TARTALOMJEGYZÉK
KIVOAT ...3
ABSTRACT ...4
AUSZUG...5
1 BEVEZETÉS ...6
2 SZAKIRODALMI ÖSSZEFOGLALÓ ...9
2.1 ALKOXIKARBONIL-METIL-TRIKARBONIL-TRIFENILFOSZFÁN-KOBALT KOMPLEXEK JELLEGZETESSÉGEI...9
2.1.1 Királis információk...9
2.1.2 „Autoszolvatáció”, spektroszkópia ...13
2.2 NANOTECHNOLÓGIA...16
2.2.1 Molekuláris propellerek, kúpfogaskerekek...16
2.3 MOLEKULAMECHANIKA...19
2.3.1 Kötő kölcsönhatások...20
2.3.2 'emkötő kölcsönhatások ...23
2.3.3 Az MM2 erőtér...24
2.3.3.1 Az MM2 erőtérben használt vegyérték jellegű kölcsönhatások... 25
2.3.3.2 Az MM2 erőtérben használt nemkötő jellegű kölcsönhatások ... 25
2.3.4 Molekulamechanikai erőterek paraméterezése ...26
2.3.4.1 Kvantumkémiai alapokon történő erőtérfejlesztés ... 26
2.3.4.2 Empirikus erőterek fejlesztése ... 27
3 KÍSÉRLETI MÓDSZEREK ...29
3.1 AZ MM2 ERŐTÉR BŐVÍTÉSE ÉS PARAMÉTEREZÉSE [KE45,KE46,KE47] ...29
3.1.1 Erőtér- és szoftverválasztás ...29
3.1.2 Paraméterkészlet bővítés ...30
3.1.2.1 Kísérleti szerkezeti információk gyűjtése ... 31
3.1.2.2 Referencia-adatbázis összeállítása... 32
3.1.2.3 A kezdeti paraméterkészlet meghatározása... 33
3.1.2.4 Kiindulási paraméterkészlet meghatározása... 36
3.2 PARAMÉTEROPTIMALIZÁCIÓ...38
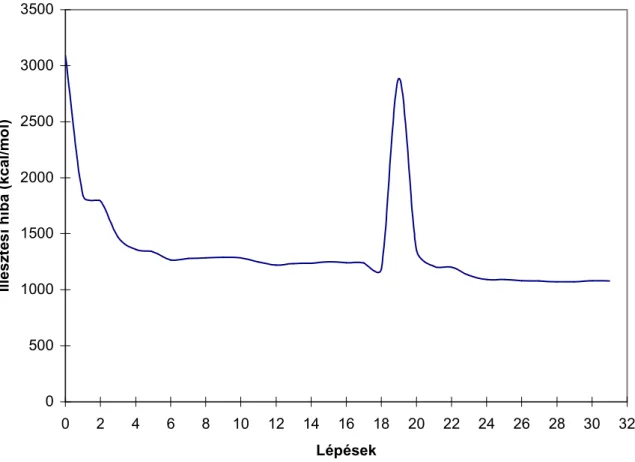
3.2.1 Az erőtér tesztelése, ellenőrzése mért vs. számított hiba ...40
3.3 KONFORMÁCIÓANALÍZIS...44
3.3.1 Egydimenziós/egyváltozós konformációanalízis...46
3.3.2 Kétdimenziós konformációanalízisek ...49
3.3.2.1 Etilészter származék (EtOC(O)CH2Co(CO)3(PPh3)) kétdimenziós konformáció-
analízise [K60, K61]... 50
3.3.2.2 A fenilgyűrűk inverziója ... 54
3.4 AZ [(ETILOXIKARBONIL)METIL]KOBALT TETRAKARBONIL KOMPLEX REAKCIÓJA TRIFENILFOSZFÁNNAL [KE63]...56
3.4.1 A komplexek szintézise...56
3.4.2 Reakcióút hipotézis ...57
3.4.3 A reakció számítása számításos kémiai módszerekkel ...58
3.4.3.1 A trifenilfoszfán ligandum támadása... 59
3.4.3.2 C komplex → D komplex átalakulás ... 64
3.4.3.3 Dekarbonileződés, D komplex → E komplex átalakulás... 66
3.4.3.4 Cisz-alkil (F komplex) → transz-alkil (A’ komplex) átalakulás... 68
3.4.4 A reakció lejátszódása ...68
3.5 A BENZILÉSZTER SZÁRMAZÉK (PHOC(O)CH2CO(CO)3(PPH3),IX) KÉTDIMENZIÓS KONFORMÁCIÓ ANALÍZISE [KE73,KE74] ...71
3.6 A FOGASKERÉK MODELL...74
4 KITEKITÉS ...76
5 ÖSSZEFOGLALÁS ...77
6 ÚJ TUDOMÁYOS EREDMÉYEK ÖSSZEFOGLALÁSA ...79
7 MAJOR RESULTS ...82
8 KÖSZÖETYILVÁÍTÁS...84
9 HIVATKOZÁSOK ...85
KIVOAT
KIRÁLIS IFORMÁCIÓK TERJEDÉSI MECHAIZMUSA ALKIL-KOBALT- TRIKARBOIL-FOSZFÁ KOMPLEXEKBE
KURDI RÓBERT
A dolgozat az alkoxikarbonil-metil-trikarbonil-kobalt-tercierfoszfán komplexek sajátos önszerveződése révén keletkező, trigonális-bipiramidális szerkezetű molekulák speciális tulajdonságainak vizsgálatával foglalkozik. A vizsgált komplexekben, a koordinációs kölcsönhatások révén, több sztereogén részegység található, amelyek gátolt rotációk révén atropizomerként, konformerként, konfigurációként viselkednek. Ennek következtében a szintézis során abszolút diasztereoszelektivitás, sőt enantioszelektivitás is érvényesül.
A szerző a királis információk terjedését vizsgálja fenti komplexekben a számításos kémia eszközeivel. Egy szerkezeti adatbázis felhasználásával empirikus molekulamechanikai erőteret készített, amellyel a vizsgált molekulaszerkezetek nagy pontossággal reprodukálhatók. Adatokat gyűjtött az enantiomerek közti átalakulás során lejátszódó intermolekuláris mozgásokra, ezek csatolt jellegére, energetikai színezetére és az indukciós mechanizmusok működésére.
Ezen eredmények felhasználásával modellezte és igazolta az enantiomerpárok egymásba történő átalakulása során lejátszódó „clockwork” mechanizmust és bővítette ezt a tercierfoszfán ligandum inverziójának 2:1 típusú mechanizmusával.
Felismerte, hogy a vizsgált vegyületek egy jól elkülönített része a közönséges fogaskerék áttétel kapcsolt rotációjának mechanizmusát követve invertálódhat.
A sztereoszelektivitás eredetét sikeresen bizonyította a komplexek kialakulásainak vizsgálatával. Ezáltal segítve a preparatív munkát végző kutatókat az eddig még nem izolált termékek geometriai és energiai adataival.
A dolgozat eredményeinek hatására bővültek az interdiszciplináris kutatási kapcsolatok. Pozitív eredményeik közé tartozik még a szponzori támogatás megjelenése is.
ABSTRACT
SPREAD MECHAISM OF CHIRAL IFORMATIOS I ALKYLCOBALT TRICARBOYL PHOSPHIE COMPLEXES
RÓBERT KURDI
This study focuses on the investigation of special properties of alkylcobalt tricarbonyl phosphine molecules. In the complexes absolute diastereoselectivity, moreover enantioselectivity was obtained during the synthesis.
The author studies the spread of chiral informations in the complexes mentioned by means of calculating chemistry. Using a structural database, an empirical molecular mechanical force field was created by which the molecule structures examined can be reproduced precisely. Clockwork- and gear-transmission mechanisms, taken place during the transformation of enantiomers into each other, were modelled and confirmed. The origin of stereoselectivity was proved successfully by investigating the formation of complexes. This could contribute to the work of preparative researchers with geometric and energetic data of compounds not isolated so far.
AUSZUG
AUSBREITMECHAISMUS DER KIRALE IFORMATIOE I DE ALKYL- KOBALT-TRIKARBOYL-PHOSPHAE KOMPLEXE
RÓBERT KURDI
Die Dissertation beschäftigt sich mit dem Untersuchen der speziellen Eigenschaften der Alkyl-Kobalt-Phosphane Molekülen.
Absolute Diastereoselektivität und sogar Enantioselektivität geht in den geprüften Komplexen während der Synthese durch.
