The Determination of the Area of the Surfaces of Solids
B Y
GEORGE JURA
Department of Chemistry, University of California, Berkeley, California
CONTENTS
Page
1. Introduction 255 2. "Absolute" Method of Area Determination 257
2.1. General Considerations 257 2.2. Theory of "Absolute" Methods 257
2.3. Experimental Methods 260
2.4. Results 262 3. Adsorption of Gases 264
3.1. Introduction 264 3.2. Experimental Methods 265
3.3. Preparation of Solids 266 3.4. Apparatus Design 267 4. Langmuir Isotherm 271
4.1. Theory 271 4.2. Applications of Langmuir Theory 276
5. Theory of Brunauer, Emmett and Teller 278
5.1. Introduction 278 5.2. Theory 279 5.3. Application 283 6. Entropy Method 289 7. Relative Method 290
7.1. Theory 290 7.2. Application of Relative Method 294
7.3. Comparison of Results obtained by B.E.T. and Relative Methods 297
8. Comparison of Area Methods 297
References 301
1. I N T R O D U C T I O N
I n m a n y respects t h e surfaces of solids exhibit t h e same general behavior as those of liquids. T h e r e is a t r e m e n d o u s a m o u n t of detailed knowledge of t h e surfaces of liquids a n d t h e interaction of these surfaces with other molecules. On t h e other h a n d t h e r e is v e r y little such knowl- edge of t h e surfaces of solids. T h i s discrepancy in available information is largely due t o t h e fact t h a t until recently no reliable m e t h o d s h a v e been available for t h e estimation of t h e surface areas of solids. F u n d a -
255
mentally, the area of t h e surface is essential for t h e comprehension of any surface phenomena, since t h e gross effect or interaction studied is directly proportional t o t h e area.
A simple illustration for t h e necessity of an area figure follows. If any solid is cleaned, i.e., foreign molecules removed from its surface, and then is immersed in a liquid, heat is usually evolved which can be measured in a sensitive calorimeter. T h e measurement is called t h e heat of immersion or wetting. Four samples of t i t a n i u m dioxide, I, I I , I I I , a n d IV, all in t h e form of anatase, were immersed in water at 25°C. Samples I I I and IV are known t o have been treated with alumi- n u m oxide. I t was found t h a t 1.15, 1.69, 1.42, and 2.21 cal. g ._ 1 were evolved. These figures are useless for comparative purposes and are of value only for t h e samples in their respective bottles or barrels. If the areas of these samples are determined it is found t h a t t h e heats of immersion are 520, 510, 650, a n d 1200 ergs c m .- 2 respectively. These figures show t h a t t h e degree of subdivision of t h e u n t r e a t e d samples has no effect on t h e surface properties for t h e range of particle sizes involved.
They also show t h a t t r e a t m e n t with aluminum oxide seriously affects the surface properties, a n d t h a t t h e t r e a t m e n t s involved in samples I I I and I V were sufficiently different t o alter their interaction radically.
The knowledge of t h e area permits not only this comparison b u t also comparisons with solids other t h a n t i t a n i u m dioxide. I t has been found t h a t in water the heats evolved for " p u r e " solids ranged from 165 £rgs c m .- 2 for graphite t o 850 ergs c m .- 2 for zirconium silicate (36).
M a n y methods have been proposed for t h e determination of t h e area of solids. Unfortunately, none has such a strong theoretical basis t h a t the results obtained b y its application can be considered as certain. T h e experimental evidence available at t h e present time does indicate strongly t h a t t h e relative areas of various solids can be determined t o within 1 0 % . For a n y comparative purpose it is only t h e relative areas t h a t are essen- tial. T h e evidence for t h e absolute areas of solids rests on t h e " a b s o l u t e "
method of Harkins a n d J u r a (39) or t h e values of t h e effective cross- sectional areas of adsorbed molecules t o be used in t h e theory of Brunauer, E m m e t t , and Teller (18). B o t h of these are discussed in detail in later sections.
T h e methods for area determination m a y be classified in three groups:
(1) Those t h a t depend on t h e determination of particle size, (2) those t h a t depend on t h e determination of t h e adsorption isotherm, a n d (3) those t h a t depend upon some special property other t h a n particle size and adsorption, which m u s t depend in some known m a n n e r on t h e available area. M o s t of t h e proposed methods fall into t h e first two groups, a n d since t h e adsorption methods are t h e simplest experimentally,
D E T E R M I N A T I O N O F A R E A O F T H E S U R F A C E S O F S O L I D S 257
most of t h e a t t e n t i o n has been given to this class. Thus, the major portion of this chapter deals with t h e methods based on adsorption.
T h e methods of particle size determination are numerous. F r o m the known density of t h e solid, t h e geometry of t h e particles, a n d t h e average particle size or size distribution it is relatively simple t o calculate an area. Particle size determinations in general are not satisfactory for obtaining t h e area. One u n c e r t a i n t y arises with every such method.
If t h e solid is porous, i.e., possesses an internal area, t h e particle size measurement cannot account for this area. Thus, t h e area obtained is t h e lowest t h a t t h e solid m a y have. M a n y particle size determinations have one other further difficulty. Small particles t e n d t o aggregate in m a n y solids. I n determinations such as sedimentation or microscopic examination, either visual or electron, an aggregate will appear as a single particle. Since a large particle has a lower area t h a n t h e same weight of smaller particles t h e area again will be low. This difficulty is not encountered when t h e particle size is determined b y t h e broadening of x-ray diffraction lines or t h e low angle scattering of x-rays. Because of these inherent difficulties, t h e determination of areas from particle size will not be considered in this chapter. M a n y methods of particle size determinations are given in other chapters of this volume.
2. "A B S O L U T E " M E T H O D O F A R E A D E T E R M I N A T I O N
2.1. General Considerations
T h e " a b s o l u t e " m e t h o d is a calorimetric m e t h o d a n d was first pro
posed b y Harkins and J u r a (39). This m e t h o d is experimentally slow and difficult, a n d its theory restricts its use t o nonporous solids. I n spite of its difficulties a n d limitations, it is considered first because (1) the theory is simple and sound, (2) it is t h e only known m e t h o d t h a t gives an area directly, a n d (3) it indicates t h a t t h e answers obtained by adsorption methods are absolute as well as relative.
I n general outline, t h e m e t h o d is based on t h e fact t h a t when t h e solid has adsorbed t h e vapor of a liquid on its surface, a n d t h e fugacity or chemical potential of t h e adsorbed molecules is equal t o t h a t of t h e liquid at t h e same temperature, t h e n t h e heat content per unit area of the surface can be related t o t h e heat content of t h e liquid. I n a n y system for which t h e m e t h o d can be used, t h e surface of t h e solid with respect t o this property is changed t o t h a t of a liquid if t h e contact angle of t h e liquid against t h e solid is zero.
