlorophenol-indophenol ( D P I P ) , methylene blue, indigo carmine, and ferrocytochrome c] by fumarate.
T h e work of Frenkel and Vernon is of particular interest, in t h a t light-induced reduction of pyridine nucleotide has been observed in intact cells. Recently Duysens and S w e e p2 8 have obtained evidence t h a t ir
radiation of intact cells of Chromatium and R. rubrum with infrared light resulted in an increase in concentration of a fluorescent substance similar to reduced pyridine nucleotide. This effect was dependent upon the presence of reducing agents in the medium.
3. C O M P A R I S O N O F R E A C T I O N S O F C H R O M A T O P H O R E C O M P O N E N T S W I T H I N T A C T B A C T E R I A L C E L L R E A C T I O N S
a. Light-Induced Oxidation-Reduction: Bacteriochlorophyll and Hemo
protein Components of the Chromatophore and of the Intact Bacterial Cell. A light-induced oxidation-reduction of components of extracts has been observed by Geller and G r e g o r y ,2 0 G e l l e r ,1 5*3 0 and Smith and B a l t s c h e f f s k y .3 1'3 2 Geller studied changes of absorption of R. rubrum chromatophore suspensions in the visible portion of the spectrum induced
4 6 6 DAVID Μ. GELLER
by infrared light. Within one second, infrared light induced an increase in absorption with a maximum a t 4 3 5 m/x (and smaller peaks a t 4 9 0 , 530, and 5 6 5 m/x) and a decrease in absorption a t 6 0 0 m/x (with a smaller trough at 3 9 5 m/x). T h e 435-m/x peak appeared to be composed of several components. Since the addition of sodium hydrosulfite to chromatophore preparations in the d a r k induced a maximum increase in absorption a t 4 3 0 m/x, it was concluded t h a t the peak at 4 3 5 m/x induced by light repre
sented photoreduction of one or more components, the most likely being R H P and cytochrome b. T h e addition of ferricyanide to chromatophores in the d a r k caused a maximum decrease in absorption at 5 9 5 - 6 0 0 ιημ. The light-induced change of absorption at 6 0 0 m/x therefore represented photo
oxidation of one or more components (perhaps bacteriochlorophyll, which has a peak at 5 9 0 m/x). T h u s light induced the simultaneous oxidation of one or more components and the reduction of others.
The significance of the 435-m/x shift has not been established with certainty, however. T h e effect of infrared light on R. rubrum cells and extracts has been examined in more detail by Smith and Baltscheffsky.3 2 I n agreement with Geller, it was found t h a t light induced the formation of a broad peak in extracts at 4 3 4 m/χ. I n contrast to this, allowing the extract to become anaerobic in the d a r k resulted in formation of a peak at 4 2 8 m/x. This would probably be the hemoprotein R H P . T h e same peak appeared if D P N H was added to a preparation in the d a r k .3 3 I n the presence of D P N H , however, the light-induced increase in absorption
(observed at 4 3 0 m/x) persisted. T h e effects of light on D P N H utilization were irregular and not reproducible. I n these experiments the D P N H added was being consumed throughout the experiment, an indication t h a t oxygen was present. T h u s the fact t h a t the light-induced increase of absorption a t 4 3 0 m/x persisted in the presence of D P N H did not rule out the possibility t h a t the light effect represented a photoreduction.
However, D u y s e n s3 4 has introduced the suggestion t h a t the 430-m/x change m a y represent bacteriochlorophyll oxidation. H e observed t h e appearance of a broad peak centered at 4 3 2 m/x on irradiation of intact cells of R.
rubrum in "aerobic distilled water." This was accompanied by shifts in the infrared indicating bacteriochlorophyll photooxidation.3 5 , 3 6 T h e conclusions concerning the significance of the infrared shifts have been supported by the fact t h a t identical spectral changes are induced by ferricyanide and reversed by f e r r o c y a n i d e .3 6 - 3 8
T h e light-induced change a t 4 3 2 m/x observed by Duysens does not, however, appear to be related to bacteriochlorophyll (Olson and K o k3 9) since the appearance of the 432-m/x peak is not synchronous with changes in the infrared bands of bacteriochlorophyll. More recently, Smith et al.40 have concluded t h a t the 435-m/x peak probably does not represent a cyto-
10. B A C T E R I A L P H O T O S Y N T H E S I S 467 chrome: the absorption band is much broader t h a n t h a t of the usual cyto
chrome pigment; furthermore, it does not appear to be related to the spectral changes attributed to the carotenoids. T h u s the significance of the light-induced absorption changes in the region of 430 to 435 m/x, and the nature of the pigment (s) involved are unknown at the present time.
b. Photooxidation of Cytochrome c2 of the Chromatophore. Two ob
servations of great interest have been made by Smith and co-workers.