Der Autor prüft mit den Methoden der rechnerischen Chemie den Ausbreiten der kiralen Informationen in den oben genannten Komplexen.
Er schaffte mit der Anwendung einer strukturellen Datenbank ein empirisches molekularmechanisches Feld, mit dem die geprüften Molekularstrukturen mit grossen Präzision reproduziert werden können. Er modellte und bestätigte den während der Ineinander-Konvertierung der Enantiomerpaaren abrollenden sogenannten Uhrwerk-Zahnrad Mechanismus.
Er bewiese erfolgreich den Ursprung der Stereoselektivität mit dem Untersuchen der Herausbildung der Komplexen. Er hilft mit den geometrischen und energetischen Daten der bis jetzt noch nicht isolierten Produkten der präparativen Arbeit der Forscher.
1 Bevezetés
A kobalt-karbonil vegyületeket gyakran használják homogén katalizátorként a fémorganikus kémiában. Tanulmányozásuk igen hasznos, mind gyakorlati, mind elméleti kémiai szempontból. 1964-től a nyolcvanas évekig zászlóshajója volt ez a Markó László nevével fémjelzett, a Pannon Egyetem (egykori Veszprémi Egyetem) Kémia Intézet, Szerves Kémia Intézeti Tanszék keretei között működő kémiai iskolának. Számos kiváló tudós szerzett nemzetközi elismerést az itt folyó kutatásokból (Markó L., Ungváry F., Heil B, Sisak A., Pályi Gy. Váradi Gy., Horváth I., Kovács I., Kollár L. stb.).
Napjainkban már alig lehet összefüggést találni az említett tudósok mai kutatási tematikáival. Egy kivétel azért van; Professzor Pályi Gyula (University of Modena and Reggio Emilia, Olaszország) tanszékén még mindig, sőt, megint folyik az egykoron kobalt katalizált hidroformilezési mechanizmus intermedierének tekintett alkil- és acil-karbonil- komplexek újabb és újabb képviselőinek szintézise. Az eredeti cél már lassan elfelejtődik. A preparatív munka hajtóereje ma már e vegyültek tulajdonságaiból és azok interdiszciplináris aspektusaiból fakad. A szintetikus munka (Prof. C. Zucchi, University of Modena and Reggio Emilia, Olaszország) és a biológiai homokiralitás eredetével kapcsolatos elméleti kutatás (Prof. Pályi Gy.) a modenai csoportban, a röntgen szerkezetvizsgálat Essenben (Prof. R.
Boese, University of Essen, Németország), a molekulák motorikus tulajdonságainak vizsgálata, pedig a Pannon Egyetem (Prof Bencze L, Kurdi R.) keretei között folyik.
A kutatási vertikum középpontjában az alkoxikarbonil-metil-trikarbonil-kobalt- tercierfoszfán komplexek sajátos önszerveződése révén keletkező, trigonális-bipiramidális szerkezetek állnak. A koordinációs kölcsönhatások révén több sztereogén részegységet hoznak létre, amelyek gátolt rotációk révén atropizomerként, konformerként, konfigurációként viselkednek. Ennek következtében a szintézis során abszolút diasztereoszelektivitás, sőt enantioszelektívitás is érvényesül. Az enantiomerek interkonverziója is a szeteroszelektivitás megmaradásával jár, ami a folyamat során rendkívül effektív intramolekuláris információcserét feltételez.
Kutatásaimat a Veszprémi Egyetem (ma Pannon Egyetem) Szerves Kémia Tanszékén, a Kémia Doktori Iskola Elméleti Kémia alprogram keretében, 1998 őszén kezdtem TDK hallgatóként. Akkor már készen állt, hazai és nemzetközi fórumokon is megmérettetett, a fent említett témakörökben összegyűlt kísérleti adatok és kémiai számítások szintézisét átfogó kompakt logikai rendszer, egy hipotézis, amelyet a helyi zsargonban „klokkvörk” (clockwork
- óramű) mechanizmusként emlegettek. Érdekes módon e hipotézis bírálata szinte kizárólag a csoporton belül született, miután a kémiai számításokkal nem lehetett értelmezni az intramolekuláris átalakulásokat, így szimulációjuk is sikertelen maradt. A vita „hatalmi szóval” végződött: a kísérleti tapasztalat az első, vissza az empíriához. Lévén a legkisebb, és még egyik fronton sem elkötelezett, ez a feladat természetesen nekem jutott. Szerencsémre.
Dolgozatomban a királis információk molekulán belüli terjedésével foglalkozom. Az adatgyűjtés elsősorban a szerkezetekre vonatkozott, de sok más egyéb között többlet információforrássá vált egy- egy rendezettlen Röntgen-spektrum és a szintetikus receptúrák olyan fizikai paraméterei is, mint a reakcióidő vagy a reakcióhőmérséklet. Munkámat főként a számításos kémia eszközeivel végeztem. Az adatbázis felhasználásával elkészítettem egy, a molekulaszerkezetek nagy pontosságú reprodukálását szolgáló empirikus molekulamechanikai erőteret. A realitáshoz közeli energiaértékeket kvantumkémiai módszerekkel számítottam. Adatokat gyűjtöttem az enantiomerek közti átalakulás során lejátszódó intermolekuláris mozgásokra, ezek csatolt jellegére, energetikai színezetére, a szintézis során észlelt királis jelenségek eredetére, (tandem jellegű) indukciós mechanizmusok működésére. Kutatási eredményeim felhasználásával modelleztem és igazoltam az enantiomerpárok interkonverziójának óraműszerű mechanizmusát és bővítettem ezt a tercierfoszfán-ligandum inverziójának 2:1 típusú mechanizmusával. Felismertem, hogy az ismert vegyületek egy jól elhatárolható része a közönséges fogaskerék-áttétel (gear) kapcsolt rotációjának mechanizmusát követve invertálódhat. Osztályozásuk a kristályfázisú konfigurációik révén „clockwork” (reP, siM) vagy „gear” (siP, reM) csoportba könnyen megoldható, de az alkoxilcsoport szerkezete alapján is jól megbecsülhető.
A kétezredik év végén még bátortalanul, de egy konferencián már megjelent a kutatási eredményeink gyakorlati hasznosíthatóságának lehetősége. Az érdeklődés meglepően élénk volt, az eredmények is szaporák. Az örömkémia (belső és külső) kommunikációs kényszere ellentétébe fordult. Külföldi partnereinket is publikációs moratóriumra, de legalább is kontrollált kéziratok benyújtására kértük. Lassan meginduló publikációink java a fiókban várakozik, és csak e dolgozat révén juthat szűkrétű szakértői nyilvánossághoz.
Pozitív eredmények közé tartozik a kutatás pragmatizmusa, a molekulatervezés és szelekció intenzitása, az interdiszciplináris kutatási kapcsolatok bővülése, és a szponzori támogatás megjelenése. A kutatási spektrum is bővült, elsősorban a molekulák kiroptikai tulajdonságainak számításával, tervezésével és vizsgálatával. A változások mértékét és minőségét jelképesen ez a kis példa is jelezheti: az etoxikarbonil-metil-trikarbonil-kobalt-
nevet kapta. Munkámnak ezt a részét már nem tekintem a dolgozat részének, ezért erre csak egy rövid kitekintést nyújtok az olvasónak.
2 Szakirodalmi összefoglaló
2.1 Alkoxikarbonil-metil-trikarbonil-trifenilfoszfán-kobalt komplexek jellegzetességei
A 80-as évek elejétől kezdve Pályi és munkatársai számos érdekes tulajdonságú komplexet állítottak elő (RO(O)CCH2Co(CO)3PR'3 R, R' = alkil-, arilcsoport) [1]. Ezek a vegyületek közti termékként fontos szerepet játszanak számos homogén-katalitikus szintézis (oxószintézis, hidroformilezés, hidrogénezés) katalitikus ciklusában.
2.1.1 Királis információk
Nagyszámú ilyen komplexet állítottak elő akirális és királis R és R' ligandumokat használva, amelyek közül 14-nek a kristály és molekula szerkezetét is meghatározták [2].