2.2. Theory of "Absolute" Method
W i t h every surface, whether solid or liquid, there is associated a free surface energy, γ, a surface heat content, h, a surface entropy, s,
etc. If t h e surface is t h a t of a liquid, t h e free surface energy is more commonly called t h e surface tension. T h e heat content a n d surface tension are simply related. T h e relationship is given b y eq. (1)
* - * -
r( S ) ,
( 1 )where Τ is t h e absolute t e m p e r a t u r e a n d ρ isUhe external pressure. A simple derivation is given b y Lewis a n d Randall (57). If a clean solid is immersed in a liquid, its free surface energy is changed from ys t o ySLf and its heat content from hs t o hSL- Thus, t h e heat evolved, AhE in the immersion process per unit area is
Ahs = hs — hsL = ys — ysL — Τ {^ψ — ~^ψ^ (2)
Equation (2) has been extensively discussed b y Harkins a n d Boyd (36).
If the solid is saturated with t h e vapor of t h e liquid, then its free surface energy is changed from yB to y8e and t h e heat evolved on immersion from AhE to AhEe, and
(
^*Vse si \ΊΪΤ βΨ/ρ ^
Equation (3) can be simplified by use of t h e Dupree equation
yse = ysL + TL cos θ (4)
where yL is t h e free surface energy of t h e liquid and θ is t h e contact angle, measured through t h e liquid, of t h e liquid against t h e solid.
If eq. 4 is substituted in eq. 3 it is found t h a t
AhEe = | T L - Τ ( | ^ ) } cos θ + TyL sin θ II (5)
If the contact angle is zero, which is usually true, eq. (5) reduces to
E q u a t i o n 6 simply states t h a t when a drop of liquid with unit area is immersed in a larger body of t h e liquid t h e heat evolved is exactly the same as t h a t obtained if a unit area of t h e solid saturated with t h e liquid is immersed, provided t h e contact angle is zero. Thus, if Σ7 is t h e area per gram of t h e solid covered with its film a n d HEe is t h e heat evolved, when one gram of t h e solid is immersed in t h e liquid the area g .- 1 of t h e film covered solid is
The total surface energies of most liquids are available in the literature.
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 259
T h e International Critical Tables, Vol. IV, McGraw-Hill, New York, for example, contains t h e necessary d a t a for nearly a n y liquid which might be used for this purpose.
If t h e solid is porous, t h e pores will be filled in as the solid absorbs t h e vapor a n d t h e area will be decreased. Since there is no independent method of determining t h e area of t h e porous p a r t of the sample, t h e area of t h e film covered solid cannot be related t o t h e area of t h e clean solid. This is a limitation which restricts t h e use of the method.
E v e n if t h e solid is nonporous one further correction m u s t be m a d e before t h e area of t h e solid is known. Fortunately, this correction is not large. This correction is m a d e necessary by the fact t h a t t h e adsorbed film is polymolecular, which increases the size of t h e particle and consequently the area. T h e relation between t h e area of t h e solid and t h e film covered solid depends upon t h e geometry of t h e individual particle, t h e film thickness, and t h e size distribution of t h e particle.
Since the entire correction for all of the factors is not large, ca. 5 % , none of these need be known with great accuracy.
T h e film thickness can be obtained with sufficient accuracy from t h e area of t h e film covered solid, t h e a m o u n t of gas adsorbed at saturation, and the assumption t h a t t h e density of t h e adsorbed material is t h e same as t h a t of t h e liquid. None of the above is strictly correct b u t no serious error is introduced by these assumptions. For example, if t h e particles are cubes of t i t a n i u m dioxide 1200 A. on edge and t h e film thickness is assumed to be 20 A. the final result is changed b y only 2 % if the film thickness is changed from 20 to 40 A. T h e adsorption work indicates t h a t at saturation the film thickness is on t h e order of 25 A. for water (38).
T h e film thickness depends upon t h e vapor as well as the solid. Films of η-heptane average about 200 A. at 25°C. Thus, there is a distinct a d v a n t a g e in t h e use of water. T h e above figures probably could be used for a n y solid without introducing too great an error. For liquids other t h a n water and η-heptane it would be necessary to determine t h e a m o u n t adsorbed at saturation. Since t h e area of t h e solid covered with the film is not m u c h different from t h a t of the clean solid, t h e film thick
ness is essential only if t h e highest accuracy is desired.
T h e geometry of t h e individual particle also affects the magnitude of t h e correction. For example, consider t h e rectangular parallelopipeds whose dimensions in t h e three normal directions have t h e ratios 1 : 1 : 1 , 1:10:10, and 1:100:100. For a sample of graphite whose area is 4.2 m .2g ._ 1, when η-heptane is the liquid, t h e necessary correction for each of these shapes would be 10, 5, and 0 %, respectively. This varia
tion in the correction factor arises from the ratio of t h e area t o t h e volume for a given geometry.
A small variation in t h e correction factors arises depending on t h e assumptions m a d e concerning t h e particle size distribution. I t is readily seen t h a t t h e smaller t h e particle, t h e greater t h e correction. T h e assumption of an average size, as determined from t h e density of t h e solid and area of t h e film covered solid leads t o t h e largest difference in area between t h e film-covered and clean solid. If t h e particle is a cube, the relation between t h e area g .- 1 Σ of the clean solid and Σ' is
where τ is t h e film thickness, d t h e edge of t h e cube, a n d a depends upon the assumed shape of t h e film-covered particle. If it is assumed t h a t the film forms a cube, a has the value of 4. On t h e basis t h a t t h e film will be cylindrical about t h e edges t h e value of a is τ. On physical grounds, t h e cylindrical shape about t h e edges is more probable, since the surface area is lower. T h e assumption of t h e formation of t h e cubical shape, yields t h e largest correction factor, a n d consequently t h e minimum area for the clean solid. If a n y reasonable, continuous size distribution is assumed, then the value for a is even less t h a n π. T h e smallest value of a t h u s far obtained is ψ on t h e assumption t h a t t h e distribution is of t h e form
Thus, for a nonporous solid, t h e minimum correction is zero, a n d t h e m a x i m u m possible correction is given by t h e use of eq. (8) when a is 4.