T h e first concerns the effect of phosphate acceptors upon light-induced absorption changes in extracts of R. rubrum.32 T h e addition of adenosine diphosphate ( A D P ) to an extract resulted in a trough in the light-dark difference spectrum at 420 χημ. I n the absence of A D P a peak at 420 τημ was induced by light. T h e difference between the light-induced effects with and without A D P was thus represented by a trough at 420 τημ. This indi
cated an increased rate of photooxidation of cytochrome c2 in the presence of A D P . Such changes in the steady state of cytochrome c2 were seen only in very active extracts. I n further work, it was established t h a t the addi
tion of 3-hydroxy-l-heptyl-quinoline-iV-oxide to the same extract deep
ened the trough induced by light a t 420 m/x, in the presence of A D P and shifted the broad 434-nni band to about 430 ηΐμ. These observations were interpreted as an increased photooxidation of cytochrome c2 and increased photoreduction of cytochrome b. T h u s this inhibitor, which effectively blocks photophosphorylation but not respiration by an R. rubrum extract, blocks electron transport between cytochrome b and cytochrome c2 . T h u s cytochromes b and c2 have been implicated as members of the electron chain involved in photophosphorylation (discussed below) and phospho
rylation is coupled to the oxidation of cytochrome c2.
c. Light-Induced Changes in the Absorption of the Carotenoid Pigments.
T h e second observation of Smith and R a m i r e z4 1 m a y lead to an unraveling of the complex light-induced absorption changes observed with extracts of the photosynthetic bacteria. I n a study of the possible relationship of the carotenoid pigments to these absorption changes seen in the intact cells, Smith and Ramirez have examined a variety of photosynthetic bac
teria, varying in kind and quantity of carotenoid pigments. W i t h intact cells of the carotenoidless blue-green m u t a n t of R. spheroides (or d a r k - grown photosynthetic bacteria, which are devoid of carotenoids), for ex
ample, the difference spectra showed only the oxidation of cytochrome pigments on oxygenation or illumination. I n bacteria containing carote
noids, illumination or oxygenation resulted in the partial loss of absorp
tion bands a t wavelengths characteristic of carotenoid pigments of the organism and the appearance of new absorption bands a t longer wave
lengths. T h e similarity between oxygenation and light, t a k e n with the shift toward longer wavelengths, would be consistent with t h e view t h a t
468 DAVID Μ. GELLER
these transformations represent oxidation of the carotenoid pigments.
More recently, however, Smith and R a m i r e z4 2 have concluded t h a t changes in the absorption spectrum of the carotenoid pigments m a y rep
resent "structural changes" resulting from the initiation of electron t r a n s fer processes rather t h a n oxidation of the carotenoid pigments. T h e spec
tral changes attributed to carotenoids have been distinguished from those related to cytochromes by differences in kinetics and differences in in
hibition by a variety of agents.
These findings m a y be related to the experiments of Stanier and co
w o r k e r s ,4 3"4 5 regarding carotenoid synthesis. These workers have reported the increased sensitivity of carotenoidless m u t a n t or diphenylamine- treated (carotenoidless) cells toward oxygen. T h e carotenoid pigments a p pear to prevent photooxidation of the photo a p p a r a t u s ; this is manifested by photodestruction of chlorophyll and eventual death of these carotenoid
less cells in the light in the presence of oxygen. Particles derived from di- phenylamine-treated cells carry out the photophosphorylation reaction in the case of Chromatium*6 T h e photochemical activity of these particles is (as expected) more sensitive to oxygen t h a n t h a t of the usual carotenoid- containing particles. As suggested by C a l v i n ,4 7 this protective effect of carotenoid pigments m a y be one result of the efficient conversion of ex
cited chlorophyll molecules into reducing and oxidizing agents by a proc
ess of conduction in the conjugate chain of the carotenoid molecules as
sociated with chlorophyll.
T h u s carotenoid pigments m a y have functions other t h a n t h a t of the transfer of absorbed light energy to chlorophyll.
d. Photooxidation of Phenazine Methyl Sulfate. I n a series of experi
ments on the activation of photophosphorylation by phenazine methyl sulfate, it was found t h a t partially reduced phenazine dye was photo- oxidized by suspensions of R. rubrum c h r o m a t o p h o r e s .1 5'2 9 T h e reaction was unlike the photooxidation of cytochrome c or D P I P in t h a t it was a completely reversible, limited photooxidation which proceeded under rigorously anaerobic conditions. T h e amount of dye photooxidized was of the order of magnitude of the cytochrome components. When the light was turned off, an equivalent amount of dye was reduced. Absence of oxygen (after thorough flushing with nitrogen or helium) was indicated by the stability of the reduced autoxidizable dye throughout the experi
ment.
T h e light-induced absorption changes of the chromatophores were m a r k edly altered by partially reduced phenazine dye. Two effects of the dye were noted. First of all, the trough at 600 τημ induced by light in the absence of reduced dye was abolished or appreciably reduced in size.