Érdekes tulajdonságokra figyeltek fel a vegyületek analitikai elemzése (IR, CD, röntgen diffrakció) során [3]. Akirális komplexek reakcióiban királis szerkezetek, „konformációk”
alakultak ki (1. táblázat), amelyek a következő tulajdonságokkal rendelkeznek (a-j):
1. táblázat: Alkoxi-karbonil-metil-trikarbonil-tercierfoszfán-kobalt komplexek (ROC(O)CH2Co(CO)3PR'3) kristályaikban kialakuló királis konformációk [4]
R R' Tércsoport reP siP reM siM
I Me PPh3 P-1 1 2
II Et PPh3 P21/n 3 4
III n-Pr PPh3 P21/n 5 6
IV i-Pr PPh3 P-1 7 8
V s-Bu PPh3 P21/n 9 10
VI t-Bu PPh3 P21/c 11 12
VII c-Hex PPh3 P21/c 13 14
VIII c-Hex PPh3 P-1 15 16
IX CH2Phh PPh3g
P-1 17(?) 18(?) 17(?) 18(?)
X (S)-Lacta PPh3 P212121 19
XI D-Menthb PPh3 P21 20 21
XII L-Menthc PPh3 P21 22 23
XIII i-Pr PPh2(n-Menth) P212121 24 XIV Gluce PPh3g
P212121 25(?)
XV (Bis-Me)f PPh3 P21/n 26 27
a (S)-{α-[EtOC(O)]CH(CH3)}; b (1S,2R,5S)-mentil; c (1R,2S,5R)-mentil; d (1S,2S,5R)-mentil; e 1,2:5,6-di-O-izo- propilidén-α-D-glükofuranóz; f 1,2-bis(metoxikarbonil)etil-kobalt (Ungváry féle komplex); g A foszfán kiralitása
nem határozható meg egyértelműen a fenilgyűrűk rendezetlensége miatt
(a) A vizsgált komplexek jellegzetesen torzított trigonális bipirimidális szerkezetében az ekvatoriális síkban három CO ligandum, a két átellenes axiális pozícióban pedig egy trifenilfoszfán1 és egy alkoxi-karbonil-metilén ligandum helyezkedik el (1. ábra).
triphenylphosphane ligandcarbonylcobalt fragment
Trifenilfoszfin ligandum Trikarbonil rész
Észter rész
1. ábra: A komplex szerkezeti egységei az R=Et példáján (II) keresztül
(b) A kristályokban egymással fedésbe nem hozható, tükörképi viszonyban lévő molekulák vannak, melyek az axiális ligandumokon legalább egy-egy sztereogén egységgel rendelkeznek.
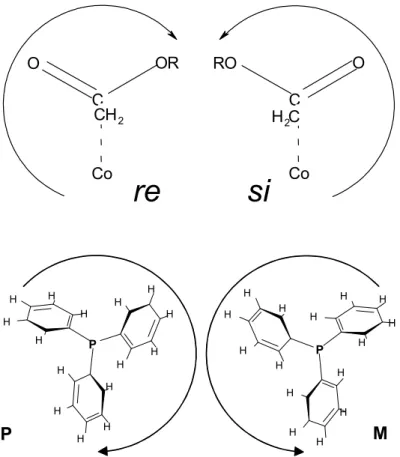
(c) A kiralitás forrása az észtercsoportnak a kobalthoz viszonyított térállása (re vagy si) és a trifenilfoszfán csavarodottsága (P vagy M), helicitása (2. ábra).
Az észtercsoport királis konformációinak meghatározásakor [4] használt elnevezések a CIP konvencióra épülnek. Ehhez azonban elengedhetetlenül szükség volt egy olyan kötés feltételezésére, mely a kobaltatom és az észtercsoport sp2-es szénatomja között alakul ki. Így az sp2-es szénatom királis atommá válik, ezáltal az egyes konfigurációk, (R)→re vagy (S)→si könnyen meghatározhatók.
1 A XIII komplex kivételével.
re
C
Co
OR O
C
Co RO O
si
CH2 H2C
H P H
H H
H H H
H H
H
H H
H H
H M
P
H P
H H
H H H
H
H H H
H H H H
H
2. ábra: A királis konformációk jelölésrendszere az alkil és a foszfán ligandumon
(d) Az észtercsoport síkja és a Co(CO)3 tárcsa síkja közel párhuzamos helyzetű. A Co-Csp2 kötés körüli elforgatással létrehozható egyéb orientációk nem kedvezményezettek.
(e) Az észtercsoport Csp2 atomjának a kobalthoz viszonyított távolsága <3 Å, ami a van der Waals erőket meghaladó mértékű kölcsönhatást "autoszolvatációt" feltételez [5]. Ezáltal mind a kobalt atom, mind pedig a karboxilcsoport Csp2 atomja királissá válik.
(f) Minden komplex esetében hiányzik a statisztikusan lehetséges izomereknek legalább az 50% -a, de a si/re és a P/M konformációk kombinációja molekulánként változó lehet.
(g) Királis R esetén még nagyobb a szelektivitás mértéke, több a lehetséges izomer. Az s- butil-észter (V) származék esetében az alkilcsoporton egy további aszimmetriacentrum is található, amely centrális kiralitását tekintve R és S lehet. Statisztikailag 23, tehát 8 szerkezet lehetséges, ezzel szemben a kristályszerkezetben csak kettő található meg (rePS, siMR) [2, 6]. A tejsav (X) és glükofuranóz (XIV) származékok esetében még nagyobb a szelektivitás. A lehetséges 8 szerkezetből csupán egyetlen módosulat (siMS az X-es illetve siPD a XIV-es
(h) Királis R’ esetén (R=i-Pr és R’=PPh2(n-Menth), XIII) szintén csak egy királis konformáció alakul ki (3. ábra).
3. ábra: R=i-Pr, R’=PPh2(n-Menth) (XIII) származék szerkezete
(i) A ciklohexil-észter (VII, VIII) származék esetében két különböző polimorf kristálymódosulat (P-1 és P21/c) alakul ki.
(j) A kristálycellák felépítése jelentős számú intra- és intermolekuláris hidrogénkötés kialakulására enged következtetni. A Pannon Egyetem Müller Laboratóriumában Tóth Gergely erre irányulóan előrehaladott munkákat végez [7].
A kobalt katalizált homogénkatalitikus reakciók az egyik legrégebben vizsgált reakciók közé tartoznak [8]. Számtalan új vegyületet szintetizáltak, azonban Pályi és munkatársai által előállítottakhoz hasonló, csak egyetlen egy található még az irodalomban.
Kobalt-katalizált hidroformilezési reakciók mechanizmusának vizsgálata közben Ungváry Ferenc egy újabb alkoxi-karbonil-metil-tercierfoszfán-kobalt származék szerkezetét írta le (4.
ábra) (R=(Bis-Met), XV) [9]. Ez a komplex is tökéletesen beleillik a vizsgált vegyületeink sorába, hiszen az összefoglalt (a)-(d) jellegzetességeknek megfelel.
4. ábra: Az R’=(Bis-Met) komplex (XV) szerkezete
A vizsgált komplexek legmeglepőbb tulajdonsága az, hogy a trifenilfoszfán ligandum egy bizonyos konformációjához (P vagy M) az észter csoportnak mindig csak egy adott konformációja (re vagy si) tartozhat. Ez alól kivétel a benzil-észter származék (IX), amelyben a kristályszerkezet „rendezetlensége” kiterjed a benziloxikarbonil-csoportra, ezáltal lehetővé téve mind a 4 lehetséges szerkezet megjelenését.
2.1.2 „Autoszolvatáció”, spektroszkópia
Kiroptikai vizsgálatok során felfigyeltek arra, hogy az RO(O)CCH2Co(CO)3PR'3 (R, R' = alkil, aril) vegyületek királis R és/vagy R' csoportokkal olyan cirkuláris dikroizmus (CD) színképeket adnak [10];
(a) amelyek gyökeresen különböznek a szabad ligandumok CD színképeitől;
(b) amelyekben olyan CD-sávok jelentkeznek, melyek a komplexekben jelenlévő átmenetifém valamely átmenetének részvételével magyarázhatók.
Ezek a megfigyelések azt jelentik, hogy a CD színképekben nem csak a klasszikus vegyérték kapcsolatok elektronátmeneteit találták, hanem egy ezeken túlmenő kölcsönhatást is, amely a fémet „királisan perturbálja”.
Ekkor a vegyületcsoportból csak a PhCH2OC(O)CH2Co(CO)3PPh3 (benzil származék) molekulaszerkezete állt rendelkezésükre. A komplexben az észtercsoportban található C(sp2)
és a kobalt távolsága 292 pm, amely némileg alátámasztja a vegyérték kapcsolatokon túli kölcsönhatás, (Pályi professzor terminológiája szerint) az „autoszolvatáció” jelenlétét is.