E q u a t i o n (8) shows t h a t t h e most accurate result is obtained b y
been obtained with water. W a t e r also has one further advantage, t h e ratio of its surface energy to specific heat is t h e highest. I t is also evident t h a t better results can be obtained b y t h e use of large r a t h e r t h a n small particles since r is dependent only upon t h e crystalline face, a n d chemical composition.
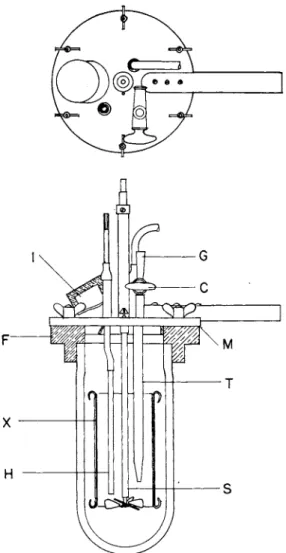
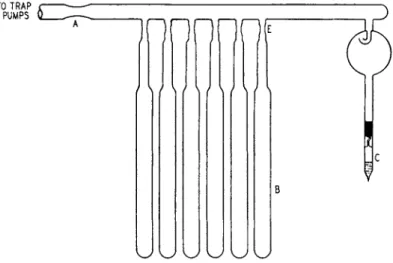
Figure 1 shows t h e calorimeter t h a t has been used for measuring t h e heats. This calorimeter is essentially the same as t h a t described b y Harkins and Dahlstrom (37) and has a sensitivity of 2 X 10~5 °C. T h e technique of making the measurements is t h a t described b y Boyd a n d Harkins (12). Figure 2 shows the a p p a r a t u s used for t h e preparation of the samples.
(8)
Ν = Ce-hd2
making the ratio as small as possible. T h e smallest values for r h a v e
2.3. Experimental Method
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 261
FIG. 1. Calorimeter. T, 36 junction thermal; H> heating element; G, inlet for liquid; C, stopcock; X, chimney to give better circulation for powder; 7, opening for introduction of powder; S, stirrer driven by synchronous motor; F and Μ brass collar and cap.
Between 10 a n d 15 g. of t h e powder is placed in each of t h e sample bulbs B, of which there are usually six, a n d 5 t o 10 c m .8 of liquid is placed in C. T h e water is degassed b y boiling away about one-half of t h e original volume a n d t h e n sealing t h e b o t t o m of t h e bulb. T h e tubes t h a t contain t h e powder are heated in an electrically heated furnace at 500° for 24 hours, after which t h e y are allowed t o cool to room tern-
perature. The p u m p s are in continuous operation during this period.
After the samples have cooled, t h e manifold is sealed off at C, a n d t h e whole a p p a r a t u s is submerged in a t h e r m o s t a t at 25.00°. After 4 hours in the thermostat, the tip of C is broken with an iron rod and a magnet.
This allows the water vapor t o penetrate t h e entire system. After about 24 hours, which is more t h a n sufficient time for t h e system to
TO TRAP
& PUMPS
m m • n n i
FIG. 2. Apparatus for the adsorption of an equilibrium film on a powder. C is the liquid, and the powder is in the tubes, B. After the degassing of the surface the system is sealed off from the pumps at A and the tubes and liquid are immersed in a thermostat. After equilibrium is reached the tubes are sealed off at E.
reach equilibrium, the manifold is raised and t h e sample tubes are sealed off at E.
2.1+. Results
Only three areas have been determined b y this m e t h o d : two samples of titanium dioxide in t h e form of anatase, one of which h a d been treated with aluminum oxide and a sample of graphite (8) containing less t h a n 0.004% ash and presumably free from any oxygen complexes. W a t e r was used with the two samples of anatase and η-heptane with t h e graphite.
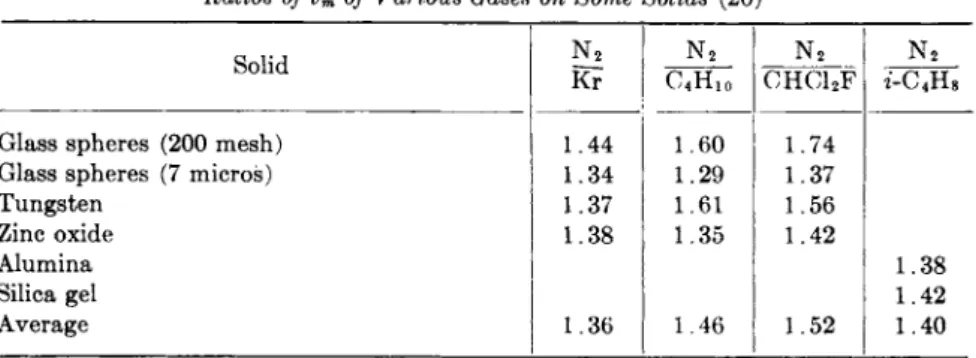
I n each case, the temperature was 25°C. W a t e r could not be used with graphite since the contact angle is not zero (30). Table I lists the areas for these three solids. The values given in Table I were obtained by using 118.5 and 50 ergs c m .- 2 as t h e heat contents of the surfaces of water
(34) and n-heptane (2), respectively.
Experimentally t h e best value is t h a t for t h e u n t r e a t e d t i t a n i u m dioxide. Much weight can not be given to the treated sample, since
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 263
Areas of Powders Obtained by the "Absolute" Method
Solid Σ' Σ
m^g."1 m .2g . - i Obtained from adsorption measurements m .2g ._ 1 T i 02 (untreated) 14.4 ± 0 . 4 13.8 13.8«
T i 02( A l203 treated) 9.1 ± 0 . 4 8.9 9.6*
Graphite 4.6 ± 0.6 4.4 4 . 1 - 4 . 3C
° This sample was used to standardize the adsorption measurements.
h Nitrogen adsorption at — 195.8°C.
c These values were obtained with nitrogen at —195.8°C, n-hexane at 0°C. and η-heptane at 25°C
electron microscope photographs. F r o m the n a t u r e of t h e instrument, this evidence cannot be considered satisfactory. T h e general theory of solids indicates t h a t cracks would not appear in individual crystals of t h e size used, ca. 1200 A. This is the point upon which t h e validity of t h e area of the clean solid rests. Until some method can be devised which will show t h a t these powders are nonporous, there will be some doubt as to whether or not t h e area of t h e clean solid can be related t o t h a t of the solid covered with a film.
The absolute method gives directly the area of the solid which has adsorbed the vapor of a liquid to saturation. Because contact angles and t h e t e m p e r a t u r e coefficients of contact angles cannot be accurately measured, the method is restricted to the use of liquids t h a t exhibit a zero contact angle against the solid. If the solid is nonporous, the area of the clean solid can be related t o t h a t of the film covered surface.