Secondly, light-induced absorption changes in the 420 to 450 τημ region were markedly altered in t h e presence of reduced dye. T h e alteration a p -
10. B A C T E R I A L P H O T O S Y N T H E S I S 469 peared to depend on the extent of dye reduction. When the ratio of re
duced dye t o oxidized dye was very low, light induced the formation of a broad trough centered a t 440 mit; at high ratios of reduced to oxidized dye, light induced a peak a t 440 χημ. W h e n the dye was completely oxi
dized, the light-dark difference spectra were identical t o t h a t observed in the absence of the dye. On the other hand, completely reduced dye abolished all light-induced absorption changes.
Photooxidation of partially reduced phenazine dye thus appears to be caused b y direct or indirect oxidation by t h e "600-mt^, , pigment of the chromatophore. T h e pigment (s) involved in the 420- to 440-mtt shifts evidently is unrelated to the 600-mtt pigment. One pigment (such as b a c teriochlorophyll) could not be responsible for both absorption changes.
C . P H O T O P H O S P H O R Y L A T I O N B Y C H R O M A T O P H O R E S
As noted above, experimental evidence has been obtained for the simul
taneous photoproduction of reducing and oxidizing agents in intact cells and in extracts of photosynthetic bacteria. I n addition, high concentra
tions of hematin compounds are present in even the most obligate anae
robes of t h e photosynthetic bacteria. These undergo oxidation-reduction changes in t h e light. T h u s a system analogous t o the oxidative phos
phorylation system of mitochondria might exist in the photosynthetic bacteria. This could be a recombination of photochemical Η and O H by electron transport across chains of electron acceptors (including the hema
tin compounds) coupled with formation of energy-rich phosphate bonds.
Evidence consistent with photophosphorylation was first obtained b y Gest and K a m e n ' s d e m o n s t r a t i o n4 8 of a light-accelerated u p t a k e of P3 2 orthophosphate by intact anaerobic cells of R. rubrum.
Photophosphorylation in bacterial extracts was first observed by F r e n k e l4 9 with extracts of R. rubrum a t about the time Arnon discovered the reaction in spinach chloroplasts.5 0 T h e reaction has since been demon
strated in cell-free preparations from R. rubrum, Chromatium,61'54 and Chlorobium limicola.51 T h e cell-free systems derived from R. rubrum and Chromatium have been examined in some detail.
F r e n k e P s4 9 first report of photophosphorylation b y R. rubrum extracts was with a particulate fraction corresponding to the chromatophores. T h e particles were inactivated b y washing; and the activity completely restored by the original supernate or b y a α-ketoglutarate. Geller1 5' 2 9 subsequently observed reactivation of washed particles b y catalytic quantities of suc
cinate, lactate, or D P N H ;1 5' 5 5 fumarate, p y r u v a t e , or D P N were ineffec
tive. Both investigators, therefore, concluded t h a t catalytic quantities of reducing agent were required for photophosphorylation.
T h e washed particles require A D P or inosine diphosphate ( I D P ) for
470 DAVID Μ. GELLER
photophosphorylation; with added supernate, which contains myokinase, adenosine monophosphate ( A M P ) will serve as phosphate acceptor. T h e product, in each case, was characterized as the corresponding triphos
p h a t e .1 5 , 5 5
On further investigation the activity of R. rubrum washed particles was found to be markedly increased by the addition of catalytic amounts of phenazine methyl sulfate.1 5 , 2 9» 3 0 Such systems were active in the a b sence of (and unaffected by) reducing agents, but if the incident light was filtered to remove the wavelengths absorbed by the photosensitive dye, a reducing agent was required for activation by the dye. Excessive reduction of the dye inhibited photophosphorylation; this inhibition was reversed by reoxidation of some of the dye. Thus, partial reduction of the phenazine dye was required for activity. Under optimum conditions the addition of this dye caused as much as an eightfold stimulation of photo
phosphorylation over t h a t observed with reducing agents alone ( D P N H , succinate, or l a c t a t e . )1 5
A similar restoration of the photophosphorylation by Chromatium53 washed particles ("chromatophore fragments") occurred on the addition of such reducing agents as thioethanol, cysteine, or hydrogen sulfide. T h e highest activity was obtained by adding phenazine methyl sulfate or ascorbate; the system was also activated by D P I P . N o filter was used in these studies with the phenazine dye, so it remains to be established whether a reducing agent is required in addition to the phenazine dye.
T h e particles were not activated by preparations of photooxidized phena
zine dye.
Photophosphorylation by spinach chloroplasts is activated by phenazine methyl sulfate.5 6 When red light was used for photophosphorylation, the phenazine dye had to be exposed to white light and air to activate chloro- plast preparations.5 7 , 5 8 Pyocyanine, one of the photooxidation products of the phenazine dye, was, however, at one-tenth the concentration, as active as the phenazine dye and required no pretreatment. Pyocyanine, or photooxidized phenazine methyl sulfate does not activate the R. rubrum system. 1 5
Photophosphorylation by phenazine methyl sulfate-activated Chroma- Hum particles was inhibited almost completely by the presence of excess reducing agent (ascorbate or t h i o e t h a n o l ) .5 3 T h e addition of D P I P or thioethanol to ascorbate-activated particles reduced photophosphorylation to t h a t level with D P I P alone.