A feltételezett kötést a Müller Laboratórium munkatársainak sikerült kvantumkémiai vizsgálatokkal is bizonyítani [11]. A röntgenszerkezetek elemzése, spektroszkópiai eredmények, és a DFT számítások egybehangzóan azt bizonyítják, hogy az α-észter csoport és a kobalt atom között egy η3-típusú parciális koordinatív kötés alakul ki, mely felelős a vegyületekben létrejövő sztereoszelektív önszerveződés és királis konfigurációk kialakulásáért.
Bencze és munkatársai már a 2000-ben leírták [12] a röntgen szerkezetekben talált enantiomerek egymás közötti átalakulásának, általuk lehetséges útját. Munkájukban tüzetesebben az etoxi származékot (II) vizsgálták. Meghatározták, hogy az észter csoport sp3- as és az sp2-es szén körüli átfordulása energetikailag sokkal kedvezőbb akkor, ha az a karbonilos oxigén atomjával fordul a CO tárcsa felé. Az átfordulás további mozgásokat gerjeszthet a molekulában. Amikor az észtercsoport re állásból jobbra fordul, eltaszítja a Co(CO)3 fragmenst, ami ezáltal az óramutató járásával megegyező irányba fordul el. A CO tárcsa viszont olyan közel található a PPh3 ligandum orto hidrogénjeihez, hogy ezeket P állásból M állásba sodorja át (5. ábra). Tehát az eredeti reP szerkezetből kialakul enantiomerje, a siM szerkezet. A molekulán belüli mozgások összehangoltságából („koncertikus” jellegéből) következtettek arra, hogy miért hiányzik a másik két kombináció.
Felfigyeltek továbbá arra is, hogy ez a molekulán belüli összehangolt mozgás nagyon hasonlít a klasszikus óraszerkezetek működéséhez, sőt ezzel lényegében azonos [13]. Eszerint az óraszerkezet és a molekula, hasonló tulajdonságú részekkel rendelkezik. Az észtercsoport az anker+ankerkerék, a CO tárcsa a potenciális energia változó irányú továbbítására szolgáló anker, míg a trifenilfoszfán ligandum az ún. billegő kerékkel rokonítható (6. ábra).
Kvantumkémiai számolásokkal bizonyították, hogy az egyensúlyi szerkezeteknél mind a négy variáns (reP, siP, reM és siM) energiája a számítások hibahatárán belül azonosnak adódott [14]. Ezzel azt feltételezték, hogy a szelektivitás oka nem pusztán a termodinamikai.
Az irodalomban számos érdekes elképzelést találhatunk molekuláris óraműszerű vegyületek előállítására [15]. Azonban a fentiekben ismertetett óramű (clockwork) modell az eddigi „legteljesebb” szerkezet, melyben a klasszikus, mechanikus óraszerkezet valamennyi
lényeges alkatrésze megvan. A kutatás, a jelenség jellege folytán természetszerűen a hasonló molekuláris szerkezetek kutatására és fejlesztésére hivatott új tudományág, a nanotechnológia területére sodródott.
Co
P O
O
O O O
H H H H H H
H
H H
H H H H
H H
H H
H H H
H H P H
Co O
O O O
O H
H H
H H
H H
H H H
H
H H
H
H H H H
H
H H
re,P si,M
5. ábra: Az EtOC(O)CH2Co(CO)3(PPh3) átalakulásának sémája
6. ábra: Az óraszerkezet és a „molekuláris óramodell” összehasonlítása
2.2 anotechnológia
1959-ben Richard P. Feynman azt mondta, hogy „egy napon a tudomány segítségével képesek leszünk egy enciklopédia tartalmát egyetlen tűhegyre felírni”. Ez a módszer a gyakorlatban már évmilliárdok óta jól működik. A természet már ennyi ideje használja a nanotechnológiát. Ha nem is atomokból, de néhány atomos molekulákból épülnek fel az élethez szükséges anyagok. Ezek a folyamatok már ismertek voltak, de csak elképzelni tudták őket.
Ahhoz, hogy Feynman tervét megvalósítsák olyan eszközre volt szükség, amivel a molekulákat, atomokat láthatják. Gerd Karl Binnig az IBM gyárral közösen 1981-re elkészítette a pásztázó alagút(áram)mikroszkópot (Scanning Tunneling Microscope, STM).
Ezért a felfedezésért 1985-ben megkapta a fizikai Nobel díjat. Kiderült, hogy nem csak az egyes atomokat, hanem a köztük lévő kötéseket is látják, sőt az atomokat is tudják vele mozgatni. Ekkor született meg a modernkori nanotechnológia.
A nanotechnológia lehetőségei határtalanok. Alkalmazási területei lehetnek például az orvostudomány (mesterséges izmok), számítástechnika (memóriák), elektronika (chipek), űrtechnológia (könnyű és stabil anyagok), nanomotorok (molekulákból felépülő kerekek, fogaskerekek, pumpák, jeltároló, jelátalakító eszközök), stb.
Nem célom a nanotechnológia összes ágának bemutatása, csak a dolgozatom témájához legközelebb álló ún. nanomotorokkal foglalkozom itt. A nanomotorok felhasználási köre sokrétű lehet az optikai kapcsolóktól egészen a mikrofolyadékokig.
2.2.1 Molekuláris propellerek, kúpfogaskerekek
Mislow és Gust [16, 17] olyan molekuláris propellereket állítottak elő, amelyekben kettő vagy több aril csoport (Ar) kapcsolódik egy egyszerű centrumhoz (Z). Az egyik arilcsoport C-Z kötés menti forgatására a molekulában a többi csoport (ArnZ) is mozog (7.
ábra).
z z
7. ábra:Ar3Z típusú molekuláris propellerek
Mislow [18] és Iwamura [19] érdekes tulajdonságokat figyeltek meg Tp2Z (8. ábra) rendszerekben, ahol a két tripticén (Tp) oxigén vagy metilén hídon keresztül kapcsolódik egymáshoz (Z=O, CH2). Meglepődve tapasztalták, hogy egy gyors belső forgás következtében a molekula úgy viselkedik, mint egy ideális kúpfogaskerék. 1H- és 13C-NMR vizsgálatokkal kimutatták, hogy a C-O kötés körüli forgás a bis(1-triptycycl) éterben -94 °C- on is kimutatható, és a rotációs gát nagysága nem nagyobb, mint 8 kcal/mól. Oxigén híd esetében (Tp2O), ha a tripticén 4-es pozícióiba egy klór atomot, vagy a Tp2CH2-nél a 3-as és a 4-es pozíciókba egy-egy metil csoportot tettek, akkor megakadályozták a „fogaskerekek”
elcsúszását egymáson, tehát egy igazi háromfogú fogaskerék-rendszert kaptak.
8. ábra: A molekuláris és a mechanikus kúpfogaskerék sémája
Mislow és munkatársai későbbi kutatatásaikban [20] három tripticil csoportot kapcsoltak hozzá egy GeCl molekulához. Olyan három fogaskerékből álló rendszert kaptak, ahol két Tp csoport egyirányba a harmadik, pedig ellentétes irányba forog (9. ábra).
9. ábra: Három tripticénből álló fogaskerék rendszer
Fuji [21] és munkatársai olyan kísérleteket hajtottak végre, amelyben a Tp helyett szubsztituált naftil csoportokat használtak. Ezáltal elsőként állítottak elő optikailag aktív kétfogú fogaskerekeket (10. ábra).
10. ábra: Kétfogú fogaskerék-rendszer
Az analóg molekula rendszerekben, a nagy amplitúdójú mozgásokat külső stimulációval (fotokémiai, kémiai, elektrokémiai stb.) érik el. Ezáltal a csoportok egyes részei valóban átrendeződnek és ezt a reverzibilis átrendeződést számos módon tudják kimutatni (abszorpció, NMR, elektrokémiai potenciál stb.) [22].
Richards és munkatársai [23] [(9-tripticil-etinil)ciklopentadienil](tetrafenil- ciklobutadién)-kobalt komplexet (11. ábra) állítottak elő. A molekulában a ciklobutadiénhez kapcsolódó fenilgyűrűk egy négyfogú, az etin csoporthoz kapcsolódó tripticén pedig egy háromfogú fogaskereket hozott létre. Azonban a két forgó csoport kapcsolt rotációját nem tudták megoldani.
11. ábra: Metallocén típusú molekuláris fogaskerék
Az eddig ismertetett vegyületek sok tekintettben hasonlítanak az általunk vizsgált alkil- kobalt-trikarbonil-foszfán komplexre.
