THESIS OF THE DOCTORAL (PhD) DISSERTATION
SURFACE CHEMISTRY OF MOLYBDENA CONTAINING CATALYSTS
Written by:
HEIDER NASZER M.Sc. environmental engineer
Supervisor: Prof. Dr. ÁKOS RÉDEY
Doctoral School of Chemical Engineering and Material Sciences
INSTITUTIONAL DEPARTMENT OF ENVIRONMENTAL ENGINEERING AND CHEMICAL TECHNOLOGY
FACULTY OF ENGINEERING UNIVERSITY OF PANNONIA
Veszprém 2008
DOKTORI (PhD) ÉRTEKEZÉS
MOLIBDÉN TARTALMÚ KATALIZÁTOROK FELÜLETKÉMIAI TULAJDONSÁGAINAK VIZSGÁLATA
Készítette: NASZER HEIDER okleveles környezetmérnök
Témavezető:
Dr. RÉDEY ÁKOS egyetemi tanár
Készült a Pannon Egyetem
Vegyészmérnöki Tudományok és Anyagtudományok Doktori Iskola Keretében
PANNON EGYETEM MÉRNÖKI KAR
KÖRNYEZETMÉRNÖKI ÉS KÉMIAI TECHNOLÓGIA INTÉZETI TANSZÉK Veszprém
2008
MOLIBDÉN TARTALMÚ KATALIZÁTOROK FELÜLETKÉMIAI TULAJDONSÁGAINAK VIZSGÁLATA
Értekezés doktori (PhD) fokozat elnyerése érdekében Írta:
NASZER HEIDER
Készült a Pannon Egyetem Vegyészmérnöki Tudományok és Anyagtudományok Doktori Iskolájához tartozóan.
Témavezető: Dr. RÉDEY ÁKOS
Elfogadásra javaslom (igen / nem) ……….
(aláírás) A jelölt a doktori szigorlaton…... % -ot ért el.
Az értekezést bírálóként elfogadásra javaslom:
Bíráló neve: …...…... (igen /nem)
……….
(aláírás) Bíráló neve: …...…... (igen /nem)
……….
(aláírás) A jelölt az értekezés nyilvános vitáján…...% - ot ért el.
Veszprém, ……….
A Bíráló Bizottság elnöke A doktori (PhD) oklevél minősítése…...
………
Az EDT elnöke
CONTENTS
CHAPTER 1. INTRODUCTION AND LITERARY OVERVIEW 8
1.1. γ-Al2O3and molybdena-alumina catalysts 8
1.1.1. The surface hydroxyls of γ-Al2O3 and molybdena-alumina 13 1.2. Current status of catalysts preparation 17
1.2.1. The impregnation method 17
1.2.2. Adsorption methods 18
1.3. Molybdena with CeO2 and SnO2 semiconductors 22 1.4. Direct conversion of methane under nonoxidative conditions 27
1.5. Characterization of the catalysts 31
1.5.1. Physical characterization 32
1.5.2. Surface characterization and IR spectroscopy 34 1.5.3. FTIR spectroscopic detection of CO adsorption on the catalysts 37
1.6. OBJECTIVES 40
CHAPTER 2. EXPERIMENTAL 41
2.1. Materials and catalyst preparation 41
2.2. Catalyst characterization methods and techniques 42
CHAPTER 3. RESULTS 44
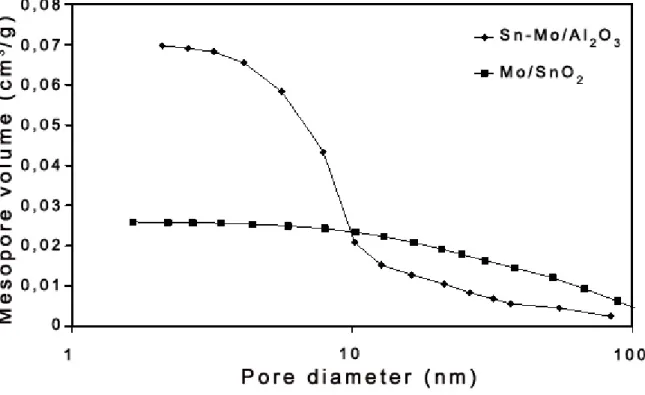
3.1. Surface texturing 44
3.2. X-ray diffraction 48
3.2.1. XRD patterns of Mo/Al2O3,Ce-Mo/Al2O3 and Mo/CeO2 48 3.2.2. XRD patterns of Sn-Mo/Al2O3 and Mo/SnO2 51
3.3. Thermal analysis 56
3.3.1. TG and DTA of Mo/Al2O3 56
3.3.2. TG and DTA of Ce-Mo/Al2O3 and Mo/CeO2 58 3.3.3. TG and DTA of Sn-Mo/Al2O3 and Mo/SnO2 60
3.4. Electron Spin Resonance (ESR) measurements 62
3.5. In situ DRIFT spectroscopy measurements 65 3.5.1. DRIFT spectra of the calcined samples under vacuum 65
3.5.1.1. DRIFT spectra of γ-Al2O3 and CeO2 65 3.5.1.2. DRIFT spectra of Mo/Al2O3,Mo/CeO2 and Ce-Mo/Al2O3 66 3.5.1.3. DRIFT spectra of SnO2, Mo/SnO2 and Sn-Mo/Al2O3 68
3.5.2. CO chemisorption 70
3.5.2.1. CO chemisorption on Mo/Al2O3 70
3.5.2.2. CO chemisorption on CeO2, Mo/CeO2 and Ce-Mo/Al2O3 72 3.5.2.3. CO chemisorption on SnO2, Mo/SnO2 and Sn-Mo/Al2O3 74 3.5.3. In situ DRIFT results on methane transformation in absence of oxygen 76
3.5.3.1. Methane transformation on Mo/Al2O3 77 3.5.3.2. Methane transformation on Ce-Mo/Al2O3 78 3.5.3.3. Methane transformation on Mo/CeO2 79 3.5.3.4. Methane transformation on Sn-Mo/Al2O3 80 3.5.3.5. Methane transformation on Mo/SnO2 81 3.5.3.6. DRIFT spectra after methane reaction under vacuum 82
CHAPTER 4. DISCUSSION 84
4.1. Surface texturing 84
4.2. X-ray diffraction 84
4.3. Thermal analysis 86
4.4. Electron Spin Resonance (ESR) 87
4.5. In situ DRIFT spectroscopy 88
4.5.1. DRIFT spectra of the calcined samples under vacuum 88
4.5.2. CO chemisorption 89
4.5.3. In situ DRIFT studies on methane transformation in absence of oxygen 93
SUMMARY AND CONCLUSIONS 104
ACKNOWLEDGMENTS 107
REFERENCES 108
THESES 113
PUBLICATIONS 115
KIVONAT
Az impregnálással és mechanikus keveréssel készült Mo/Al2O3, Mo/CeO2, Mo/SnO2, Ce-Mo/Al2O3 és Sn-Mo/Al2O3 katalizátorok struktúrájának, termikus stabilitásának, a hordozó és a Mo közötti kölcsönhatásoknak, aktivitásuknak, a Mo ionok diszperziójának és a felületi tulajdonságoknak vizsgálatára Induktív Csatolású Plazma (ICP), N2 adszorpció/deszorpció (BET módszer), Termikus analízis (TG-DTA), X-ray diffrakció (XRD), Elektron Spin Rezonancia (ESR) és Diffúz-reflexiós Fourier Transzformációs Infravörös (DRIFT) módszereket alkalmaztam. A katalizátorok aktivitásának összehasonlítása CO adszorpció és a CH4 konverziója alapján történt.