T h e effects of various inhibitors upon these photophosphorylation sys
tems have been studied in detail. R. rubrum photophosphorylation is rela
tively unaffected by cyanide, azide, hydroxylamine, arsenite, and 100%
carbon monoxide.1 5 T h e effects of the latter, even though precautions have been taken to remove all visible light a t wavelengths below 700 m/x, are in
10. B A C T E R I A L P H O T O S Y N T H E S I S 471 some doubt because of the extreme photosensitivity of the carbon monoxide compound of R H P .1 7 Photophosphorylation is inhibited b y agents which uncouple phosphorylation from oxidation. R. rubrum preparations require rather high concentrations of 2,4-dinitrophenol (10~s Μ for 50% inhibi
t i o n ) .1 5- 4 9 Chromatium particles appear to be more sensitive to dinitro- phenol.5 3 M u c h lower concentrations (3 Χ Ι Ο- 5 M) of the butyl ester of 3,5-diiodo-4-hydroxybenzoate5 9 inhibit R. rubrum photophosphoryla
t i o n .1 5
I n all the inhibition studies with the R. rubrum system described above, the effect of inhibitors was the same regardless of the activator used for photophosphorylation. This was not the case in two instances: R. rubrum particles were inhibited by low concentrations of antimycin A and com
pound S N 5 9 4 9 ;1 5-3 0 preparations (either crude extracts or particles) supplemented with the phenazine dye were relatively insensitive to either inhibitor. Both inhibitors are known to block electron t r a n s p o r t in mito
chondrial systems between cytochromes b and c ;6 0' 6 1 hence, both m a y block electron transport in the R. rubrum system at the same site a t which the phenazine dye provides a bypass. T h e stimulation by the dye would then be explained by the electron transport step bypassed being the r a t e - limiting step.
D . C H R O M A T O P H O R E R O L E I N C A R B O N D I O X I D E F I X A T I O N
Both chloroplast and bacterial chromatophore systems have been shown to catalyze a light-induced incorporation of carbon dioxide into reduced carbon compounds. I n t a c t chloroplasts alone fix carbon dioxide, whereas chloroplast fragments require in addition a soluble extract of chloro
p l a s t s .6 2 Isolated chromatophores do not catalyze carbon dioxide fixation;
they t h u s correspond to chloroplast fragments.
Fuller and A n d e r s o n6 3 have observed light-induced incorporation of radioactive carbon dioxide into phosphoglyceric and aspartic acids by cell-free preparations from Chromatium. Isolated chromatophores were inactive b u t could be reactivated b y addition of the colorless supernatant fraction obtained b y centrifugation of such extracts. Similar observations have been made with R. rubrum e x t r a c t s .6 4 T h e enzymes involved in C 02 fixation appear to be in the supernatant fraction and the chromatophores to be solely the source of light-derived reducing power. T h u s again the chromatophore corresponds to the "chloroplast fragment" rather t h a n the whole chloroplast.
E . C H R O M A T O P H O R E R O L E I N T H E P H O T O P R O D U C T I O N O F H Y D R O G E N
T h e photoproduction of molecular hydrogen b y purple bacteria sup
plied with certain accessory hydrogen donors, such as p y r u v a t e and C4- d i - carboxylic acids, has been studied in detail b y Gest and his colleagues,6 5' 6 6
472 DAVID Μ. GELLER
who find biological H2 production mediated by a multienzyme electron transport system with hydrogenase t h e terminal catalyst.
Of interest in this connection are recent experiments of K a r u n a i r a t n a m and G e s t6 7 which disclose complete localization of R. rubrum hydrogenase of extracts in the chromatophores.
III. A General Formulation of Bacterial Photosynthesis
T h e bacterial chromatophore and the plant chloroplast have much in common. Both contain high concentrations of hematin pigments, aside from the usual photosynthetic pigments. Both catalyze photochemical oxidation-reduction reactions, i.e., Hill reactions or variants thereof. I n both photophosphorylation is activated by oxidation-reduction dyes; there still m a y be some question of the reducing agent requirement for the chloroplast system.
T h e chief difference between the photochemical activity of the bacterial chromatophore and the plant chloroplast appears to be the ability of the latter to evolve molecular oxygen; the substrate requirement for growth of the photosynthetic bacteria is related to the absence of the oxygen evo
lution mechanism.1 One is led to wonder whether the requirement of chro
matophore photophosphorylation for reducing agents m a y be directly re
lated to the substrate requirement of the intact cells.