2.3 Molekulamechanika
Az informatika fejlődésével a számításos kémia lehetőségei kitárultak. Sokféle módszert alkalmazhatunk a molekulaszintű motorok, masinák tulajdonságainak feltérképezéséhez. Az általunk vizsgált molekulacsalád legkisebb tagja is több mint 50 atomból áll. Ilyen nagy molekulákra, a konformációs viszonyok számolásához ezért célszerű volt a molekulamechanika eszközeit választani.
A molekulamechanika témakörében számos érdekes és értékes könyv [24], valamint cikk jelent már meg. Nem célom ezen írások részletes összefoglalása - egy pontos, részletes munka meg is haladná a dolgozat kereteit –, inkább kiemelném a munkám számára fontos részeket, részleteket és lehetőségeket.
A molekulamechanika nem kvantumkémiai szerkezetvizsgáló módszer. A molekulamechanikai módszerek a magok helyzetét úgy vizsgálják, hogy az elektronokat csak az általuk létesített erőtér révén veszik figyelembe. Egy rendszer leírásának két fontos pillére van. Az egyik az erőteret leíró egyenletek matematikai alakja, a másik pedig az ezekben szereplő paraméterek megválasztása. Mindkét esetben a választás kritériuma az, hogy minél pontosabban tudjuk leírni a vizsgált modellek kísérletileg meghatározott jellemzőit.
Az erőtér módszer alapötlete abból adódott, hogy a kísérleti módszerek fejlesztésével a kisméretű molekulákról egyre több adat állt rendelkezésre. Ismertek lettek az atomok közti kötéstávolságok, kötésszögek, valamint még számos molekulageometriai és energetikai
jellemző. A kérdés az volt, hogy leírható-e egy nagy molekula a kisebbekkel modellezett szerkezeti egységek kombinációjaként. Erre szolgál az erőtér eljáráson alapuló molekulamechanikai számítási módszer.
Az erőtér eljárás alapjait Andrews 1930-ban közölt mechanikai modellje [25] képezi, amely szerint a molekulák merev, gömbszerű atomokból állnak. Az atomok közti kötéseket úgy szemléltetik, mintha rúgókkal kapcsolnánk őket össze.
A molekulát felépítő atomok állandóan mozognak. A fellépő szerkezeti változások pedig, a molekula teljes energiájának a megváltozását vonják maguk után. Ezek az egyszerű erőtörvények alapján számíthatóak.
A molekulamechanikában használatos erőterekben alkalmazott energiatagok kötő és nemkötő kölcsönhatások leírására szolgálnak,
kötő nem vegyérték
összes
E E
E = +
− .Vegyérték jellegű- vagy kötőkölcsönhatások közé többek közt az egymással közvetlen kötésben lévő atomok között fellépő kötésnyújtási, a geminális (1-3 vagy kötéshajlítási) és a vicinális (1-4 vagy torziós) kölcsönhatások tartoznak,
+ ...
+ +
=
nyújtási hajlítási torziósvegyérték
E E E
E
.Az egymáshoz közel lévő atomok között tapasztalható taszítás, az egymástól távol lévő atomok között kialakuló diszperziós vonzás, valamit az elektrosztatikus effektusok a nemkötő kölcsönhatások közé tartoznak,
kötés H Coulomb vdW
kötő
nem
E E E
E
−= + +
− .2.3.1 Kötő kölcsönhatások
Andrews molekulamechanikai modellje alapján a kovalens kötés irányában és az arra merőlegesen ébredő erőket a Hooke-törvény segítségével számolhatjuk. Minden egyes kötéstávolságnak és kötésszögnek definiálhatunk egy egyensúlyi értéket (kötéstávolság: két egymással kötésben lévő atom távolságának leírására szolgál; kötésszög: a központi atomtól a szomszédos atomok felé húzott szakaszok (=kötések) által bezárt szög leírására szolgál). Ha
egy kötéstávolság vagy egy kötésszög eltér annak egyensúlyi értékétől, akkor ez energianövekedést eredményez. Ezt, a kötéstávolságnál az
(
0)
22
1 k r r E
r=
r−
,kötésszögeknél pedig az
(
0)
22
1
θθ θ
θ
= k −
E
egyenlettel írhatjuk le. Ahol r0 és θ0az egyensúlyi, r és θ a számított kötéstávolság, ill.
kötésszög értékek. Nagy deformációk esetén alkalmazható lenne a Morse-függvény [26], de ezt a nagy időszükséglete miatt nem alkalmazzák. A számítás pontosítására mind a kötésnyújtási, mind a kötéshajlítási tagot kiegészíthetjük egy harmadrendű taggal. Ez azonban veszélyekkel járhat olyan molekulák esetében, ahol a geometriai jellemzők (kötéstávolság, kötésszög, stb.) jelentősen eltérnek az egyensúlyi értékektől.
Ha egy molekula részei a kötések kölcsönös taszítása miatt egymáshoz képest nem fordulhatnak el szabadon, akkor torziós gátról beszélünk. Kezdetben ennek a leírására a van der Waals paramétereket használták, de ez megnehezítette a van der Waals kölcsönhatások leírását. Ezért vezették be a torziós gátra jellemző energiatagot, a torziós energiát [27].
Első lépésben definiálni kellett egy függő változót. Ez a torziós szög. Az A,B,C,D atomok által meghatározott torziós szög az AB és CD egyenesek által bezárt szög a BC tengelyen keresztül nézve (12. ábra) [28].
12. ábra: A torziós szög definíciója
Mivel a torziós energiagátat leíró görbe a torziós szög koszinuszával arányos, ezért a molekulamechanikában az
( ω )
ω
ω
= k 1− 3 cos E
egyszerű képlettel írják le az energiát. Bonyolultabb rendszereknél, szerkezeteknél ez az egyenlet csak részben alkalmas. Részletesebben az alábbi Fourier-sorral felírva az
( )
( )
∑ −
=
j
j
j
E
E
ω1 cos ω
2 1
egyenletet kapjuk. Ha az első három tagra kifejtjük, akkor megkapjuk a molekulamechanikában használt egyenletet:
( ) ( ) ( )
∑ + + − + +
= ω ω ω
ω
1 cos 3
2 2 cos 2 1
cos 2 1
3 2
1
E E
E E
.Az egyes tagoknak szemléletes fizikai értelmet is lehet adni. Az első tag a dipólus-dipólus kölcsönhatásból származik. A második tag telített kötés körüli gátolt rotáció esetén hiperkonjugációra, telítetlen kötés esetén, pedig konjugációra utal. A harmadik tag a térbeli hatásoknak tulajdonítható.
A molekula egyes részei, amelyekben sp2-es hibridállapotú atomok találhatóak, planáris elrendeződést mutatnak. Ha a trigonális centrumban elhelyezkedő atom a sík alatt vagy felett helyezkedik el, akkor ez energianövekedést eredményez a síkbéli szerkezethez képest. Ez a kihajlás szögével (χ) arányos és a következő egyenlettel írható le [29]:
2
2 1
χχ
χ
k
E =
.A
χ
koordináta definíciója az irodalomban nem egyértelmű, többféle leírás ismert. Egy gyakori definíció szerint, egy a centrumból kiinduló kötés körüli ún. hamis torzió segítségével írja le (13. ábra).13. ábra: A síkból történő kihajlásra vonatkozó koordináták definíciója
2.3.2 emkötő kölcsönhatások
Molekulamechanika a molekulaszerkezet pontosabb energetikai jellemzésére a nyújtási-hajlítási taggal közelített 1,3 nemkötő kölcsönhatás mellett egyéb intramolekuláris kölcsönhatásokat is figyelembe vesz. Ezek közül a van der Waals kölcsönhatás [30] a legfontosabb.
A van der Waals kölcsönhatás három részből tevődik össze, melyek a következők. Az első a dipólusnyomatékkal és töltéssel nem rendelkező molekulákra jellemző diszperziós kölcsönhatás. A második a dipólusnyomatékkal rendelkező atomok dipól-dipól kölcsönhatásának energiája, és végül az effektív töltések között fellépő Coulomb-erők energiája. A van der Waals-jellegű másodlagos kölcsönhatások egyidejű számítására több összefüggés is alkalmas. Használják az ún. Hill-összefüggést [31], de a molekulamechanikai eljárásokban az általánosabb érvényű Lennard-Jones potenciált [32] alkalmazzák:
∑
∋
−
=
vdW
ij ij
ij ij
ij
vdW
R
B R
E A
12 6 .Ennek oka az is, hogy a Hill-összefüggésben található exponenciális kifejezés számítása jóval időigényesebb.