A BET, XRD és DRIFT eredmények összevetése alapján megállapítható, hogy a kalcinálás hőmérséklete és időtartama, az oldat pH-ja és a hordozó izoelektromos pontja hatással vannak a Mo ionok felületi diszperziójára. Másrészt, a cérium növelte a polimer Mo ionok felületi koncentrációját (főleg hordozóként) cérium-molibdénátok formájában, illetve Mo-O-Ce kötések megjelenésével (630, 875 cm-1), és újabb kettős O=Mo=O (995 és 1035 cm-1) kötések jelentek meg, amelyek polimer MoO3 alakulatokra jellemzők.
A termikus analízis alapján megállapítható, hogy a Mo/Al2O3 termikusan a legstabilabb (900°C-ig), ugyanakkor a Mo/CeO2, Mo/SnO2 és Ce-Mo/Al2O3 minták 700°C feletti hőmérsékleten történő hőkezelése során morfológiai és kristályszerkezeti változások következnek be, így lehetővé válik a fémionok diffúziója és kation csere játszódhat le köztük.
A szén monoxid 100°C-on történő kemiszorpciója során a karbonátok különböző formái és fém-karbonil kapcsolódások jelentek meg, amelyek vákuumban stabilak maradtak. Viszont ezek kialakulásához szükséges a katalizátorok redukált formája, továbbá a redukció során kialakult koordinatívan telítetlen helyek (CUS), a kristályrácsban lévő oxigén ionok és a felületi hidroxil csoportok jelentős szerepet játszanak. Másrészt, a Mo0 atomokat tartalmazó alakulatok 700°C-on történő redukció után tűnnek fel, amelyekhez terminális, illetve hídkötésű kapcsolódó szénmonoxid DRIFT sávjai jelentek meg (2025, 2002, 1994 cm-1).
A DRIFT vizsgálatok igazolták, hogy 800ºC-on redukált katalizátorokon a metán 700ºC-os bontása során a felületi karbonátok különböző formái jelentek meg, illetve ezek bomlásából származó CO és CO2 fejlődése megfigyelhető volt. Ez a rácsbeli oxigén reakcióképességének és a hidrogén redukció során kialakult Lewis savas helyeknek köszönhető. Azonban, a Mo/CeO2 és Mo/SnO2 katalizátorok jelentős aktivitást mutattak (főleg Mo/SnO2) a metán reakcióban, ami feltehetően a nagymértékben diszpergált MoO3 klaszterek, illetve Ce+3/Ce+4 és Sn2+/Sn4+ redox párok együtthatásával magyarázható.
ABSTRACT
Mo/Al2O3, Mo/CeO2, Mo/SnO2, Ce-Mo/Al2O3 and Sn-Mo/Al2O3 catalysts were prepared by impregnation and co-precipitation methods. The catalysts were characterized by Inductive Coupled Plasma (ICP), Thermal analysis (TG and DTA), X-ray diffractometry, Electron Spin Resonance (ESR), Diffuse reflectance Infrared (DRIFT) spectroscopy as well as N2
adsorption/desorption (BET method) techniques to examine their structural characteristics, thermal stability, mutual interactions between Mo and the support, catalytic activity as well as the dispersity of Mo ions and surface structure. The samples were compared to determine what kinds of adspecies participate efficiently during CO adsorption and CH4 decomposition.
Combining the results obtained from BET, XRD and DRIFT investigations one may suggest that beyond the Mo dispersion the formation of MoO3 clusters was found to respond to the calcination temperature and time as well as to the solution pH and the isoelectric point of the solid support. On the other hand, the introduction of ceria resulted in different molecular formulae with Mo (particularly as a support). This led to the increase of polymerized surface Mo species so as to forming Mo-O-Ce (bands at 630 and 875 cm-1) linkages, besides the formation of coupled O=Mo=O bonds at 995 and 1035 cm-1 indicative of polymeric MoO3. From thermal analysis, it can be inferred that Mo/Al2O3 is the thermally most stable material in the temperature range used in the experiment (up to 900°C). Whereas Mo/CeO2, Mo/SnO2
and Ce-Mo/Al2O3 samples undergo morphological and structural modifications above 700°C resulting in lattice defects, which motivate the mobility of metal ions and thus enhance the possibility of cation exchange between them.
Additionally, the formation of metal-carbonyl species and various types of carbonates through CO chemisorption at 100°C needs reduced catalysts containing coordinatively unsaturated sites (CUS), oxygen vacancies and hydroxyl groups. On the other hand, the bands protruding at 2025, 2002 and 1994 cm-1 are very likely associated with the terminally and bridged CO σ- bonded to metallic Mo(0) species appearing after reduction at 700°C. Thus, CO being provided as weakly adsorbed metal-carbonyls migrating towards the oxides through interfacial sites to form carbonates being stable even under vacuum at room temperature.
Methane is retained at 700ºC by the catalysts reduced at 800ºC and generates various carbonate species which decompose to CO and CO2 implying the existence of reactive lattice oxygen in addition to Lewis acid sites possessed by hydrogen reduction. However, the Mo/CeO2 and Mo/SnO2 materials presented marked activity (especially Mo/SnO2) in CH4
decomposition. This activity is presumably related to highly dispersed MoO3 clusters besides the Ce3+/Ce4+ and Sn2+/Sn4+ redox couples as further emphasized by means of DRIFT results.
1. INTRODUCTION AND LITERARY OVERVIEW
1.1. γ-Al2O3 and molybdena-alumina catalysts
Molybdena containing catalysts have been the subject of numerous studies due to their potential role in different important reactions such as hydrodesulphurization (HDS), hydrogenation (HYD), oxidative dehydrogenation (ODH), hydrodeoxygenation (HDO) hydrodenitrogenation, hydrometallization, isomerization, epoxidation, partial oxidation of alkanes and alkene metathesis reactions [1-5]. Presently, there is a continuous interest in Mo- containing catalysts directed toward understanding the catalytic properties, beside extensive studies have been addressed to characterize the structure of the catalyst surface. The chemical studies of molybdena-alumina catalysts include reduction or sulphidation. These two treatments are generally needed to activate the catalyst for most of the catalytic reactions [6].