Indications of this possibility are demonstrated by the catalysis by R.
rubrum chromatophore suspensions of: (1) the photoreduction of D P N by succinate,2 3 (2) carbon dioxide fixation in the presence of acceptor en
zymes by the supernatant fraction of a cell-free extract.6 4 Succinate is capable of inducing photophosphorylation by washed R. rubrum chro
m a t o p h o r e s ;1 5 succinic dehydrogenase is localized in the chromato
phores.6 8 T h u s chromatophores should carry out the photoreduction of carbon dioxide by succinate in the presence of pyridine nucleotide and carbon dioxide-fixing enzymes. For photophosphorylation the quantity of succinate required is catalytic, whereas stoichiometric amounts would be required for carbon dioxide fixation. Several schemes relating carbon dioxide fixation, respiration, and photophosphorylation in the photosyn
thetic bacteria have been a d v a n c e d .1 1 , 3 2
T h e requirement of catalytic amounts of reducing agents for photo
phosphorylation remains unexplained. Newton and K a m e n5 3 have sug
gested t h a t , since the photophosphorylation system must consist of a coupled system of electron acceptors, an optimal steady state relationship must exist between the reduced and oxidized forms of the interacting elec
tron carriers. Alteration of this steady state—for example by isolation of the chromatophores—would decrease the activity. T h e adjustment of the steady states of the carriers toward the "optimal relationship" m a y be ac-
10. BACTERIAL PHOTOSYNTHESIS 473 complished by "reductants 'driving' the system a t low potential level," and b y "mild oxidants, e.g., phenazine methyl sulfate and ascorbate, 'pulling' the chain at higher potential levels." Related to this proposal is the possi
bility t h a t catalytic amounts of reducing agent would effectively remove oxygen, which is an inhibitor of photophosphorylation.5 3' 6 9 One of the functions of the accessory hydrogen donor has been suggested to be pre
vention of destructive peroxidation resulting in photooxidation of the electron transport chain components.6 5
A second explanation of the reducing agent requirement for photophos
phorylation is based on the proposal of F r e n k e l5 5 t h a t the activation ef
fects are due to reduction of one or more of the electron carrier components, enabling the system to react with the "short-lived photochemical oxidant."
F o r example, one m a y consider t h e reduction of Y by X H2 : γ + χ π2 ~ = i - Y H2 + X
T h e reduction of Y by X H2 is not a spontaneous process; a t equilibrium little or no X or Y H2 m a y be found. T h e reaction is, however, driven in direction a by light; and in the dark, proceeds backward spontaneously in direction 6. One m a y then suppose t h a t another p a t h w a y for the reduc
tion of X by Y H2 exists, p a t h w a y c:
γ + χ π2
^=^~
Y H2 + X ο\ c /
P a t h w a y c consists of a complex of enzymes composed of cytochromes, etc; phosphorylation is coupled t o electron transport via p a t h w a y c. This p a t h w a y is assumed to be such an efficient t r a p for the reducing power of Y H2 t h a t oxidation of Y H2 by p a t h w a y b does not occur.
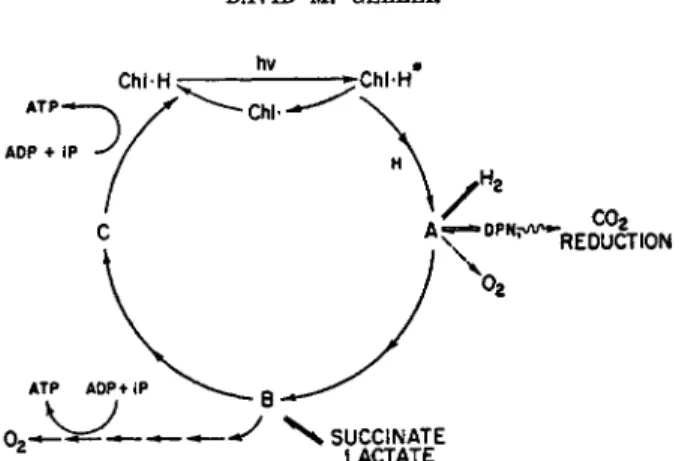
If one assumes bacteriochlorophyll is X H2 and the initial component of p a t h w a y c is Y, t h e scheme indicated by Fig. 1 m a y be formulated. Follow
ing the suggestion of D u y s e n s ,7 0 this scheme depicts the action of light as resulting in a photoreduction of t h e lowest potential member of the elec
tron transport chain, rather t h a n of photolysis of water. T h e effect of light is solely to change the potential of (unexcited) bacteriochlorophyll, C h l - H , from an estimated 400 to 500 m v .3 7»3 8 to a potential (of excited bacteriochlorophyll, C h l - H * ) sufficiently low to reduce pyridine nucleo
tide (about —300 m v . ) . T h e reduction of A, t h e lowest potential electron carrier (pyridine nucleotide?), would produce unexcited oxidized bac
teriochlorophyll (Chi) which would react either with reduced A or elec
tron carrier C (which has the highest potential in t h e system, aside from bacteriochlorophyll). Reduced excited and oxidized unexcited bacterio
chlorophyll thus correspond to t h e Η and O H of other schemes.