A töltések között kialakuló nemkötő kölcsönhatások számolására az erőterek többségében a Coulomb-potenciált használják:
∋
∑
=
Coulomb j
i ij
j i Coulomb
DR q E q
,
,
ahol qi és qj atomi töltések, Rij a köztük lévő távolság és D a dielektromos állandó.
Az intra- vagy intermolekuláris H-kötés leírására a következő egyenletet használják [33]:
∑
−∋
−
−
=
kötés H j
i ij
ij ij
ij kötés
H
R
D R
E C
,
10
12 .
Ezt a tagot nem minden molekulamechanikai erőtérbe építették be. Kvantumkémiai számításokkal bizonyították, hogy az ilyen másodlagos kötések leírhatóak egyszerű elektrosztatikus kölcsönhatásokkal is [34].
2.3.3 Az MM2 erőtér
Az előzőekben leírtak alapján elmondható, hogy egyes kölcsönhatások kiszámítására több módszer ismeretes. Szinte minden erőtér más és más formában használja ezeket az egyenleteket.
A szervetlen vegyületekre végzett molekulamechanikai számítások a szimulációs eszközök fejlődésével a 80-as évek közepétől kerültek be a kutatói gyakorlatba. Az átmenetifém atomokat tartalmazó vegyületek molekulamechanikai kezelése két jól definiált csoportra osztható, a koordinációs- és a (számunkra fontosabb) fémorganikus vegyületek szimulációjára.
1995-ben egy átfogó tanulmány [35] jelent meg fémorganikus komplexek molekulamechanikai kezeléséről. Több mint 250 hivatkozás segítségével tekintik át a 90-es évek elejétől megindult erőtérfejlesztéseket.
Az első igazán jól alkalmazható módszer a PCMODEL [36] programkörnyezetében megvalósított MMX erőtér, amelyben az átmenetifém komplexek modellezése vegyérték jellegű kölcsönhatásokkal történik. A paraméterkészletet a vas atom különböző vegyületeire fejlesztették ki. Más fémek komplexeinek kezeléséhez szükséges paramétereket a fématomok van der Waals sugarainak különbsége alapján határozza meg. Nagy előnye, hogy a hiányzó paramétereket meg tudja becsülni. A becslés pontossága azonban kapcsolatban van a hiányzó paraméterek számával.
Munkám során a Cerius2 program MM2 [37] erőterét alkalmaztam. (Döntésemet a kísérleti részben indokolom.) Az MM2 erőteret Allinger és munkatársai fejlesztették ki. Ez volt az első általánosan is alkalmazható erőtér. Dolgozatomban az egyes energiatagok számítási módszereit ismertettem röviden.
2.3.3.1 Az MM2 erőtérben használt vegyérték jellegű kölcsönhatások Kötésnyújtási kölcsönhatás
Ez az energia csak a két atom távolságától függ. Ennek számolására a program a „köbös”
potenciál függvényt használja,
)]
( 1 [ ) (
2 /
1 K R R0 2 d R R0
Enyújtási = b − + − , (E1)
ahol R0 a kötéstávolság (Å), Kb az erőállandó ((kcal/mól)/Å2) és a d pedig a „köbös”
korrekciós tag (1/Å).
Szöghajlítási kölcsönhatás
Az MM2 szög potenciált, vagy más néven Allinger potenciált használja a program ennek a kölcsönhatásnak a számítására.
] ) ( 1 [ ) ( 2 /
1 θ θ−θ0 2 + θ −θ0 4
= K d
Ehajlítási , (E2)
ahol θ0 a bezárt szög (º), d „hatos” term (1/rad4) és Kθ az erőállandó.
Torziós kölcsönhatás
A torziós energia számítása a következő egyenlettel történik,
[ ( ) ]
∑
=−
= p
n
n
torziós K d n
E
1
, 1 cos
2 /
1 θ θ , (E3)
ahol Kθ,n a rotációs gát (kcal/mól), n a periodicitás (n=1,2,3,4,5,6) és d a fázis faktor (±1).
2.3.3.2 Az MM2 erőtérben használt nemkötő jellegű kölcsönhatások Van der Waals kölcsönhatás
Az MM2 erőtérben a van der Waals kölcsönhatás számítására egy módosított Lennard-Jones potenciált használnak,
( ) ( )
− −
= −
−
=
−
−
−
6 0 1
0 6
exp 6 6
6 0
R D R
BR Ae
R
E R
R CR
vdw γ
γ γ
γ
, (E4)
ahol a γ rendszerint 12, hogy a Lennard-Jones 12-6 potenciál esetében a nagy atomtávolságoknál fellépő hibákat kiküszöböljék [38]. R0 a kötéserősség (kcal/mól), D0 pedig a kötéstávolság (Å).
Coulomb kölcsönhatás
A teljes elektrosztatikus energiát a következő egyenlettel tudjuk leírni,
∑∑
>=
i j i ij
j i coulomb
R Q C Q
E 0 ε , (E5)
ahol Qi és Qj az atomok töltése, Rij az atomok közti távolság (Å), ε a dielektromos állandó és C0 a konverziós faktor (C0=332,0637).
2.3.4 Molekulamechanikai erőterek paraméterezése
Az erőterek paraméterezésén a bennük előforduló paraméterek beállítását értjük. Bár a molekulamechanikában használatos általános erőterekben a szerzők a lehető legszélesebb paraméterkészlet fejlesztésére törekednek, az alkalmazások sokszínűsége folytán egyes paraméterek hiányára hamar fény derül. Az ilyen esetekben gondoskodni kell a hiányzó paraméterek pótlásáról.
A molekulamechanikai erőtér pontosságát, az erőteret alkotó potenciálegyenleteken túl, a kísérleti szerkezetek száma is biztosítja. A korábban végzett manuális elemzések alkalmazása már technikai problémákat vet fel, miszerint a sok paraméter és belső koordináta együttes kezelése már csak számítógépes célszoftverek segítségével oldható meg.
A molekulamechanikai paraméterek fejlesztésére az irodalomban két fő irányt tartanak megfelelőnek. Az egyik módszer lényege az, hogy a paraméterezéshez csak kvantumkémiai számításokat használnak fel. A másik módszer, ezt használtuk mi is, csupán kísérleti eredményeket használ a fejlesztéshez.
2.3.4.1 Kvantumkémiai alapokon történő erőtérfejlesztés
Ezen módszeren alapuló molekulamechanikai paraméterkészlet előállítására számos példa található az irodalomban [39]. A fejlesztések során volt, amikor még együtt alkalmazták a kísérleti és elméleti adatokat, de a sokak által ismert és használt Merk erőtér [40] már csak tisztán kvantumkémiai számításokon alapul. A szerzők a paraméteroptimalizációhoz közel 1500 HF és MP2/6-31G* szinten optimalizált molekulaszerkezetet használtak fel. A
kvantumkémiai számításokon alapuló erőtérfejlesztés előnye, hogy a számításokkal lényegében minden olyan adat előállítható, ami a paraméteroptimalizációhoz kell. Azonban ahhoz, hogy egy pontos, használható paraméterkészletet elkészítsünk, rengeteg kvantumkémiai számításra van szükség. Ez, a számítástechnika rendkívüli mértékű fejlődésével is, csak egyedi, kis molekulacsoportok részére kidolgozott erőterek esetében lehetséges.
2.3.4.2 Empirikus erőterek fejlesztése
A molekulamechanika, eredeti definícióját is alapul véve, szinte kizárólag kísérleti adatokat használ a paraméterezéshez. Ennek a módszernek a legfontosabb és a legidőigényesebb része a kísérleti adatok összegyűjtése és azok elemzése.
Két olyan nagy kutatócsoportot ismerünk, akik tisztán kísérleti alapokon keresztül készítik el a molekulamechanikai paraméterek optimalizációját.
Allinger és munkatársai a 1991-ben megjelent munkáikban elsősorban az MM2 és MM3 erőterek eltéréseit mutatják be, viszont magáról a paraméterezésről kevés szót szólnak.
Kidolgoztak viszont egy olyan becslési módszert, amellyel a hiányzó paramétereket kvantitatív úton tudják pótolni [41].
Rasmussen és munkatársai a molekulamechanikai paramétereket a legkisebb négyzetek módszerével optimalizálták a Lewenberg-Marquardt algoritmus alkalmazásával [42]. Ezzel a módszerrel egyszerre lehet optimalizálni a belső koordinátákat, kondenzált fázis cellaparamétereit és rácsenergiáját, valamint a dipólus momentumokat.