The sulphidation occurs readily above 300°C, and the extent of sulphidation increases with temperature. However, the catalyst sulphur content is limited at a given temperature. The major sulphiding reaction is through the exchange of oxygen associated with molybdenum for sulphur. The molybdena phase of the sulphided catalyst was found stoichiometrically close to MoS2 [3, 6-10]. Perhaps, the most detailed analysis of the reduction of molybdena-alumina catalyst is the works of Hall, Massoth, Wang and Rédey [3-12]. In their work, the overall average oxidation state was determined from the gas consumed on oxidation or reduction and an attempt was made to separate this average into percentages of Mo6+, Mo5+, Mo4+ and Mo3+
on the basis of H+ retained and vacancies created upon reduction with H2. It was found that the reducibility of the catalyst after 2 hours reduction at 500°C under 1 atm H2 increased from approximately e/Mo = 0.5 to e/Mo = 2 (e/Mo: electrons gained per Mo atom) as the Mo loading increased from 2% to 10%, while the extent of reduction of 25% Mo loading catalyst reached e/Mo = 2.7 in just one hour reduction. It was also noted that the extent of reduction at short times for the 10% and 25% catalysts were actually greater than for bulk MoO3 implying the presence of a MoO3 phase on the catalysts but with a smaller particle size than the low surface area of bulk MoO3. In view of all reduction studies, it has been generally supposed that from the amounts of H2 consumed and water generated in the reduction step, an amount of hydrogen retained on the reduced catalyst could be calculated. Two types of adsorbed hydrogen were characterized: (1) reversibly adsorbed (HR) which is removable as H2 by evacuation above 450°C, and (2) irreversibly retained (HI) which could not be removed by evacuation. The latter could only be removed as H2O by either evacuation at higher temperature or by reoxidation. In addition to the retained hydrogen, water was produced
during the reduction. The amount of water increased with increasing the extent of reduction.
The formation of water was attributed to the removal of oxygen from the molybdena and consequently created anion vacancies on the surface. On reoxidation, two processes occurred:
the anion vacancies were filled and the HI was reacted to H2O. Given these surface processes, the following material balance equations were written:
[H2] = [WR] + HR +HI (Eq-1) [O] = [WR] + HI (Eq-2)
[Wo] = HI – WA (Eq-3)
Where WR and Wo are the quantities of water produced in the reduction and reoxidation steps, respectively, and WA is water retained by alumina after the reoxidation step (ideally WA =0) [3, 7, 12].
The redox study was extended by Hall and Lo Jacono who calculated HI per molybdenum (HI/Mo) and vacancies per molybdenum (□/Mo) at various extents of reduction (Hc/Mo) all measured as atom/atom [13]. The quantity of hydrogen (Hc) consumed during the reduction process can be expressed by the equation:
Hc = WR + HR + HI (Eq-4)
At low extents of reduction (Hc/Mo ≤ 1), two HI were present per vacancy created. However, as reduction was further increased, HI reached a limiting level of about HI/Mo = 0.45 while
□/Mo increased more rapidly. The formation of HI and vacancies was interpreted by assuming that molybdena forms an epitaxial monolayer on Al2O3. Accordingly, HI was taken as a measure of Mo5+ and each vacancy was equated to one Mo4+. The reduction process was pictured as follows:
Fig. 1. The reduction process of molybdena according to Hall and Lo Jacono
Later, FTIR study of the surface hydroxyl groups of the same catalyst was conducted by Millman [14]. These results led to a minor modification in the reduction process of Mo/Al2O3
(Fig. 2). The spectra from catalysts reduced with H2 were indistinguishable in the OH region from the alumina support or from the unreduced catalysts. Indeed, all five alumina OH bands were found and no new ones were detected. Thus, the intensity increased on reduction was
probably due to the recoveries of Al-OH rather than the formation of Mo-OH. Accordingly, the authors presented a modified picture for the reduction process of Mo/Al2O3:
Fig. 2. The reduction process of molybdena according to Millman
The implication is, however, that reduction reverses, the process of monolayer formation as evidenced by the increased intensities of the OH region.
Other studies have suggested that patches of molybdena may be present on the surface, particularly at high Mo loading. Giordano et al. studied molybdena-alumina catalysts containing up to 30% Mo by various chemical and physical techniques [15]. These included
differential thermal analysis (DTA), thermogravimetric analysis (TGA) infrared and diffuse reflectance spectroscopy. The authors deduced that below 4% Mo loading the Mo ions are highly dispersed in the form of MoO42-. At increasing Mo contents (up to 10-15%), a progressive increase of structures with bridged oxygen atoms in mixed tetrahedral and octahedral environment (schemes IV and V surrounding Mo6+) were found.
Scheme 1.
They obtained MoO3-rich catalysts at Mo loading higher than 15% prevailing octahedral configuration attributed to species like schemes V and VI. It was clearly demonstrated by these authors that the molybdena surface structure could be altered by the Mo concentration and by the calcination procedures. The Mo concentration was found to play an important role in the nature of the molybdena surface structure. They suggested that tetrahedral MoO42- is the main species in the catalysts with less than 5% Mo. Octahedrally coordinated polymolybdates were formed with more than 5% becoming the dominant species as the loading increased, while crystalline Al2(MoO4)3 phase was observed in the catalysts calcined at elevated temperatures (>600°C) and MoO3 clusters were formed when the loading reached 16% and 20%, respectively. The degree of the polymerization becomes higher and higher as the Mo loading is further increased. After the surface has been saturated (high Mo content), the formation of bulk MoO3 and Al2(MoO4)3 occurs at different extents depending on the preparation and calcination conditions.
Another surface polymolybdate structure was proposed by Medema et al. to explain their Raman results:
Scheme 2.
It was suggested that supported molybdena species are aligned as one-dimensional chains which result in Mo-O-Mo bonds, yielding octahedral coordination for Mo6+ [16, 17].
Despite the variations made by various authors, a consistent pattern has emerged. At low Mo loading, tetrahedral MoO42- species dominate the surface. Increasing loading is generally accompanied by the formation of a polymolybdate phase. On the basis of these studies, it is generally known that the surface of molybdena remains mainly containing monomeric Mo species. Moreover, polymeric molybdate species and free MoO3 can be formed at high Mo contents taking into account the calcination temperature and time as well as the solution pH, the isoelectric point and surface area of the solid support [12-17].
1.1.1. The surface hydroxyls of γ-Al2O3 and molybdena-alumina
γ-Al2O3 has a defect spinel structure, a high surface area, a certain degree of acidity, and forms solid solutions with transition metal oxides such as NiO and CoO. Above 900°C, γ- Al2O3 is transformed into α-Al2O3, which has hexagonal structure and smaller surface area.
Even at lower temperatures a slow phase transition occurs, which shortens the catalyst lifetime. Therefore, the incorporation of small amounts (1-2%) of SiO2 or ZrO2 in γ-Al2O3
shifts the γ → α transformation to higher temperature and increases the stability of the catalyst [1, 2]. The alumina surface hydroxyl groups have been extensively studied by infrared spectroscopy. Thermal dehydroxylation studies of γ-Al2O3 by IR spectroscopy were first executed by Peri who concluded that Al-OH groups close to each other could form water, and desorbed from the surface [18, 19]. The spectra displayed five different OH frequencies at 300°C for Al-OH groups. Assuming that the (100) crystal face is dominantly exposed on
γ-Al2O3 surface, Peri proposed a model predicting the existence of five types of Al-OH groups. The model attributes the different OH frequencies to differing numbers of surrounding surface oxide sites. In other terms, the difference of Al-OH frequency depended upon the number of the nearest neighbour oxygen anions surrounded a particular Al-OH group. Peri surmised that the high wavenumber band at 3800 cm-1 was an OH group with four neighbouring oxygen anions. Whereas the low wavenumber band at 3700 cm-1 was an OH group with no oxygen neighbours, but four aluminium ions. The other three OH groups giving rise to the intermediate stretching frequencies at 3730, 3745, 3780 cm-1 were supposed to have one, two and three nearest neighbouring oxygen anions, respectively. As the temperature increased, the lower frequency associated (H-bonded) hydroxyl groups, being closest together, were removed first, leaving isolated Al-OH groups to be removed at higher temperatures after heating to 700°C. Upon heating to 900°C no hydroxyl groups were detected by IR.