474 DAVID Μ. GELLER
ην *
Chl-H ς— -^ChlH
. co2
REDUCTION
FIG. 1. Scheme of photophosphorylation of bacteriochlorophyll and its associated electron transport system.
Formation of reducing power by light would thus be a function of the concentration of reduced bacteriochlorophyll. I n the light bacteriochloro
phyll would be oxidized by A. T h e light excitation and oxidation reactions are assumed not to be rate limiting; the rate-limiting reaction must lie somewhere within the electron transport chain, such as Β - » C. T h e light steady states of bacteriochlorophyll and terminal electron acceptor C would thus be more oxidized, and t h a t of electron acceptor A more reduced, with respect to their levels in the dark. Addition of reducing power to the system, to pull the light steady state of C toward reduction would increase the concentration of reduced unexcited bacteriochlorophyll ( C h l * H ) , the substrate of the light reaction, leading to increased electron transport and thus to photophosphorylation.
Bacteriochlorophyll and its associated electron transport system repre
sent, according to Fig. 1, a system which m a y function either solely as a
"closed circuit" or as an "open circuit" for electrons. T h e first case is represented by photophosphorylation. T h e phosphorylation is coupled to electron transport; but without loss of electrons from the system. T h e only over-all change measurable is the accumulation of adenosine triphosphate.
T h e second case is represented by the reduction of pyridine nucleotide and carbon dioxide, or by hydrogen evolution, at the expense of an externally supplied hydrogen donor. Electrons flow into the system at one point and emerge at another. Light energy is utilized to alter the potential of elec
trons flowing through the system. F o r example, electrons from the suc
cinate-fumarate couple m a y enter the system so as to leave it at a lowered potential to reduce pyridine nucleotide or to produce molecular hydrogen.
Photophosphorylation m a y occur simultaneously with these oxidation- reduction processes, as demonstrated by Arnon and his co-workers, who
10. BACTERIAL PHOTOSYNTHESIS 475 have shown t h a t photophosphorylation m a y be coupled to T P N H and oxygen production by chloroplast fragments.7 1
Several features favor the scheme just described, as shown in Fig. 1. I t is consistent with the activation of photophosphorylation by a catalytic q u a n t i t y of reducing agent, i.e., with the system operating solely by photo
phosphorylation, electrons circulating in a closed circuit. T h e quantity of reducing power to stimulate photophosphorylation would be consider
ably less than is required for reduction of all of the components of the system; the reducing power furnished to the system would not be con
sumed.
T h e scheme is also consistent with inhibition of photophosphorylation by excess reduction or oxidation. F o r example, excessive reduction of any carrier would slow reduction of t h a t carrier; i.e., t h e r a t e of reduction of A would be decreased by "pulling" t h e steady state level of A t o a more reduced level. Complete reduction of pigment A by an external electron donor would block oxidation of bacteriochlorophyll. T h e system is thus poisoned by excess reducing agent.
Evidence has been obtained implicating bacteriochlorophyll directly in light oxidation-reductions. D u y s e n s3 5 and Duysens et αϊ.3 6 have demon
strated the photooxidation of bacteriochlorophyll in intact R. rubrum cells deprived of substrates. G o e d h e e r3 8 and D u y s e n s3 7 have observed identical changes in infrared absorption of intact cells and of extracts on adding ferricyanide. Recently Vishniac and R o s e7 2 have reported a light-induced incorporation of tritium into chlorophyll from tritium-labeled water by Chromatium chromatophores and acetone powders of spinach chloroplasts.
With the latter preparation the experiments indicate a light-induced t r a n s fer of tritium from chlorophyll to T P N without loss of tritium to water.
T h e energy requirement for bacterial photosynthesis has been calcu
lated by D u y s e n s .7 0 T h e m a x i m u m efficiency calculated is 6 8 % : on this basis, only 21 kcal. per einstein absorbed by bacteriochlorophyll could be utilized by bacterial systems. Assuming t h a t bacterial pyridine nucleotide is reduced, with the simultaneous oxidation of a substance (presumably bacteriochlorophyll) with an EQ value of 0.44 volt, Duysens has calculated t h a t 35 kcal. of free energy are required per mole of reduced pyridine nu
cleotide; or sufficient energy if properly coupled for t h e production of a maximum of three energy-rich phosphate bonds (assuming a 60 to 70%
efficiency for this process). Furthermore, according to Duysens' calcula
t i o n s ,7 0 two einsteins of q u a n t a would be required per mole of pyridine nucleotide reduced; t h u s a maximum of three A T P molecules produced per two quanta. T h e experimental d a t a with Chromatium cells range from three to ten q u a n t a required to reduce one molecule of pyridine nucleotide.
Olson and C h a n c e7 3 have estimated t h a t two q u a n t a per electron are r e quired for the oxidation of Chromatium cytochrome.