This relation is not accurate, b u t since it is small, equal to or less t h a n 5 % for t h e solids and liquids t h u s far used, this source of error is not serious. T h e entire difficulty with the application of t h e method lies in demonstrating t h a t the solid is nonporous. There is no direct experi-
the evolution of heat was too slow for t h e t y p e of calorimeter used. W i t h this slow evolution of heat, low results are generally obtained. T h e results with graphite are t h e best t h a t could be hoped for with t h e low area and low heat evolved per unit area.
T h e agreement between the results obtained by adsorption measure
ments and t h e absolute method is excellent.
Before t h e values obtained b y this m e t h o d can be considered as certain, it is essential to show t h a t these solids are nonporous. There is no known method for showing this. T h e only direct evidence t h a t t h e powders are nonporous is t h a t none obviously appear t o be so in
TABLE I
mental evidence t o demonstrate this point. T h e theory of t h e structure of solids does indicate t h a t crystals as small as those used in these experi- ments would not exhibit any cracks t h a t contribute t o t h e area.
3. AD S O R P T I O N O F GA S E S
3.1. Introduction
The most general and reliable methods for t h e determination of the areas of solids are based on t h e determination of t h e adsorption isotherms of gases on t h e surfaces of solids. T h e isotherm is t h e determination of the q u a n t i t y of gas adsorbed as a function of t h e equilibrium pressure of t h e adsorbed gas a t constant pressure.
The adsorption of gases is one of t h e oldest known phenomena.
Scheele (65) is credited with t h e .discovery in 1773. Since this time thousands of papers have appeared in t h e literature with reports of experimental results, and m a n y theories have been proposed in explana- tion. T h e complexity of the phenomena t h a t m a y occur has been such t h a t it is only in t h e last half century t h a t there has been a n y real progress in t h e elucidation of the observed facts.
When a gas is adsorbed a n u m b e r of possibilities exist. T h e gas m a y dissolve in t h e solid. This solution m a y or m a y not lead t o com- pound formation. For example, hydrogen is soluble in iron at high t e m - peratures (67). Here, there is no evidence of compound formation.
If paladium is substituted for iron, solution occurs with t h e formation of the interstital compound, paladium hydride (68). Alternately t h e gas molecules m a y form a compound with molecules on t h e surface of t h e solid. T h e n a t u r e of m a n y of their compounds is unknown. For example, if carbon monoxide is adsorbed on iron at temperatures as low as 77°K., t h e a m o u n t of gas adsorbed which is sufficient t o form a mono- molecular layer is apparently bound differently from t h e remainder of the adsorbed gas (27). This first layer cannot be desorbed at low tem- peratures as can the remainder of t h e gas. This gas can be removed at high temperatures, not as carbon monoxide, b u t as iron carbonyl. T h e experiments indicate t h a t there is one such carbon monoxide molecule adsorbed for each iron a t o m in t h e surface. I t is t r u e t h a t no iron car- bonyl is known t h a t has t h e formula Fe(CO), b u t it is evident t h a t there is some sort of chemical binding between t h e gas a n d solid. W h e n a reaction occurs between the gas molecules a n d t h e surface of t h e solid, the n a m e used t o describe t h e process is chemisorption. T h e process of surface compound formation m a y be irreversible as in t h e example cited, or reversible.
T h e molecule m a y undergo a n y n u m b e r of chemical reactions on t h e
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 265
surface, a n d t h e products m a y be described. T h e solid itself apparently is unchanged. This is, of course, t h e well-known process of hetergeneous catalysis. T h e role of t h e surface is not thoroughly understood at present.
Finally, t h e adsorption m a y occur, due to t h e intermolecular forces between t h e molecules of t h e solid in t h e surface region and t h e mole- cules of gas. T h e n a t u r e of these forces depends of course on t h e n a t u r e of t h e molecules in b o t h t h e gas a n d t h e solid. This process occurs because there is a decrease in free energy of t h e system, particularly t h a t of t h e surface of t h e solid.
Of all t h e various possibilities, t h e one t h a t will always occur is t h e last. Therefore, a n y general m e t h o d of area determination m u s t be based on t h e intermolecular interaction. Since Van der Waals' forces are always present, t h e theory should be founded on this t y p e of inter- action. Another requirement, of course, is t h a t all other effects be absent. T h e adsorption methods of area determination are based upon t h e above assumptions.
Only a few theories of adsorption t h a t involve area determination are discussed. Other theories a n d proposals are excellently reviewed in a recent book b y Brunauer (14). A survey of t h e earlier literature is to be found in McBain (59).
3.2. Experimental Methods
T h e determination of t h e adsorption isotherm involves t h e measure- m e n t of t h e a m o u n t of gas adsorbed as a function of t h e pressure. A large n u m b e r of m e t h o d s a n d techniques h a v e been devised. M a n y of these h a v e been built for obtaining information concerning a specific system. I n this review, t h e factors t h a t affect t h e final results are discussed a n d a detailed description is given for a volumetric method t h a t gives results of t h e highest precision. I t is hoped t h a t this is suffi- cient so t h a t changes can be m a d e t o fit individual needs.
One of t h e most i m p o r t a n t factors in a successful area determination is t h e choice of t h e gas a n d t e m p e r a t u r e at which t h e determination is t o be made. T h e gas should have t h e following characteristics: it m u s t not react with t h e solid, it m u s t not dissolve in t h e solid, t h e solid should not catalyze a change in t h e chemical n a t u r e of t h e gas, t h e gas should be pure, or readily purified, a n d a gas molecule without a permanent electric m o m e n t is preferable.
There are two factors t h a t dictate t h e choice of temperature. A t e m p e r a t u r e should be chosen such t h a t t h e b a t h can be maintained at constant t e m p e r a t u r e . This is essential since t h e effect of t e m p e r a t u r e on t h e equilibrium pressure of t h e adsorbed gas is marked. Convenient
temperatures are the melting and boiling t e m p e r a t u r e of pure compounds.
Those in most general use are liquid nitrogen at 77.3°K. for use with nitrogen, oxygen, argon, carbon monoxide, etc. Ice is generally used at 0°C. for determinations with η-butane. At room temperatures simple water t h e r m o s t a t s can be used.