Knözinger and Ratnasamy, assuming that all of crystallite faces of Al2O3 (100, 110 and 111) have an equal chance of projection on γ-Al2O3 surface, proposed a model that assigned the different OH frequencies of the five types of Al-OH groups to tetrahedrally and octahedrally coordinated Al3+sites in terminal and bridged configuration [20, 21]. These assignments are shown in Fig. 3.
Fig. 3. Assignments of OH groups on γ-Al2O3 surface according to Knözinger and Ratnasamy
Despite the studies mentioned above, none has attempted to correlate directly the extent of dehydroxylation with the production of Lewis acid sites as a function of the treatment temperature. Nevertheless, in the spirit of the work of Zaki and Knözinger who undertook a low temperature IR study of the interaction between Al-OH groups and adsorbed CO, five bands of different types of hydroxyl groups were assigned on Al2O3 dehydroxylated at 500°C, two types of associated Al-OH were removed first, leaving three types of isolated Al-OH groups to be removed at higher temperature. They observed two low temperature CO adsorption sites at -193°C on Al2O3 and they attributed the first adsorption site (2140-2150 cm-1) to CO hydrogen-bonded to Al-OH groups and a physisorbed CO layer, while the second CO adsorption site (2195-2213 and 2238 cm-1) was assigned to tetrahedrally and octahedrally coordinated Al+3 sites [22, 23].
Later, John and Ballinger undertook a low temperature (-196°C) IR study of the interaction between Al-OH groups and adsorbed CO, in order to examine the acid-base properties of the hydroxyl groups on Al2O3 after dehydroxylation at elevated temperatures [24]. They found an approximately direct correlation between the elimination of surface hydroxyl groups and the increase in integrated absorbance of CO on Lewis acid sites of Al2O3 produced during the dehydroxylation. Furthermore, they observed two Al3+ adsorption sites: the first develops following mild dehydroxylation, and the second appears only after dehydroxylation at 600°C and higher due to the presence of a mixture of Lewis acid sites on highly dehydroxylated Al2O3 surface. From these studies, it has become apparent that the acidity of Al2O3 develops when it is dehydroxylated, not because of the OH groups themselves. Consequently, the removal of water and/or hydroxyl groups (surface ligands), coordinatively unsaturated (CUS), anions (oxygen ions) and cations (exposed Al3+, anion vacancies) are created.
Numerous IR studies reported that both tetrahedrally and octahedrally coordinated Mo6+ are present in impregnated Mo/Al2O3 catalysts, and the octahedral/tetrahedral ratio increased with increasing Mo loading. Infrared spectra of molybdena-alumina have also shown that alumina surface hydroxyls are eliminated by the addition of molybdena [25-29]. These results provided strong evidence of bonding between molybdena and the alumina surface. The formation of tetrahedrally coordinated Mo6+ on the surface of alumina was pictured as the replacement of the terminal hydroxyl groups as shown in the equation below [12, 30]:
(Eq-5)
Rédey and Millman independently measured the loss of OH groups on alumina in the formation of molybdena-alumina catalyst by determination of the total hydrogen contents by exchange with D2 using the isotope dilution method [3, 14]. The number of hydroxyls eliminated in the formation of 8% Mo/Al2O3 catalyst (∆OH/Mo) was determined to be about 1.7 0.6, which is in agreement with the epitaxial monolayer model (Eq-5). ±
Interestingly, the hydroxyls, which are replaced by molybdena when the catalyst is formed (calcined), reappear when the catalyst is partly reduced in H2.
The fragments arising from the dissociative adsorption of water on the surface of metal oxides give rise to hydroxyl groups that are potentially more or less active Brönsted acid sites. Such surface hydroxyl groups can be detected, directly, recording the IR spectra of the oxide catalyst powders in the region 3800-3000 cm-1, where the O-H stretching modes (νOH's) occur [12-24]. Although the position and shape of the ν(OH) bands of such surface hydroxyl groups is informative on their coordination, these data do not give straightforward information on their Brönsted acidity. In fact, as for example, the position of the OH band over a basic catalyst like MgO, of a weakly acidic catalyst as amorphous silica and of a strong Brönsted acidic catalyst like silica-alumina is almost the same (3745±3 cm-1). Moreover, very acidic catalysts like, for example, sulphated zirconia and titania do not present any definite sharp ν(OH) band, while others, like zeolite ZSM-5 and silica-alumina, show sharp ν(OH) bands.
These facts are due to the following main reasons:
1. The ν(OH) frequency depends not only on the O-H bond strength, but also on the nature of the M-O(H) bond, i.e. the element(s) to which the OH is bonded.
2. In any case, even for OH's bonded to the same element, the function ν(OH) versus acidity is not necessarily linear but can present a maximum.
3. The state of the OH groups on the surface also depends on the basic strength of the oxide ions. In fact, in very covalent structures, like for silica-alumina and zeolites, where oxygens are almost not basic, the acidic OH's are responsible for rather sharp and well-defined bands, while when the nearest oxygens are more or less basic, the acidic OH's give rise to H-bonding, with a shift down and a broadening of the ν(OH) band [25-31].
1.2. Current status of catalysts preparation
Different methods have been described in the literature for the preparation of these catalysts.
Among these methods, the impregnation (IW) and the equilibrium adsorption (EA) methods have been widely used [1, 2, 3].
1.2.1. The impregnation method
On most of the previous studies, the molybdena-alumina catalysts were prepared by the impregnation method, which is the most widely used preparation technique for supported heterogeneous catalysts. This procedure involves making a solution of the active component having a volume equal to the pore volume of the support material [3-15].
The solution is then impregnated into the support material and mixed evenly where it is completely taken up. The solid is first dried at about 150°C and then calcined at elevated temperatures, in general, between 400 to 600°C (Fig. 4). Consequently, the resulting preparations have varied somewhat depending upon the initial molybdenum compounds used, the initial pH value of the molybdenum solution, the pore structure of the support and the preparation skill. One obvious disadvantage of this preparation method has been that molybdena cannot be uniformly distributed over the support surface.
Knözinger et al. studied molybdena-alumina catalysts prepared by impregnation at pH = 6 and 11 and concluded from Raman results that the catalyst prepared at pH = 6 contained a large amount of bulk MoO3 while that prepared at pH = 11 did not [32]. Moreover, the same results showed that this technique does not lead to uniform coverage. Accordingly, it can be argued that this technique is not well defined. For instance, the pH value of the solution is not usually specified when using this technique in addition to the pre-treatment conditions including calcination temperature, time and ambient atmosphere. For example, it has been reported by Hercules and co-workers that increased calcination temperature and time favour the formation of Al2(MoO4)3, which is believed to be a sub-surface species, while bulk MoO3 was observed at low calcination temperature and short time [33-35].
In summary, the final catalyst made by the impregnation method may be affected by the solution pH value, the mesh size of the support used, and other variables such as calcination temperature and time. Therefore, it is difficult to reproduce the catalyst by this method from one batch to another.