Ezzel egyidőben az Allinger-csoport egy új kvalitatív becslési módszert dolgozott ki, amellyel az új paramétereket lehet kiválasztani [43]. A közlemény nagy értéke, hogy tartalmazza az MM2 és MM3 erőterekben alkalmazható van der Waals paramétereket az egész periódusos rendszerre nézve. Így ezeket a paramétereket nem kell feltétlenül optimalizálni a későbbi erőtérfejlesztésekben. A tanulmányban részletesen elemzik, hogy a vegyérték jellegű kölcsönhatások paramétereit milyen kísérleti geometriákból lehet pontosabban megbecsülni. A legalkalmasabb a jó minőségű elektrondiffrakciós szerkezet, amennyiben ez nem áll rendelkezésre akkor használhatók a mikrohullámú és a röntgendiffrakciós szerkezetek is. Fémorganikus komplexeknél általában csak az utóbbiak ismertek. Mivel mi elsősorban kísérleti szerkezeteken alapuló molekulamechanikai erőteret kívánunk létrehozni, a röntgendiffrakciós szerkezeteket részesítjük előnyben.
A szakirodalmat átvizsgálva megállapíthatjuk, hogy a gyakorlatban is használható, jól reprodukálható optimalizálási módszer - egyet kivéve - nincs publikálva. A Veszprémi
Egyetem (Pannon Egyetem) Müller Laboratóriumában Szilágyi Róbert dolgozott ki [44] egy olyan paraméterezési eljárást, amelyet már sikerrel alkalmaztak más molekulacsoportok paramétereinek előállítására is. A paraméterezési algoritmus előnye az, hogy kémiai és számítógépes környezettől független és nem tartalmaz a felhasználótól függő algoritmus- paramétereket. A módszert a dolgozat kísérleti részében mutatom be a vizsgált vegyületcsalád példáján keresztül.
3 Kísérleti módszerek
A kutatások, a bevezetésben említettek szerint, egy nemzetközi konzorcium keretében folynak. Ezen belül, a munkánk célja és jellege, a számítási kémia eszközrendszerével végzett molekulaszerkezet-vizsgálat, a molekuláris tulajdonságok számítása, modellezése és a molekulatervezés.
Konkrétabban: a korábbi munkákban főleg DFT és PM3 számításokat végeztek el, Gaussian és Spartan programokkal. A kvantumkémiai számítások eszközrendszerét és alkalmazását változatlanuk megtartottuk. A molekulák mérete miatt azonban szükségünk volt egy gyorsabb számítási módszerre, amely eléri a műszeres vizsgálatok, pl. Röntgen- spektroszkópia pontosságát. Ennek megfelelően egy „host” (gazda) szoftverből és egy új erőtérből felépített molekulamechanikai számítást vezettünk be, amelynek gyorsasága három nagyságrenddel haladja meg a korábban alkalmazott számítási módszerekét és geometriai pontossága is megfelel az eredményes munka követelményeinek. (A konformációanalízis alatt előállítottunk majdnem 15 ezer szerkezetet. Molekulamechanikai módszerrel 1 geometriai optimalizáció ~1 perc alatt lesz kész, kvantumkémiai módszerrel, a rendelkezésünkre álló
„számítókapacitás” maximális kihasználásával is, ez az idő megközelítette az 1 napot.)
Munkánk során többféle szoftvert és számítógépet használtunk. A molekulamechanikai paraméterezést egy INDY R4000-es munkaállomáson, Cerius2 szoftverrel végeztük el. Az eredmények kiértékeléséhez Microsoft EXCEL makrót használtuk. A reakcióút számításához a Spartan ’02 program Windows-ós változatát használtuk.
3.1 Az MM2 erőtér bővítése és paraméterezése [KE45, KE46, KE47]
3.1.1 Erőtér- és szoftverválasztás
Ahhoz, hogy molekulamechanikai módszerrel tudjuk vizsgálni az alkoxikarbonil- metil-trikarbonil-trifenilfoszfán-kobalt típusú komplexeket, olyan erőteret kellett választani, amelyik tartalmazza az összes szükséges paramétert. A rendelkezésünkre álló számítógépes modellező szoftverek közül (Spartan 5.0 [48], Cerius2 3.0 [49]) mindegyik tartalmaz MM erőtereket, de sajnos ezeknek az erőtereknek egyike sem tartalmazta az összes szükséges paramétert. Döntenünk kellett, hogy melyik szoftver, melyik erőterét bővítjük, paraméterezzük.
A szoftver választásánál két okból döntöttünk a Cerius2 mellett. Az első ok az volt, hogy a Veszprémi Egyetemen Bencze és munkatársai már korábban végeztek ezzel a programmal paraméterezési munkákat. A második, hogy a programcsomagban az erőtér nyitott az energiafüggvények és paramétereik szabad változtatására, mint ezt már a neve is mutatja: Cerius2 Open Force Field (C2-OFF).
A Cerius2 Anyagtudományi Szakértői rendszerben jelentős számú molekulamechanikai erőtér található, többek közt a Dreiding I és II [50], az Universal FF [51, 52], az AMBER [53] és az MM2, valamint számos kisebb paraméterkészlet a speciális munkákhoz ( például: bks [54] szilikát és alumino-foszfátok kvantumkémiai vizsgálata, msxx [55] halogénezett polimerek). Programkörnyezetében csak egyetlen erőtér képes az átmenetifém komplexek szerkezetét modellezni. Ez az Universal FF, amely a teljes periódusos rendszerre alkalmazható atomi paramétereket tartalmaz. A lefedett tartomány növelésével növekszik a számolási hiba is. Ebből is következik, hogy ezzel az erőtérrel nem lehet olyan pontosságot elérni, amilyet a kutatások megkívánnak. A Pannon Egyetem Müller Laboratóriumában korábban már bebizonyították [56], hogy az MMX második generációs erőtér [57] alkalmas átmenetifém komplexek szerkezetének leírására. Mivel a Cerius2 programban megtalálható MM2 erőtér, ami az MMX erőtér alapja, ezért munkám során az MM2 erőteret használtam.
3.1.2 Paraméterkészlet bővítés
A szakirodalom többféle paraméterezési eljárást említ. Az irodalmi összefoglalóban már többet bemutattam. Választásunk – a már említett okokból - a Szilágyi Róbert által kidolgozott eljárásra esett. Az eljárás algoritmusát az 1.sémán mutatom be, majd az egyes lépéseket fejtem ki részletesebben.
START
Kísérleti szerkezeti információk gyûjtése
Kezdeti paraméterkészlet meghatározása
Kiindulási paraméterkészlet meghatározása
Atomtípusok ellenörzése
Konvergencia Erõállandókszerinti skálázás Kiindulási szerkezetoptimalizáció
Eltolási mûvelet
Szerkezetoptimalizáció
Illesztési hibák elemzése
STOP Referencia-adatbázis összeállítása
nem megfelelõ megfelelõ
nem igen
1. séma: Paraméteroptimalizálás algoritmusa
3.1.2.1 Kísérleti szerkezeti információk gyűjtése
Molekulamechanikai számítások paramétereit szerkezeti adatokból származtatjuk. A megfelelő kísérleti adatbázis létrehozásához elegendő mennyiségű és minőségű szerkezeti információ kell.
Összegyűjtésükhöz számos forrás és módszer áll rendelkezésünkre. Többek közt a rezgési-spektroszkópia (IR), mágneses magrezonancia (NMR), elektron- (ED) és röntgendiffrakciós (XD) eljárások. A munkánk során a kristályos, szerves és fémorganikus anyagok szerkezeti adatbázisát, a Cambridge Crystallographic Database-t (CSD) használtuk.
A kristályos anyagok szerkezete – így a fémorganikus komplexeké is – röntgen diffrakciós technikával vizsgálható. A mérés a nem-hidrogén atomok pozícióját 0,1 Å pontossággal adja meg. Ez megegyezik a molekulamechanikai módszer általánosan elfogadott, maximálisan megengedett hibahatárával [58], ezért jól használható e molekulák paraméterezésekor.
3.1.2.2 Referencia-adatbázis összeállítása
Mivel más szerkezeteket nem találtunk, azon komplexek szerkezeteit gyűjtöttük ki, amelyeket Pályi és munkatársai, valamint Ungváry és munkatársai állítottak elő (1. táblázat).
Mivel a ciklohexil származék (VII, VIII) esetében megjelent a polimorfia, a kétféle tércsoport közül csak az egyiket (P21/c) használtuk fel a paraméterezés során. A teljes adatbázis végül 25 alkoxi-karbonil-metil-kobalt-trikarbonil-tercier foszfán molekulaszerkezetre duzzadt (2. táblázat).