T h e other i m p o r t a n t factor determining t h e choice of t e m p e r a t u r e is the area of the solid. T h e temperature is used to control the vapor pressure of t h e stable three-dimensional phase of t h e gas. For materials of small area, it is necessary t h a t the vapor pressure be low so t h a t the a m o u n t of gas adsorbed is a reasonable fraction of t h e a m o u n t of gas measured if a volumetric method is used, and t h a t the change in bouyancy correction does not overshadow the increased weight of t h e sample if gravimetric methods are used. For economy of time, it is essential t h a t as high a t e m p e r a t u r e as possible for a given gas be used, because the time required to obtain equilibrium between t h e adsorbed and unadsorbed gas is materially reduced for m a n y solids as t h e t e m p e r a t u r e is increased.
3.3. Preparation of Solids
One of the important features in the determination of t h e area of a solid is the "degassing." Degassing consists in the removal of all gases from the surface preparatory to t h e actual determinations. T h e general method is to heat the sample in a v a c u u m until these gases are removed. N o specific rules can be given to cover all possible solids.
Frequently, t h e exact conditions are determined b y t h e solid. I n general, as high a t e m p e r a t u r e and v a c u u m as possible should be used.
T h e t e m p e r a t u r e chosen should be such t h a t t h e structure of t h e solid is undisturbed. These disturbances are of several kinds. If t h e solid has more t h a n one crystalline form, t h e t e m p e r a t u r e should not exceed t h e transformation temperature. An example of this t y p e of behavior is tin, which changes its crystalline form at 185°C. (44). A second factor which m a y determine the m a x i m u m t e m p e r a t u r e is sinter
ing. Most crystallites have a minimum t e m p e r a t u r e below which crystal growth will not occur, regardless of the time maintained at a given temperature. Above this t e m p e r a t u r e crystal growth can occur.
M a n y noncrystalline materials exhibit a similar effect, a decrease in area with an increase of temperature. Table I I exhibits t h e effect of tempera
t u r e on t h e area of a silica-alumina cracking catalyst. T h e maximum t e m p e r a t u r e used in degassing should be below t h e sintering t e m p e r a t u r e for crystals, and below the maximum t e m p e r a t u r e to which a noncrystallite has been previously heated.
Frequently these conditions cannot be met, and a compromise m u s t
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 267
T°C. Area (m^g."1)
100 455 500 434 600 409 700 395 800 321 pressure of the liquid or solid is 760 m m . the error is not greater t h a n
3 t o 5 % if t h e v a c u u m maintained is 10~3 m m . H g for a period of 12 hours.
B y t h e time t h e vapor pressure is 100 mm., a v a c u u m of 10~5 m m . H g m u s t be used. W h e n t h e vapor pressure is below 1 m m . t h e above conditions are no longer satisfactory. Three to four days at 5 0 0 ° C , and a v a c u u m of 1 0- 6 m m . is absolutely essential. Also, it is necessary to bake out t h e entire system.
8.4. Apparatus Design
There are numerous methods t h a t can be used for pressure determina- tion. T h e exact m e t h o d chosen will depend upon t h e pressures t o be measured. T h e device used should be such, if possible, t h a t each pressure measured is determined t o within 1 %. For pressures between 35 a n d several hundred millimeters of mercury, an ordinary mercury in glass m a n o m e t e r is suitable. For pressures ranging from .2 t o several millimeters, a wide bore mercury m a n o m e t e r m a y still be used if t h e difference in height is measured with a traveling microscope sensitive t o .001 m m . (46). For intermediate pressures mercury manometers of suitable bore, used in conjunction with cathetometers of sufficient accuracy, are suitable. T h e factor t h a t determines the bore of t h e tubing for t h e m a n o m e t e r is t h a t t h e change in capillary depression due to variations in t h e internal diameter be less t h a n the precision desired in t h e measurement of height. For pressures below 0.2 mm., recourse m u s t be h a d to other devices, t h e McLeod a n d Pirani gauges, etc. F o r descriptions of these and other gauges used for these low pressures see Strong (69), Hoag (43), or Ostwald-Luther (61).
T h e methods for t h e determination of t h e a m o u n t of gas adsorbed be made. If this is done, it should be borne in mind t h a t t h e observed result m a y not be the correct result, a n d m a y depend on t h e actual time and t e m p e r a t u r e used.
One other factor determining t h e degree of degassing is the range of pressures of t h e adsorbed gas. T h e lower t h e gas pressures t h e more drastic are t h e conditions needed for degassing. For example, assume t h a t the solid can be heated t o 500°C. for any desired time. If t h e vapor
TABLE II
The Change of Area of a Cracking Catalyst as a Function of Activation Temperature
are of two kinds, gravimetric a n d volumetric. I n the gravimetric methods the gas adsorbed is determined b y t h e change in weight of t h e sample of solid. Two general methods are available, t h e extension of a spring and a beam balance.
The extension-spring method was used b y McBain and B a k r (60).
One end of a quartz spiral spring is a t t a c h e d t o the t o p of a long tube.
T h e solid, in a platinum bucket, is suspended at t h e other end of t h e spring, and its extension is determined with a cathetometer, or traveling microscope. T h e spring is calibrated by placing weights in t h e bucket and measuring t h e extension. For sufficiently high pressures it is also essential to m a k e a buoyancy correction, which depends on t h e actual pressure and molecular weight of t h e gas surrounding t h e system.
This method is useful for high area materials b u t does not possess sufficient sensitivity for determination of solids of low specific area.
Boyd and Livingston (13) applied t h e method to low area materials b u t the precision of t h e a m o u n t adsorbed is not as high as desirable. T h e area of these solids can be conveniently determined with m u c h higher precision b y a volumetric method.
T h e quartz beam balance on the other h a n d can be m a d e as sensitive as desired. W i t h sufficient care t h e balance can be m a d e sensitive t o
1 0- 1 1 t o 10~1 2 g. This method is useful for determinations of t h e lowest
areas. I n theory an isotherm could be determined on a few square centimeters of solid. T h e calibration of t h e quartz beam is essentially the same as t h a t for the quartz spiral. A satisfactory a p p a r a t u s is described b y Barrett et al. (7).
In t h e volumetric methods, the volume and pressure of the gas is determined before a n d after exposure to solid. F r o m t h e thermodynamic behavior of t h e gas, t h e a m o u n t of gas t h a t is adsorbed is calculated from the measured initial and final pressures. This m e t h o d is rapid, and capable of high precision. B y t h e judicious choice of gas, temperature, and a p p a r a t u s design, it is possible t o determine areas of a n y extent.
T h e m e t h o d can be m a d e as precise as desired b y an increase in t h e accuracy of t h e determinations of "both volume a n d pressure.