Fig. 4. Production of supported molybdena catalysts by impregnation method
1.2.2. Adsorption methods
These methods for preparation of molybdena-alumina catalysts were first used by Sonnemans and Mars in both liquid and gas phases [36]. In the preparation from liquid phase a fresh solution of 1% ammonium paramolybdate (pH 1-9) was flowed through an alumina bed for a period of 4 hours (until the concentration of the effluent solution was found the same as that of the entering reagent). The final Mo content was found to depend on the solution pH value.
It reached 21% in MoO3 when prepared at pH = 1, but decreased as the pH was increased. At pH = 9, the catalyst contained only 2% Mo. They also reported that the concentration of the solution had an effect on the amount adsorbed (the concentration range studied was in the range between 0.2 and 1%). They concluded that Mo/Al2O3 with monolayer coverage could be achieved by either gas phase or liquid phase adsorption method. The authors suggested that the pH effect on the molybdenum content resulted from a change in the mean size of the polymolybdate ions, which is a function of the pH.
In contrast, Iannibello and co-authors who showed the same pH dependency interpreted the pH effect as due to a higher fraction of protonated alumina hydroxyls at lower pH leading to a higher adsorption of anions [37]. They observed that the pH decreased rapidly at first and then increased slowly to a steady value. The authors attributed the fast initial pH decrease to
exchange of ammonium ions with protons of alumina and the slow subsequent increase to exchange of molybdate ions with surface hydroxyl groups as represented by Eq-6 as [MoO4]2- was adsorbed, the equilibrium (Eq-7) would shift toward the right:
Al-OH + [MoO4]2-
⇔
Al-O-MoO3- + OH- (Eq-6) [Mo7O24]6- + 4H2O⇔
7[MoO4]2- + 8H+ (Eq-7) Actually, at pH values above 7 or 8, Mo4+ occurred as the tetrahedral monomeric molybdate anions, but polymerization occurred at concentration in excess of 10-4 M at somewhat lower pH values. The two major equilibriums [3, 12, 37], which may occur in the solution, may be written as follows (Eqs. 7 and 8):8MoO42- + 12H+
⇔
Mo8O264- + 6H2O (Eq-8) The major molybdate species at concentration above 10-3 M that are present at various pH ranges can be roughly illustrated by the scheme below [11, 12]:Scheme 3.
However, the well-understood principle of colloid chemistry can provide an explanation for the variations in Mo loading with pH and the reversibility of the adsorption process. The surface of a solid oxide in an aqueous solution is generally electrically charged. This charge may be attributed to: (a) dissociation of surface hydroxyl groups or (b) adsorption of protons formed by hydrolysis of H2O. These two mechanisms can qualitatively explain the pH dependence of surface charge and the existence of a pH resulting in zero net charge, called the isoelectric point of the solid (IEPS) or zero point of charge (ZPC) [38-40].
Based on the concept of IEPS, the surface chemistry of an oxide with an aqueous solution can conveniently be expressed as written by Parfitt [41]:
M-OH2+ ⇔ M-OH ⇔ M-O– + H+ (Eq-9)
Decreasing pH ⇐ IEPS ⇒ Increasing pH
The equilibrium indicates that at IEPS of the oxide, the net surface charge is zero, although this does not necessarily mean that all the surface hydroxyls are in the form of M-OH because they vary in acidity depending on their coordination. The net zero surface charge simply means the concentrations of M-OH2+ and M-O– are equal at IEPS. When the pH is adjusted lower than the IEPS, the concentration of M-OH2+ will be higher than M-O–, consequently the surface will carry a net positive charge:
+
+ ⇔−
+
−MOHsurf H (aq) MOH2surf (Eq-10)
The situation is reversed when the solution pH is adjusted higher than the IEPS, and the oxide surface will carry a net negative charge:
) (
2
aq H MO
MOH or
O H MO
OH MOH
surf surf
surf surf
+
−
−
−
+
−
⇔
−
+
−
⇔ +
−
(Eq-11)
The same factors play an important role in the adsorption of metal oxyanions on the oxide support. Consequently, the fact that the charge on the surface can be adjusted by varying the solution pH. Thus, the amount adsorbed can be varied by changing the zeta potential of the surface. Accordingly, the IEPS has a great influence on the band position of the adsorbed species. The IEPS of several oxide supports is shown in Table 1.
Table 1. Comparison between IEPS of several oxide supports Oxide supports Isoelectric point (IEPS)
SiO2 1-2
γ-Al2O3 6-8
TiO2 4.7-5 (Rutile), 5.7-6.2 (Anatase)
MgO 12.1-12.7
ZrO2 6.6-7.1
CeO2 (cerianite) 6.7-6.8
SnO2 (cassiterite) 4.2
J. Sarrin and co-workers compared the reducibility and activity of two series of molybdena- alumina catalysts prepared by impregnation (IW) and adsorption methods (EA) [42]. Their results showed that for both series of catalysts, the reducibility of the molybdenum species increases as the Mo loading increases, in agreement with the literature [12-15]. For the catalysts with similar loading, the (IW) series showed a higher surface coverage of the Mo phase and higher degree of reduction than for the corresponding (EA) preparations. The reducibility data were consistent with the catalytic results and oxygen chemisorption results.
The (IW) preparations (with similar Mo loading) were more active in the isomerization of 1-butene and chemisorbed larger amount of oxygen than their (EA) counterparts. The differences in reducibility can be ascribed to a nonuniform repartition of the molybdenum species between the external and internal surfaces of alumina for (IW) preparations, which may contain a greater fraction of easily reducible polymeric Mo species than their more uniform (EA) counterparts. Another possible explanation may stem from the decoration effect of the Mo species by Al3+ ions. The latter may arise from the dissolution of alumina, which is favoured on the (EA) series due to the long contact time between the Mo solution and alumina.
Other studies indicate that the preparation method does not influence the molecular structure of the Mo species present on Al2O3. Thus, for a given Mo loading, the nature of the Mo species is independent of the preparation method [43-47]. However, it is not clear, how a given preparation method may induce the Mo speciation.
1.3. Molybdena with CeO2 and SnO2 semiconductors
Promoters are the subject of great interest in catalyst research due to their remarkable influence on the activity, selectivity and stability of industrial catalysts. It is sometimes difficult to define precisely the function of the promoters that has not been elucidated [1, 2].
Ni and Co promoters in Mo/Al2O3 catalysts are well-known for their success in the hydrodesulphurization (HDS) of petroleum feedstock and coal liquefaction products [48]. The promoting role of both promoters was found to increase the Mo dispersion and reduction, in addition to the increase in H2 mobility, an intercalation effect with MoS2, a decrease in deactivation and an increase in surface segregation of mixed sulphide phases. For instance, when Co was introduced into Mo/Al2O3, various effects occurred. Free MoO3, as well as Al2(MoO4)3, was converted into CoMoO4, and the Co addition resulted in a decrease of the isolated Mo tetrahedral concentration and favoured the formation of the polymeric form.
Moreover, Topsøe et al. reported direct evidence of Co and Mo existing in the active form as a Co-Mo-S surface phase [49, 50].
The purpose of doping a semi conductive carrier in order to enhance the catalytic activity of supported metal catalysts has recently been applied in developing “three-way” catalysts [51].