476 D A V I D Μ . G E L L E R
IV. Concluding Remarks
T h e m a n y gaps in our knowledge reduce t h e foregoing presentation t o b u t a sketchy picture of t h e mechanism of bacterial photosynthesis.
T h e most successful approach t o this problem has been through t h e rec
ognition of reaction steps of t h e photosynthetic process in i n t a c t cells a n d t h e demonstration of these reactions in cell-free extracts. B y this means, t h e chromatophore h a s been recognized as t h e photochemical center of t h e bacterial cell, j u s t as t h e chloroplast is for t h e p l a n t cell.
ACKNOWLEDGMENTS
The author wishes to express his thanks to Dr. Albert Frenkel for sending him a copy of his review article on this subject, prior to its publication in Annual Review of Plant Physiology (1959). He also is indebted greatly to Dr. Lucile Smith for copies of several articles prior to publication.
R E F E R E N C E S
1C . B. van Niel, Advances in Enzymol. 1, 263 (1941).
aH . K. Schachman, A. B. Pardee, and R. Y. Stanier, Arch. Biochem. Biophys. 3 8 , 245(1952).
8 A. B. Pardee, Η. K. Schachman, and R. Y. Stanier, Nature 169, 282 (1952).
*A. E. Vatter and R. S. Wolfe, Bactenol. Proc. {Soc. Am. Bactenologists) p. 30 (1957).
5 A. E. Vatter and R. S. Wolfe, J. Bactenol. 7 5 , 480 (1958).
6 A. Frenkel, D . Hickman, and L. Smith, Bactenol. Revs. 21,256 (1957).
7 J. W. Newton and G. A. Newton, Arch. Biochem. Biophys. 7 1 , 250 (1957).
8 J. W. Newton, G. A. Newton, and M. D . Kamen, Intern. Congr. Microbiol., 7th Congr., Stockholm, 1958, Abstr. p. 75 (1958).
9 J. A. Bergeron, I. C. Anderson, and R. C. Fuller, Plant Physiol. 3 2 , Suppl. xvi (1957).
1 0 J. W. Newton and M. D . Kamen, Bactenol. Proc. (Soc. Am. Bactenologists) p. 115 (1956).
1 1 A. W. Frenkel, Brookhaven Symposia in Biol. 1 1 , 276 (1959).
MD . D . Hickman and A. W. Frenkel, J. Biophys. and Biochem. Cytol. 6, 277 (1959);
A. W. Frenkel and D . D . Hickman, J. Biophys. and Biochem. Cytol. 6, 285 (1959).
1 8 M. D . Kamen, in "Research in Photosynthesis" (H. Gaffron, A. H. Brown et al., eds.), p. 524. Interscience, New York, 1957.
1 4 L. P. Vernon and M. D . Kamen, J. Biol. Chem. 2 1 1 , 643 (1954).
1 5 D. M. Geller, Ph.D. Thesis, Harvard University, Cambridge, Massachusetts, 1957.
1 6 R. Hill and M. D . Kamen, Unpublished results (1956).
1 7 R. G. Bartsch and M. D . Kamen, Λ Biol. Chem. 2 3 0 , 41 (1958).
1 8 C. S. French, J. Biol. Chem. 123, xxxviii (1938).
1 9 C. S. French, / . Gen. Physiol. 2 3 , 469 (1940).
2 0 L. P. Vernon and M. D . Kamen, Arch. Biochem. Biophys. 4 4 , 298 (1953).
2 1M . D . Kamen and L. P. Vernon, / . Biol. Chem. 211,663 (1954).
2 2 R. Hill, Proc. Roy. Soc. B 1 2 7 , 192 (1939); Symposia Soc. Explt. Biol. No. 6, 222 (1951).
8 8 A. W. Frenkel, / . Am. Chem. Soc. 8 0 , 3479 (1958).
10. BACTERIAL PHOTOSYNTHESIS 477
2 4 L. P. Vernon, J. Biol. Chem. 233 , 212 (1958).
2 5 L. P. Vernon and Ο. K. Ash, Λ Biol. Chem. 234,1878 (1959).
2 6 A. San Pietro and Η. M. Lang, / . Biol. Chem. 231 , 211 (1958).
2 7 L. P. Vernon, J. Biol. Chem. 234,1883 (1959).
2 8 L. Ν. M. Duysens and G. Sweep, Biochim. et Biophys. Acta 25,13 (1957).
2 9 D . M. Geller and J. D . Gregory, Federation Proc. 15 , 260 (1956).
8 0 D . M. Geller, Intern. Congr. Microbiol. 7th Congr., Stockholm, 1958. Abstr. p. 73 (1958).
8 1L . Smith and M. Baltscheffsky, Federation Proc. 15 , 357 (1956).
8 2 L. Smith and M. Baltscheffsky, J. Biol. Chem. 234,1575 (1959).
8 8 B. Chance, M. Baltscheffsky, and L. Smith in "Research in Photosynthesis" (H.