There are two characteristics of gas behavior which m u s t be con- sidered. M a n y gases cannot be considered ideal and corrections m u s t be m a d e for t h e deviation of t h e gas from t h e perfect state. Since extensive fugacity d a t a are not available for most gases, approximations must be made. T h e method for making these corrections recommended by Giauque (33) is t o use t h e Berthelot equation,
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 269
where t h e critical t e m p e r a t u r e a n d pressure, Tc a n d pc, are the constants in t h e equation.
T h e other property of gases creates a problem only if t h e pressure measuring device and t h e pressure t o be measured are at widely different temperatures. T h e possible effect of t h e thermal transpiration (51) brought on by these conditions m u s t be considered. If t h e pressures are such t h a t t h e m e a n free p a t h is small compared t o t h e diameter of t h e tubing t h e effect can be neglected. If t h e mean free p a t h of t h e gas is long compared t o t h e diameter of t h e tubing, the correction is simple, since t h e pressure desired, ph is related to the measured pressure p2 by t h e formula
where Ti a n d T2 are t h e corresponding absolute temperatures. If the mean free p a t h is of t h e order of t h e diameter of t h e tubing, tedious measurements are necessary t o obtain t h e correct pressure. Thus, t h e t e m p e r a t u r e should be chosen so t h a t all of t h e necessary measurements are m a d e when t h e mean free p a t h is small or large compared to t h e diameter of t h e tubing.
A p p a r a t u s design for volumetric methods depends on t h e area t o be determined. For t h e very smallest areas, t h e a p p a r a t u s described by Wooten a n d Brown (71) or Armbruster a n d Austin (3) is satisfactory.
For areas on t h e order of one square meter, t h e method of Beebe and co-workers (9) is satisfactory. For several square meters t o about 40 m .2, t h e a p p a r a t u s described b y J u r a and Harkins (46) is suitable, while for t h e largest areas, t h e a p p a r a t u s described b y E m m e t t and Brunauer (24) is usable. A modification of t h e latter is described in detail. Needless t o say m a n y special variations a n d modifications are possible which are useful for special needs. For example, if all of t h e solids have approximately t h e same area, then t h e a p p a r a t u s suggested b y Krieger (53) is economical of b o t h space and time.
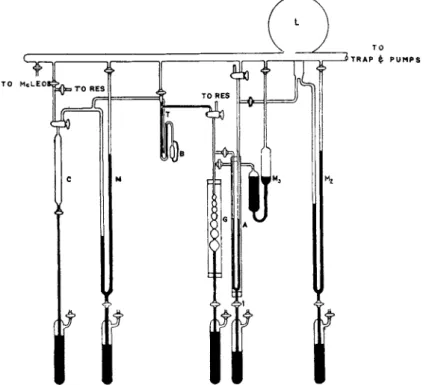
I n t h e following paragraphs a variation of t h e design of Brunauer a n d E m m e t t is illustrated in Fig. 3. T h e a p p a r a t u s has been designed primarily for determinations with nitrogen at — 195.8°C, its normal boiling point. T h e gas reservoirs a n d purification trains are not shown.
T h e t e m p e r a t u r e is determined b y means of a vapor pressure ther
mometer which is composed of t h e compression chamber C, t h e m a n o m eter Μ, and t h e t u b e T, which is next t o t h e adsorption bulb B. After t h e b a t h is placed around t h e adsorption bulb, the gas is a d m i t t e d into t h e evacuated compression chamber t o t h e approximate vapor pressure of t h e gas at t h e b a t h t e m p e r a t u r e . T h e gas is then condensed by filling, (9)
or partially filling the compression chamber with mercury. T h e vapor pressure is read off t h e manometer M.
A n y gas which has an easily measurable pressure at t h e b a t h t e m perature m a y be used. I n t h e event t h a t t h e gas used in t h e ther-
FIG. 3. Apparatus for the determination of adsorption isotherms. This system is designed for work over two ranges: to 1 0 0 mm. and to 7 6 0 mm. For the low pres
sure range the manometer Μ ζ is used. The U tube A and the manometer Μ 2 are removed from the working system by stopcocks. For the higher pressure range manometer M3 is closed out of the system and the U tube A and manometer M>2 used as indicated in the text.
mometer is different from t h a t used in the adsorption the measured vapor pressure of t h e thermometer gas is used t o calculate t h e temperature, from which t h e vapor pressure of t h e adsorbent is calculated. Since for t h e purpose of area determinations, t h e vapor pressure determination is more important t h a n the temperature, it is preferable t o use the adsorbent in t h e thermometer and measure its vapor pressure directly.
T h e b u r e t t e is the series of glass bulbs m a r k e d G. Calibration m a r k s are either cut or etched. T h e volume of each bulb is determined by the
D E T E R M I N A T I O N O F A R E A O F T H E S U R F A C E S O F S O L I D S 271
weight of mercury it holds. Duplicate determinations agree to within 0.005 cc.
T h e pressure in t h e b u r e t t e system is read on manometer M2. T h e pressure read on m a n o m e t e r M2 is t h a t of t h e air in t h e 5-liter flask marked L. T h e pressure in L is m a d e equal to t h a t in t h e b u r e t t e in the following manner.
T h e mercury in t h e cut-οίϊ A is pulled down a n d the cut-off is evacu
ated. After this t h e mercury is allowed t o rise. On the left side the meniscus of t h e mercury is brought exactly t o one of t h e graduation marks. T h e a m o u n t of mercury in t h e cut-off is maintained constant b y closing stopcock 1. As long as the t e m p e r a t u r e remains constant, the pressures on t h e right and left h a n d sides are equal when the mercury is at the chosen graduation mark. W h e n this method is used it is possible t o reproduce pressures to 0.05 m m . with a maximum deviation of 0.10 m m .
This m e t h o d of determining pressures is chosen because t h e small internal diameter of t h e b u r e t t e means t h a t small variations in diameter have a m a r k e d effect on t h e level due t o capillary depression. In a given system t h e difference in height of t h e mercury in t h e two arms with a v a c u u m on b o t h sides is —3 t o about + 3 m m . as the mercury is moved u p . Thus, if it were assumed t h a t t h e pressure on both sides is t h e same when t h e arms are level, an error of as much as 3 m m . might be made. T h e per cent error would of course depend on the absolute value of t h e pressure. Working with nitrogen at —195.8° at a relative pressure of 0.2, t h e error in t h e pressure reading could be as high as 4 % . This error would obviously also be reflected in the calculation of t h e adsorbed volume of gas.