The conductivity of semiconductors is generally low but can be considerably changed by either incorporating with other oxides or upon pre-treatments. Their crystal lattices tend to release or take up oxygen. Therefore, alloying the metals with semiconductors can increase or decrease the activity. This effect has some industrial relevance since can both accelerate desired reactions and suppress undesired reactions [52, 53]. For instance, the addition of Sn to Pd gives selective catalysts for the removal of acetylene from ethylene streams [54].
In the case of n-type semiconductors (e.g. ZnO, TiO2, CeO2, SnO2), a pre-treatment in a reducing atmosphere generates electron-donor levels (oxygen vacancies VO, metal under a lower oxidation state), which increases the free electron concentration [55-57].
The presence of electron-donor levels gives rise to electronic transitions, which may occur in the infrared region:
M(n−1)+ → Mn+ + e− (Eq-12)
VO → VO+ + e− (Eq-13)
VO+ → VO2+ + e− (Eq-14)
For instance, on heating or by reaction with reducing gases such as H2, CO for n-type semiconductors such as CeO2, the release of oxygen can be described as below:
CeO2 ↔ CeO2-x + ½ xO2(g) (Eq-15) Oox ↔ Vo + ½ O2(g) (Eq-16) Ce4+ + Vo ↔ Ce3+ + Vo+ (Eq-17) Where VO, VO+ and VO2+ are the neutral and ionized oxygen vacancies, respectively, OOx is the lattice oxygen [2].
On the other hand, the adsorption of oxygen on a nonstoichiometric oxide, containing oxygen vacancies VO, generates lattice oxygen OOx. At the same time, metal ions are oxidized at the surface and the conductivity is lowered for n-type semiconductors because the oxygen acts as an electron acceptor:
½ O2(g) + Vo ↔ Oox (Eq-18) Ce3+ + O2 ↔ Ce4+ + O2– (Eq-19) The adsorption of oxygen results eventually in complete coverage of the surface by O– or O2- and the heat of the adsorption remains practically constant while the surface becomes saturated with oxygen and negatively polarized.
Considering the adsorption of hydrogen on n-type semiconductors, it has been shown that H2
mainly undergoes heterolytic dissociation [2, 58-60]:
M2+ + O2- + H2 → M+ H + OH– (Eq-20)
On heating, the hydroxyl ions are decomposed to water and anionic defects, and a corresponding number of metal cations are reduced to atoms. In this strong chemisorption, a free electron or positive hole from the lattice is involved in the chemisorptive bonding. This changes the electrical charge of the adsorption center, which can then transfer its charge to the adsorbed molecule. Thus, chemisorbed hydrogen acts as an electron donor and increases the conductivity of n-type semiconductors. Furthermore, the change in the electrical charge density on the surface can hinder the further adsorption of the same gas. A decrease in the heat of adsorption with increasing degree of coverage is then observed, and hence a deviation from Langmuir adsorption isotherm occurs [2, 61-65].
However, when a metal is applied to an appropriate n-type semiconductor, its electron density increases [1, 2]. The general behaviour of some nonstoichiometric semiconductor oxides is summarized in Table 2.
Table 2. Behaviour of nonstoichiometric semiconductor oxides
n-type p-type
For instance TiO2, SnO2, CeO2 NiO, CoO, FeO Type of conductivity Electrons Positive holes
Addition of M21+ O oxides Lowers conductivity Increases conductivity Addition of M23+O3 oxides Increases conductivity Lowers conductivity Adsorption of O2, N2O Lowers conductivity Increases conductivity Adsorption of H2, CO Increases conductivity Lowers conductivity
The optical absorption of semiconducting oxides arises from five different phenomena: (i) intrinsic absorption, corresponding to transitions between (full) valence bands and (empty) conduction bands, which occur often in the UV–visible range and sometimes in the near- infrared (NIR) (for narrow gap semiconductors); (ii) transitions between valence bands, called intervalence transitions, only observed in p-type materials, which may appear in the NIR; (iii) free carrier absorption, arising from transitions within one band; (iv) transitions of an electron to or from a localized state; (v) lattice vibrational absorption. Their mid-infrared examination offers special difficulties due to mainly the transition types (iii) and (iv), which involve the absorbance due to free carriers and electron- or hole-donors, whose concentration depends on the semiconduction type, the surrounding atmosphere and the temperature [62].
These difficulties are seriously enhanced when the sample under study is a metal supported on an n-type semiconducting support, the reduction of the support is then greatly favoured by the metal, e.g. through activation in vacuum and spill over of hydrogen or CO. For example, this has been observed in the case of metals supported on ZnO and ceria [63-65].
Lanthanide ions of variable valence particularly Ce3+/4+ usually lead to nonstoichiometric CeO2-x. It has been reported earlier that the latter aspect and the defect structure on ceria was due to oxygen vacancies accompanied by triply and/or quadruply Ce interstitial to maintain the electrical neutrality. However, lately, oxygen vacancies have been finally affirmed as the prevailing defects neglecting the negligible effect of Ce interstitials in such a fluorite- structured oxide system [66-72]. The availability of these defect sites on the surface is probably related to their high bulk concentration. The oxygen anions (O2-) on ceria surface may be one, two or three coordinated to cerium cations. From what has been conferred for CeO2 in addition to its role as either oxygen storage and release or thermal stabilizer. It has been used either as a promoter or as a support for metal catalysts in many applications since
ceria has a beneficial effect for CO oxidation and NOx reduction under both stoichiometry and excess oxygen beside for CO/NO reaction, CO and CO2 hydrogenation. Since the catalytic oxidation of CO has acquired tremendous attraction lately particularly in connection with world-wide endeavors to curb the detrimental impacts of automotive emissions on the atmosphere. Although the detailed mechanism of the reactions mentioned above is still unknown, the researchers clearly assigned the promotional effect of the catalysts to the role of ceria in creating Ce3+/4+ redox couple [73-75]. However, due to the limited supply of precious metals and some impractical properties, an attention has been given to transition metals and their oxides as catalysts supported on CeO2 or doped with CeO2, since the ability of ceria to donate oxygen to supported metals is also a key feature in other catalytic reactions like for example, catalytic combustion and water gas shift reaction [76-78].
M. Mokhtar investigated the influence of ceria on Mo/TiO2 and found that the presence of ceria leads to increase the concentration of polymerized surface Mo oxide species, and rather initiated the formation of MoO3 over-layers. Additionally, the involvement of ceria, on the other hand, retarded the strong association rendered between Mo and Ti and thus stimulated the formation of discrete amounts of the corresponding oxides. More specifically, ceria was found to work as a mediator between Mo and Ti [79, 80].
Lucia and co-workers found that the presence of cerium in the Mo-Sn system increases the rate of ethanol dehydrogenation as well as the selectivity to acetic acid and acetaldehyde. In addition, it caused changes in the distribution of Mo species and in the textural properties, but mostly increasing the basicity of the catalyst [81].
Stannic oxide thin films are attractive for many applications due to their unique physical properties such as high electrical conductivity, high transparency in the visible part of spectrum, and high reflectivity in the IR region. In particular, tin oxide films are stable at high temperatures, have excellent resistance to strong acids and bases at room temperature, are resistant to mechanical wear, and have very good adhesion to many substrates [82-85]. Thus, transparent and electrically conductive stannic oxide films are widely used for a variety of applications. Briefly, these applications include: as electrodes in electroluminescent displays, imaging devices, protective coatings, antireflection coatings, gas and chemical sensors, transducers applications based on transparent conductors and other optoelectronic devices.