Gaffron, A. H. Brown et al., eds.), p. 195, 199. Interscience, New York, 1957.
8 4 L. Ν. M. Duysens in "Research in Photosynthesis" (H. Gaffron, A. H. Brown, et al., eds.), p. 164. Interscience, New York, 1957.
8 5 L. Ν . M. Duysens, Nature 173,692 (1954).
8 0 L. Ν. M. Duysens, W. J. Huiskamp, J. J. Vos, and J. M. van der Hart, Biochim.
et Biophys. Acta 19,189 (1956).
8 7 L. Ν. M. Duysens, Brookhaven Symposia in Biol. 11 , 10 (1959).
8 8 J. C. Goedheer, Brookhaven Symposia in Biol. 11,325 (1959).
8 9 J. M. Olson and B. Kok, Biochim et Biophys. Acta 32 , 278 (1959).
4 0 L. Smith, M. Baltscheffsky, and J. M. Olson, / . Biol. Chem. 235 , 213 (1960).
4 1L . Smith and J. Ramirez, Arch. Biochem. Biophys. 79 , 233 (1959).
4 2 L. Smith and J. Ramirez, J. Biol. Chem. 235 , 219 (1960).
4 8 M. Griffiths, W. R. Sistrom, G. Cohen-Bazire, and R. Y. Stanier, Nature 176 , 1211 (1955).
4 4 G. Cohen-Bazire and R. Y. Stanier, Nature 181,250 (1958).
4 5 R. Y. Stanier and G. Cohen-Bazire, Intern. Congr. Microbiol., 7th Congr., Stockholm, 1958. Abstr. p. 76 (1958).
4 6 R. C. Fuller and I. C. Anderson, Nature 181 , 252 (1958).
4 7 M. Calvin, Nature 176,1215 (1955).
4 8 H. Gest and M. D . Kamen, J. Biol. Chem. 176 , 299 (1948).
4 9 A. W. Frenkel, J. Am. Chem. Soc. 76 , 5568 (1954).
8 0 D. I. Arnon, Μ. B. Allen, and F. R. Whatley, Nature 174 , 394 (1954).
6 1 A. M. Williams, Biochim. et Biophys. Acta 19 , 570 (1956).
6 3 J. W. Newton and M. D . Kamen, in "Research in Photosynthesis" (H. Gaffron, A. H. Brown et al., eds.), p. 311. Interscience, New York, 1957.
8 8 J. W. Newton and M. D . Kamen, Biochim. et Biophys. Acta 25 , 462 (1957).
5 41 . C. Anderson and R. C. Fuller, Plant Physiol. 32 , Suppl. xvi (1957).
6 5 A. W. Frenkel, / . Biol. Chem. 222 , 823 (1956).
M A. T. Jagendorf and M. Avron, J. Biol. Chem. 231 , 277 (1958).
6 7D . A. Walker and R. Hill, Biochem. J. 69 , 57P (1958).
6 8 R. Hill and D . A. Walker, Plant Physiol. 34,240 (1959).
5 9 F. L. Hoch and F. Lipmann, Proc. Natl. Acad. Sci. U. S. 40 , 909 (1954).
8 0 V. R. Potter and A. E. Reif, / . Biol. Chem. 194 , 287 (1952).
8 1E . G. Ball, C. B. Anfinsen, and O. Cooper, J. Biol. Chem. 168 , 257 (1947).
" F . R. Whatley, Μ. B. Allen, L. L. Rosenberg, J. B. Capindale, and D . I. Arnon, Biochim. et Biophys. Acta 20 , 462 (1956).
6 8 R. C. Fuller and I. C. Anderson, Plant Physiol. 32 , Suppl. xvi (1957).
8 4 A. W. Frenkel, Unpublished observations (1954).
8 5 H. Gest and M. D . Kamen in "Handbuch der Pflanzenphysiologie" (W. Ruhland, ed.), Vol. V, Chapter IV. Springer, Berlin, 1958.
4 7 8 DAVID Μ. GELLER
8 8 Η. Gest, in "Proceedings of the International Symposium on Enzyme Chemistry, Tokyo and Kyoto, 1957, p. 250. Academic Press, New York, 1958.
0 7 M. C. Karunairatnam and H. Gest, Intern. Congr. Microbiol. 7th Congr., Stockholm, 1958. Abstr. p. 74 (1958).
0 8 B. R. Woody and E. S. Lindstrom, Λ Bactenol. 6 9 , 353 (1955).
8 9 A. W. Frenkel, Plant Physiol. 3 1 , Suppl. xxx (1956).
7 0 L. Ν. M. Duysens, Ann. Rev. Plant Physiol. 7, 25 (1956).
7 1D . I. Arnon, F. R. Whatley, and Μ. B. Allen, Science 127, 1026 (1958).
7 8 W. Vishniac and I. A. Rose, Nature 182,1089 (1958).
7 3 J. M. Olson and B. Chance, Biochim. et Biophys. Acta 2 8 , 227 (1958).