4. L A N G M U I R I S O T H E R M
4.1. Theory
One of t h e earliest a t t e m p t s t o obtain a theoretical relationship between t h e area of a solid and t h e adsorption of a gas on t h e surface is t h a t of Langmuir (55). T h e application of the results of this theory have met with restricted success. T h e fundamental lack of generality of this t r e a t m e n t lies in one of the assumptions of t h e theory, namely, t h a t the adsorption of gases on the surfaces of solids is monomolecular.
Actually, if t h e structure of the solid does not restrict t h e adsorption t o t h e thickness of a single layer, polymolecular films are formed. I n a later section, it is shown t h a t t h e Langmuir theory is a special case of the theory of Brunauer, E m m e t t , and Teller. Although several deriva-
tions of the Langmuir equation have been formulated, the original t r e a t m e n t of Langmuir is followed in this chapter.
T h e t r e a t m e n t of Langmuir represents a simple application of t h e kinetic theory of gases. If μ molecules impinge per second on a surface, some are adsorbed while others are elastically reflected. If a is t h e absorbed fraction of molecules t h a t strike t h e surface, then t h e total number of molecules adsorbed in unit time on unit area is αμ. However, a certain number of molecules per unit area, v, will obtain sufficient energy t o leave t h e surface and re-enter t h e gas phase. When equilib
rium is reached, t h e n u m b e r of molecules leaving t h e surface is t h e same as t h a t being adsorbed. T h u s
αμ = ν (10)
The result expressed b y eq. 10 is completely general, and no assumptions t h a t limit t h e validity of t h e equation have as yet been made. T h e assumptions t h a t limit t h e usefulness of the final result are m a d e in t h e evaluation of t h e quantities in eq. 10 in t e r m s of pressure, p, and t h e fraction of t h e surface, 0, covered b y adsorbed molecules.
B o t h sides of eq. 10 can be evaluated with t h e aid of certain assump
tions. Only those molecules on t h e surface t h a t have an energy equal t o or greater t h a n t h e heat of adsorption, g, can evaporate. If it is assumed t h a t there is no interaction between t h e adsorbed molecules, then t h e only energy change in t h e process is t h a t due to t h e interaction between t h e atoms, ions, a n d / o r molecules of the solid a n d t h e gas.
q is then a constant and is independent of t h e surface concentration of the adsorbed molecules. T h u s if v\ is t h e rate of evaporation where t h e surface is completely covered, t h e n
ν = ΡΙΘ (11)
where θ is t h e fraction of the surface covered by the adsorbed molecules.
Also t h e n u m b e r of molecules on t h e surface t h a t have an energy equal t o a greater t h a n q is proportional t o vxe~q,ltT. T h u s t h e rate of evapora
tion is
Vl = k0e-«/kT (12)
where fc0 is a proportionality constant, k the Boltzmann constant, and Τ t h e absolute temperature. T h e complete expression for ν is,
fcoMie-"'*7, (13)
I n t h e evaluation of μ, the rate at which molecules are adsorbed, another assumption is m a d e : the adsorption is monomolecular, those molecules which strike t h e surface where molecules are present are elastically reflected, and only those which come into contact .with t h e
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 273
bare surface can be adsorbed. From this assumption it follows that,
αμ = αο(1 - θ)μ (14)
F r o m the kinetic theory of gases
μ = , V - (15)
V2TimkT
where ρ is the gas pressure and m the molecular mass. T h u s
«o(i - e)p
\/2*rnkT
E q u a t i n g eqs. (16) and (13) and solving for 0, t h e result is
(16)
9 - i ^ T S1 -f ap ( 1 7 )
where
aoh'kT
k0 \/2%mkT
If vm is t h e number of molecules required to completely cover the surface and ν c m .3 of gas have been adsorbed, then
9 = f (18)
V
T h e substitution of — for θ in eq. (17) gives the final result
- = ϊ - ^ - (19) vm 1 + ap
For t h e purposes of calculation eq. (19) is written
(20)
V aVm Vm
E q u a t i o n (20) is now in a linear form with respect t o t h e variables p/v a n d p. If t h e Langmuir equation is valid, a plot of ρ vs. p/v results in a straight line whose slope is l/vm and whose intercept is l/avm. Assump
tions other t h a n t h e kinetic approach of Langmuir, csCn be used to derive equations which possess t h e same form as t h a t of eq. (19). T h e most i m p o r t a n t of these is t h a t of Fowler, who used t h e methods of statistical mechanics (31).
T h e statistical m e t h o d does have t h e a d v a n t a g e in t h a t t h e assump
tions underlying t h e derivations are clearly defined. I n t h e t r e a t m e n t of Fowler and Guggenheim (32), t h e following assumptions are essential:
(1) t h e adsorption is monomolecular, (2) there is no interaction between
the adsorbed molecules, and (3) the adsorbed molecules have a fixed position on the surface, t h a t is, there is no motion of t h e adsorbed molecules in t h e plane of the surface. T h e third assumption required in t h e statistical t r e a t m e n t does not appear in t h e kinetic derivation.
The equation derived by Fowler is
where
a(T) = — et/KT
I n the definition of a(T), h is Planck's constant, fa(T) is the partition function of t h e molecule in the adsorbed state, fg t h e partition function in the gas state, a n d e is the energy difference of t h e lowest energy states in t h e adsorbed a n d gaseous phases.
Other derivations are those of Volmer (70) a n d Laidler et at. (54).
Volmer obtained his result b y a thermodynamic t r e a t m e n t applied t o an assumption of a Van der Waals t y p e equation of state for t h e adsorbed molecules. Laidler et al. obtained t h e equation from a consideration of the absolute reaction r a t e theory of Eyring (29).
For t h e purpose of area determination, t h e theory of Langmuir, if applicable, yields vm, t h e number of molecules required t o cover t h e surface with a monomoleeular layer of gas. Before t h e area can be obtained it is essential t o determine in some m a n n e r t h e effective cross sectional area, σ, of t h e molecules in t h e adsorbed state. T h e specific area, Σ, then is,
VmNoa
Σ-
( 2 2 )where No is Avogadro's n u m b e r a n d Vm is t h e gaseous molar volume.
Any number of methods can be used t o approximate σ. None of these are really satisfactory. This problem is discussed in detail in a later section.
T h e assumptions m a d e in t h e derivation are rather sweeping, and t h e consequences are a serious limitation on t h e applicability of t h e theory.