Furthermore, tin oxide films are more stable than the other transparent conducting oxide (TCO) films such as zinc oxide (ZnO). Moreover, they have a lower material cost. Recently, the synthesis of ultra fine tin oxide particles is of great technological and scientific interest
owing to their superior physical and chemical properties and their use as either catalysts for the oxidation of organic compounds or gas sensors [83-86].
As the electrical conductivity of SnO2 derived from the variable valence on the Sn atomic center is very sensitive to oxidative and reducing atmospheres, tin oxides as gas sensors detecting a trace amount of the gases have been applied to processes in chemical, heretical and fermentation industries to control the amount of the harmful wastes discharged from the plants, the explosion of the combustible gases and incomplete combustion, exhaust gases from automobiles [87-90]. However, molybdenum–tin thin films seems to be promising gas sensors. It has recently been stated that addition of MoO3 to SnO2 increases the sensor response to CO and NO2 [91-94].
All the above properties have led to intense research of SnO2 coatings over the past few decades. Currently, numerous techniques exist for the preparation of tin oxide films such as chemical vapor deposition, spray pyrolysis, sputtering, and sol–gel deposition [95-98].
Nevertheless, the ability of SnO2 to generate defects has been only recently shown to induce interesting performances for supported Pd catalysts, e.g. in deNOx reactions [99]. Conversely, tin dioxide has received limited attention in the catalysis field and the use of Mo-Sn oxides in selective oxidation appears to be unique industrial application [100-109].
On the other hand, Mo/SnO2 catalysts have been used for selective oxidation reactions due to their high activity. Niwa et al. [100] studying the methanol oxidation with several supported molybdenum catalysts found the following sequence of activity:
Mo/SnO2 > Mo/Fe2O3 > Mo/ZrO2 > Mo/TiO2 > Mo/Al2O3
Goncalves et al. [101] and Medeiros et al. [102] have shown that acetic acid can be obtained from ethanol oxidation in only one-step with high yield when Mo/SnO2 catalysts prepared by precipitation procedure are used.
Recently, Liu et al. [103] have shown that Mo/SnO2 catalysts are very active for the oxidation of dimethyl ether although they are more selective for formaldehyde than Mo/Al2O3 catalyst.
V. Lochar claimed that the activity of MoO3/SnO2 catalyst for methanol oxidation could be associated with its Brönsted and Lewis acidity as the result of the catalyst reduction [104].
Other catalytic activity results suggest the existence of synergy between the apparently pure phases of MoO3 and SnO2. Therefore, MoO3 in close contact with SnO2 has shown to be much more active and selective than the individual pure phases. The high dispersion of
molybdenum species on the highly reducible SnO2 support was suggested to be responsible for the exceptional activity of these catalysts [105-109].
However, the information available in the literature on the interpretation of infrared and Raman spectra of Mo/Sn and Mo/Ce compounds prepared on different surfaces is rather limited. In assigning vibrational spectra, some DFT (Discrete Fourier Transform) calculations or some vibrational spectroscopic data relating to Mo/Sn and Mo/Ce systems can be relied on [65, 80, 88, 91, 97, 104-109]. On the other hand, few papers can be found in the literature on IR and TG studies related to either Mo/Sn or Mo/Ce system in contrast to publications relating to noble metals with ceria and tin.
Anyhow, a lot of debates concerning the role played by ceria and tin oxides necessitate further studies in order to explore the influence of CeO2 and SnO2 on the structure and surface characteristics of molybdena for better understanding the nature, structure and the physico- chemical properties of these oxides, since the nature of the interactions between metal oxides and supports are often attributed to the complexity of these systems and differences in the preparation and experimental conditions adopted.
1.4. Direct conversion of methane under nonoxidative conditions
An important task confronting catalytic chemists is how to realize direct conversion of methane to versatile fuels and valuable chemicals by building up the desired C–C (or C–O) bond. Thermodynamic constraints on the reactions in which all four C–H bonds of CH4 are totally destroyed, such as CH4 reforming into synthesis gas or CH4 decomposition into carbon and hydrogen, are much easier to overcome than the reactions in which only one or two of the C–H bonds are broken under either oxidative or nonoxidative conditions [110-117]. Direct conversion of CH4 with the assistance of oxidants is thermodynamically more favourable than that under nonoxidative conditions. Therefore, the direct conversion of CH4 under the aid of oxidants has received much more attention than that under nonoxidative conditions, especially when considering the production of fuels and valuable chemicals from CH4 [118-123].
With the urge to quest for renewable energy and cleaner fuels, it is recognized that hydrogen energy will inevitably replace fossil fuel energy in the near future due to the fact that the burning of hydrogen is pollution free. However, it is a practical way to produce H2 from CH4
due to its high H/C atomic ratio and great abundance in reserves. Therefore, the direct conversion of CH4 under nonoxidative conditions into H2 and/or H2 accompanied with basic chemicals is closely related to the effective utilization of CH4-containing resources and thus to sustainable progress and development of the living conditions of humankind [124-127].
The direct conversion of CH4 under nonoxidative conditions is thermodynamically unfavorable. Nevertheless, as an alternative approach, it has still attracted the attention of many researchers. In heterogeneous catalysis, various metals have been discovered that can chemisorb CH4 at moderate temperatures and that can decompose CH4 to C and H2 at higher temperatures [128-137].
Amariglio and co-workers reported a “two-step” process on Pt, Ru, and Co in isothermal experiments [128-130]. In a series of publications, the authors suggested that C–C bonding could take place between H-deficient and CHx formed during the first step of methane chemisorption, while H2 saturated the alkane precursors in the second step and removed them from the surface. In view of the fact that hydrogenation at a temperature lower than that of CH4 chemisorption is favorable for lessening hydrogenolysis. The authors reported a nonoxidative conversion of methane to higher hydrocarbons through a dual temperature two- step reaction on Pt/SiO2 and Ru/SiO2 catalysts. Indeed, when chemisorption of methane was set at a fixed temperature (usually lower than 320°C), the selectivity to heavier alkanes increased with the lowering of hydrogenation temperature on both catalysts. On the other hand, when the hydrogenation temperature was less than 120°C, hydrogenolysis was negligible, and thus the variations of the products can only be attributed to the changes affected by the adlayer formed during the chemisorption of methane at a certain temperature.
It was discovered that the products of C2+ hydrocarbons at every hydrogenation temperature displayed a maximum versus the methane chemisorption temperature on both catalysts. In the case of the Pt/SiO2 catalyst, mainly C2H6 and n-C5H12 were produced during the first minute of the reaction. This illustrates that C–C bonds could form during CH4 adsorption, and the authors assumed a surface intermediate of C5 precursor bonded on dispersed and coordinately unsaturated Pt atoms.
Van Santon et al. suggested that CH4 first dissociated on a precious metal to form carbide and H2. Then, the carbide was hydrogenated by H2 to produce higher hydrocarbons. C–C bonds were supposed to be created during the hydrogenation step. Since the reactivity of the CHx surface intermediates formed from CO and CH4 was quite similar. The authors suggested that the chain-growth probability would depend on the metal–carbon bond strength and that the mechanism of C–C bond formation in the two-step route should be related to that occurring in the Fisher–Tropsch reaction. They also demonstrated that the homologation of olefins (C2H4, C3H6, etc.) with methane could occur over Ru/SiO2 and Co/SiO2 catalysts [131, 132].