2. táblázat: A paraméterezéshez felhasznált röntgenszerkezetek (ROC(O)CH2Co(CO)3PR'3)
R R' °°°°
Me PPh3 1, 2
Et PPh3 3, 4
n-Pr PPh3 5, 6
i-Pr PPh3 7, 8
s-Bu PPh3 9, 10
t-Bu PPh3 11, 12
c-Hex PPh3 13, 14
CH2Ph PPh3g 17, 18
(S)-Lact PPh3 19
D-Menth PPh3 20, 21
L-Menth PPh3 22, 23
i-Pr PPh2(n-Menth) 24
Gluc PPh3 25
(Bis-Me) PPh3 26, 27
3.1.2.3 A kezdeti paraméterkészlet meghatározása
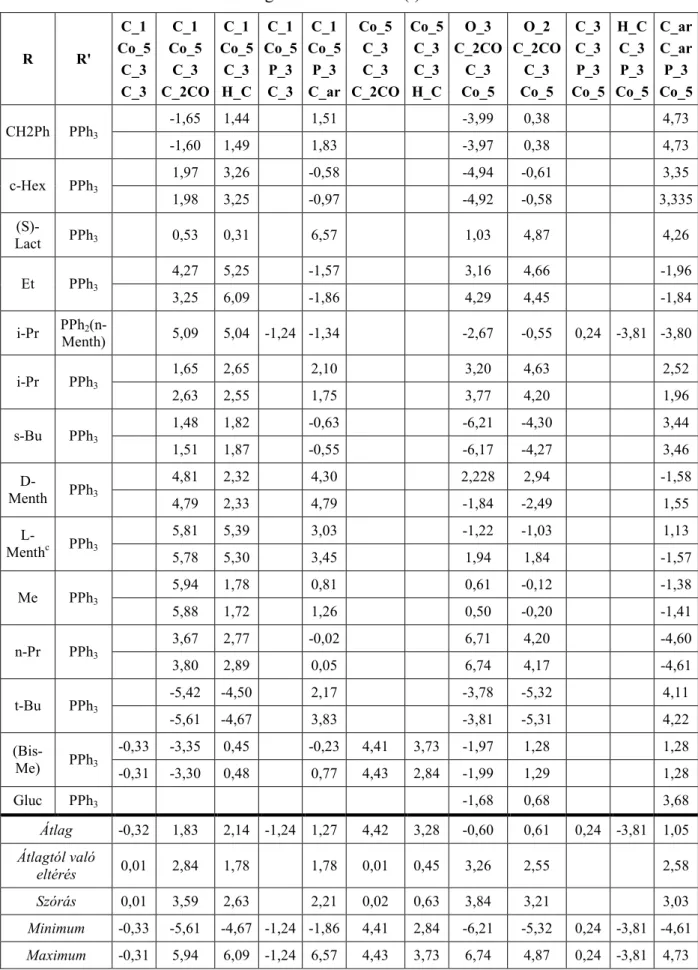
A gazdaerőtérnek választott C2-MM2 molekulamechanikai paraméterkészlet nagy pontossággal képes leírni a fémorganikus komplexeket. Tesztszámításokat végeztünk annak a kiderítésére, hogy melyek azok a belső koordináták, amelyeket a fejlesztendő MM2-es paraméterkészlet nem tud kezelni. Az adatok elemzését statisztikai módszerrel végeztem el. A hiányzó paraméterek 1 kivételével (C_1 – Okarbonil kötéstávolság) az átmenetifém környezetét leíró belső koordinátákhoz kapcsolódnak (3. táblázat).
3. táblázat:A C2-MM2 paraméterkészletből hiányzó belső koordináták
Kötéstávolságok Kötésszögek Torziósszögek
Co_5 – C_1 Co_5 – C_3 Co_5 – P_3 C_1 – Okarbonil
C_1 – Co_5 – C_1 C_1 – Co_5 – C_3 C_1 – Co_5 – P_3 C_ar – P_3 – Co_5 C_2CO – C_3 – Co_5
C_3 – Co_5 – P_3 Co_5 – C_1 – Okarbonil
Co_5 – C_3 – H_C C_3 – C_3 – Co_5 C_3 – P_3 – Co_5
C_2CO – C_3 – Co_5 – C_1 H_C – C_3 – Co_5 – C_1 C_1 – Co_5 – P_3 – C_ar O_3 – C_2CO – C_3 – Co_5 O_2 – C_2CO – C_3 – Co_5 C_ar – C_ar – P_3 – Co_5
Kötéstávolságok
A kapott értékek statisztikai elemzéséből kiderül, hogy az egyes kötéstávolságokhoz tartozó értékek megközelítően azonosak, a szórás nagyon kicsi (4. táblázat).
4. táblázat:A paraméterkészletből hiányzó kötéstávolságok statisztikai elemzése Kötéstávolságok (Å)
C_1 - Co C_1 - Okarbonil C_3 - Co_5 Co_5 - P_3
Átlag 1,784 1,139 2,086 2,217
Eltérés 0,006 0,005 0,008 0,006
Szórás 0,008 0,007 0,011 0,009
Minimum 1,771 1,130 2,065 2,205
Maximum 1,803 1,156 2,116 2,239
Darabszám 25 25 25 25
Kötésszögek
A molekulák szerkezeti tulajdonságaiból következik, hogy a C(sp3) - C(sp3) – Co és a C(sp3) – P – Co szög nem mindegyik molekulában szerepel (5. táblázat). Az előbbi a (XV, R=1,2-bis(metoxikarbonil)etil-kobalt; 4. ábra) komplexben, az utóbbi, pedig abban, amelyikben az egyik fenilgyűrű helyén mentil csoport található (XIII, R’=PPh2Menth; 3.
ábra).
5. táblázat:A kobalt környezetében található kötésszögek statisztikai elemzése Kötésszögek (º)
C_1 Co_5
C_1
C_1 Co_5
C_3
C_1 Co_5
P_3
C_ar P_3 Co_5
C_2CO C_3 Co_5
C_3 Co_5
P_3
Co_5 C_1 Okarb.
Co_5 C_3 H_C
C_3 C_3 Co_5
C_3 P_3 Co_5 Átlag 119,82 87,59 92,44 114,39 110,29 176,84 177,62 109,34 114,09 112,84 Eltérés 0,04 0,28 0,28 0,36 1,63 1,10 0,74 0,27 0,00
Szórás 0,05 0,36 0,36 0,46 2,02 1,32 0,98 0,39 0,00 Min. 119,69 86,83 91,75 112,95 106,04 174,82 175,15 108,58 114,09 Max. 119,91 88,27 93,20 114,99 113,46 179,16 178,59 110,10 114,09
Darab 25 25 25 25 25 25 25 25 2 1
Torziósszögek
A torziós szögek a legkisebb energia-befektetéssel változtatható belső koordináták, amelyeknél a kísérleti és a számított értékek közötti tökéletes egyezést szinte lehetetlen elérni.
A torziós kölcsönhatások lépésenkénti elemzése feleslegesen terheli az optimalizációt, ezért csak az illesztési hiba jelentős növekedésekor kell ezeket korrigálni.
A torziós paraméterek megadásánál fontos szempont a periodicitás meghatározása.
(Periodicitás: A 360°-os forgatás alatt felvett maximumok száma, ismétlődése) A röntgen szerkezeti adatokból kigyűjtöttük a számunkra fontos torziókra vonatkozó értékeket és a gyakoriságokból következtettünk a periodicitásra. Mivel az összes torzió részletes elemzésének bemutatása túlságosan megnövelné a dolgozat méreteit, ezért itt csak a Co-P-C(ar)-C(ar) torziós szögek elemzését mutatom be.
A meghatározást úgy végeztük, hogy a röntgenszerkezetekben megmértük az összes említett torziós szöget. Mivel a fenilgyűrűk aromás szén atomjai nincsenek megkülönböztetve egymástól, ezért gyűrűnként kettő, molekulánként pedig hat torziósszög értéket kaptunk.
Ezeket az értékeket ábrázoltuk gyakoriság szerint (1. diagram). A diagramon négy gyakoriság maximum látható, -130º, -40º, +50º és -140º környékén. Ez nem meglepő, hiszen ez a komplexek tulajdonságaiból egyértelműen következik. A négy maximum értékből tehát négyes periodicitásra következtettünk.
0 2 4 6 8 10 12 14 16 18 20
Gyakoriság
Rekesz
Hisztogram
Gyakoriság
1. diagram: A Co–P-C(ar)-C(ar) torziósszög értékeinek gyakorisága