The two-step route is also feasible over a number of oxide- or zeolite-supported transition metal and bimetal catalysts. Solymosi and Cserenyi illustrated that over a Cu-promoted Rh/SiO2 catalyst, the enhanced formation of C2H6 and higher hydrocarbons could be observed in the two-step process [133, 134].
Guczi et al. reported that the chemisorption of CH4 at 250°C and the subsequent hydrogenation of the CHx species at 250°C over Co–Pt/NaY and Co–Pt/Al2O3 performed the best of all the catalysts tested. The chemisorbed CHx species had the highest concentration, and all CHx species were hydrogenated in the second step, giving a selectivity of C2+ close to 84%. They found that there was a correlation between the hydrogen content of the surface CHx species (the optimum value of x being around 2) and the chain length of the hydrocarbons produced in the hydrogenation step in their mechanistic study of the two-step process [135]. Later, they reported that the two-step process could be simplified into a one- step process with a C2+ hydrocarbons production higher than that obtained in the two-step process over Co–Pt/NaY bimetallic catalyst. These results could be obtained if the CH4 was pulsed with H2/He mixture at 250°C [136].
Bradford reported the results of the isothermal, nonoxidative, two-step conversion of CH4 to C2+ hydrocarbons over supported and unsupported Pt and Ru catalysts at moderate temperatures and elevated pressures. It was shown that an increase in reaction pressure increased the branching and molecular weight distribution of the product [137].
Several researchers suggested the preparation of a multifunctional catalyst to avoid the use of a two-step process. Furthermore, it has been reported that dehydrogenative coupling of CH4
without any oxidant could be carried out over Pt–SO4/ZrO2 catalysts. A steady conversion of 0.2% (the equilibrium conversion of CH4 into C2H6 and H2 is estimated to be 0.6%) was observed after the catalyst was reduced in H2 at 500°C [138, 139].
On the other hand, in order to overcome the thermodynamic limit and to enhance the reactivity for obtaining high yields in direct conversion of CH4 under nonoxidative conditions, plasma excitation has also been attempted. The product distribution is dependent on the method by which plasma excitation is produced. For example, in pulsed corona discharges at atmospheric pressure, C2 hydrocarbons (mainly C2H2) were obtained with a high selectivity of around 70 to 90%. In microwave plasmas, the product distribution shifted from C2H6 to C2H4
and finally to C2H2 with an increase in power density. By introducing a proper catalyst into the microwave plasma reactor, CH4 could be converted to higher hydrocarbons at atmospheric pressure. In addition, with a CH4 and H2 mixture as the feed gas, the selectivity to C2H2 was 88% and that to C2H4 was 6% at a CH4 conversion of 76% [140, 141]. Here, again, the main
drawback is the low energy efficiency to drive this thermodynamically unfavorable reaction.
Thermodynamically, the transformation of CH4 under nonoxidative conditions is more favorable to aromatics than to olefins. The direct conversion of CH4 to aromatics was tested on several catalysts in either a pulse or a flow reactor. Wang et al. reported on the dehydroaromatization of methane (MDA) for the formation of aromatics (mainly C6H6) and H2 under a nonoxidizing condition in a continuous flow reactor on Mo/HZSM-5 catalysts [142]. More detailed studies on the reaction revealed that the channel structure and acidity of the HZSM-5 zeolite, as well as the valence and location of the Mo species, are crucial factors for the catalytic performance of the Mo/HZSM-5 catalysts. In addition, W/HZSM-5 and Re/HZSM-5 are also reported to be active elements for MDA [142-144].
Solymosi and co-workers [147-152] and Lunsford and co-workers [153-155] characterized the Mo/HZSM-5 catalyst by means of XPS and found that during the initial induction period, the original Mo6+ ions in the zeolite were reduced by CH4 to Mo2C, accompanied by the depositing of carbonaceous cokes. They suggested that Mo2C provides active sites for C2H4
formation from CH4, while the acidic sites catalyze the subsequent conversion to C6H6. The Mo2C species probably are highly dispersed on the outer surface, and some of them reside in the channels of the zeolite. Meanwhile, the spectra of Mo/HZSM-5 samples reacted with CH4
at 700°C for one and 24 hrs were basically identical to the Mo2C reference spectrum, except for a partial contribution from the Mo oxide, given rise by MoOxCy. These authors claimed that the Mo oxide species dispersed in the HZSM-5 framework might migrate onto the external surface of the HZSM-5, be converted by CH4 to Mo2C, and disperse on the support surface. Therefore, the carbonaceous deposits created in MDA are in various forms and play different roles. First, Mo2C and/or MoOxCy, which are possibly active species for CH4
activation, are formed during the induction period. Second, the formation of the active intermediates, the CHx species, follows the activation of CH4 on Mo2C and/or MoOxCy. The last one to be formed is coke leading to the deactivation of the catalyst. It is understandable that there are some similarities between the carbonaceous species formed in MDA and those formed in the first step of the two-step process, since both reactions are carried out under nonoxidative conditions, and Mo2C shows some precious metal-like properties.
In spite of the fact that the reaction is thermodynamically unfavorable under pressurized conditions and that 10% CO2 added to the feed totally suppresses the activity of the 2 wt%
Mo/HZSM-5 catalyst. Ichikawa and co-workers found that an increase in CH4 pressure and the addition of small amounts of CO and CO2 (less than 3%) to the CH4 feed enhanced the catalyst stability in the reaction [156, 157]. By increasing the CH4 pressure, the formation
rates of C6H6 and hydrocarbons could be moderately increased. This kind of pressure relationship may be related to a sufficient supply of H2 from CH4 and a suitable concentration of surface carbon species CHx for the formation of aromatic products. By using a CO and CH4
mixture as the feed to conduct the reaction, the authors suggested that CO dissociated on the Mo sites to form the active carbon species CHx. The dissociated oxygen species [O] from CO might react with the surface inert carbon species to regenerate CO, resulting in the suppression of coke formation on the catalyst. These results imply that although the Brönsted acid sites are necessary, excess Brönsted acid sites are detrimental for the reaction, since severe coke formation will occur on them [157-159].
Considerable efforts have been devoted to developing active and selective catalysts and understanding the bifunctionality of Mo/HZSM-5 catalysts and the nature of carbonaceous deposits formed during the reaction. However, neither new active and selective catalysts nor a thorough understanding of the mechanism of the reaction has been achieved.
However, despite all substantial research efforts into nonoxidative two-step or one-step CH4
homologation, its low efficiency is the main problem to further developing it as a commercial process. In any case, these studies enhanced our knowledge in direct conversion of CH4 under nonoxidative conditions, particularly methane dehydroaromatization, and stimulated chemists to explore new methane conversion processes.
1.5. Characterization of the catalysts
Both of the physical and the chemical properties of a catalyst must be known if relationships between the structure and activity, selectivity, and lifetime are to be revealed. There are many techniques commercially available for the analysis of catalysts opening up new possibilities for fundamental catalyst research. In this section, I will encounter some methods for characterizing catalysts and not discuss their capabilities and limitations that have been described in the literature [1, 2].