T h e assumptions discussed in t h e following paragraphs are those m a d e in t h e statistical t r e a t m e n t of Fowler. T h e most important assumption m a d e is t h a t t h e adsorption is monomoleeular. I t is true t h a t Langmuir in later discussions of t h e theory (56) does m a k e allowance for t h e fact t h a t in special cases t h e adsorbed layer m a y become more t h a n mono- molecular, b u t this does not occur in the t r e a t m e n t until high relative pressures are reached, i.e., for values of p/po near unity. Since in general
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 275
t h e adsorbed films are polymolecular, it is apparent t h a t t h e equation will not be generally valid. T h e only systems known t o t h e a u t h o r for which t h e Langmuir equation holds for solids in which t h e structure does not limit t h e adsorption t o a thickness of a unimolecular layer is for t h e adsorption of η-propyl alcohol on barium sulfate and t i t a n i u m dioxide in t h e form of anatase. These systems were investigated b y Boyd and Livingstone (13). Since, in general, t h e adsorption of gases is polymolecular, t h e Langmuir isotherm does not possess general utility.
Brunauer, E m m e t t , and Teller, have succeeded in removing this restric
tion. This theory is discussed in t h e following section.
T h e second assumption m a d e is t h a t t h e energy of adsorption is independent of t h e concentration of molecules on t h e surface, i.e., there is no contribution t o t h e energy of adsorption due t o t h e interaction of t h e adsorbed molecules. F o r low surface concentrations t h e measured energies of adsorption v a r y greatly with t h e n u m b e r of molecules adsorbed per square centimeter, while a t high surface concentrations there is usually only a small variation in t h e energy of adsorption. Thus, from t h e consideration of t h e energy relations, t h e equation should be valid only at high surface concentrations. Actually this is found t o be true.
When t h e Langmuir equation is valid over an extended region, it is always at high surface concentrations t h a t agreement is obtained between t h e observed pressure-volume relations a n d those demanded b y theory.
One other consequence of t h e assumption t h a t t h e adsorbed molecules do not interact with each other is t h a t t h e theory demands t h a t t h e . volume adsorbed is a continuous function of t h e pressure, a n d also t h a t all of the derivatives of t h e volume with respect t o t h e pressure are continuous. T h e physical consequence of this assumption is t h a t t h e adsorbed phase must be in a single two dimensional phase for a n y value of the pressure. Recent experiments b y the a u t h o r and co-workers show t h a t two dimensional phase changes do exist in films adsorbed on
„the surfaces of solids (49, 50). Theoretically, t h e interaction between t h e adsorbed molecules has been considered b y Fowler and Guggenheim for films of non mobile molecules (32). These isotherms exhibit dis
continuities t h a t are changes of phase. T h e equations, however, are two complicated for ordinary use. This restriction can be removed from the theory; the results are such, however, t h a t nothing is gained for the purpose of area determination.
T h e final assumption, concerning t h e mobility of t h e adsorbed molecules also can be removed. This has been accomplished b y Hill (42). On t h e basis of a simple model Hill has come t o t h e conclusion t h a t in t h e absence of ionic or dipole intermolecular forces t h e molecules are mobile at temperatures above 250°K. and are partially mobile and
partially localized at 77°K., t h e lowest t e m p e r a t u r e normally used in adsorption work. T h e consideration of this effect also has a negligible effect on t h e determination of t h e area.
Of all t h e assumptions m a d e in t h e theory t h e one t h a t seriously affects t h e utility of t h e theory is t h a t which restricts t h e adsorption t o a thickness of a single layer. T h e other assumptions, though unsound, do not cause a serious error.
4.2. Applications of Langmuir Theory
Since t h e Langmuir theory assumes t h a t adsorption is monomolecular, it is evident t h a t t h e theory is applicable only if t h e adsorption is indeed
3 2 k
ρ - mm. H g
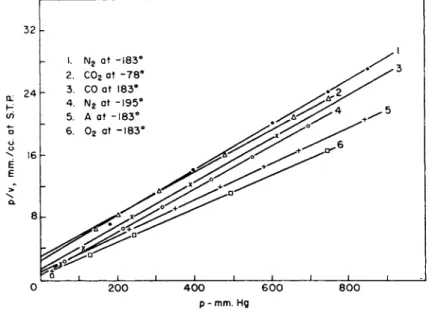
FIG. 4. The adsorption of gases on charcoal. The data are represented by the linear form of the Langmuir equation.
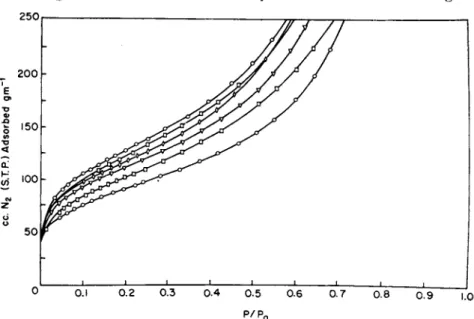
monomolecular or if t h e adsorbed film remains monomolecular t o high pressures. T h e first class contains charcoals and gels whose pore diam
eters are small. I n general, t h e structure of these solids is such t h a t only monomolecular adsorption can occur. At t h e present time knowl
edge of t h e second class is limited t o t h e adsorption of η-propyl alcohol on t i t a n i u m dioxide a n d barium sulfate at room temperatures.
As an example of t h e application of t h e Langmuir equation t o porous solids, Fig. 4 exhibits t h e d a t a for t h e adsorption of several gases on charcoal in t h e linear form of t h e equation. T h e d a t a are those of Brunauer and E m m e t t (17). I t is readily seen t h a t t h e points fall on
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 277 TABLE III
Area of a Charcoal as Determined with Several Gases Gas T°C. cm.3 STP g."Vm 1
m^g."1
N2 - 1 9 5 . 8 1 8 1 . 5 7 9 5
N2 - 1 8 3 1 7 3 . 0 7 9 5
A - 1 9 5 . 8 2 1 5 . 5 8 0 4
A - 1 8 3 2 1 5 . 5 8 3 9
C 02 - 1 8 3 2 3 4 . 6 8 9 4
C 02 - 1 8 3 1 7 9 . 5 8 2 0
C 02 - 7 8 1 8 5 . 5 8 5 3
good straight lines except at low pressures. Table I I I gives t h e values for vm calculated for each isotherm a n d t h e area obtained from vm.
T h e average area from all these determinations is 829 m .2g ._ 1, t h e average deviation 29 m .2g ._ 1 (3.5%), a n d t h e m a x i m u m deviation
FIG. 5. The adsorption of π-propyl alcohol on T i 02 (anatase) at 25°C.
65 m ^ g .- 1 (7.8%). T h e agreement is excellent. There are m a n y char
coals for which such good agreement is not obtained; for m a n y charcoals t h e Langmuir equation does not give agreement over a n y region of pressures. I n these cases, it is not possible t o calculate an " a r e a . "
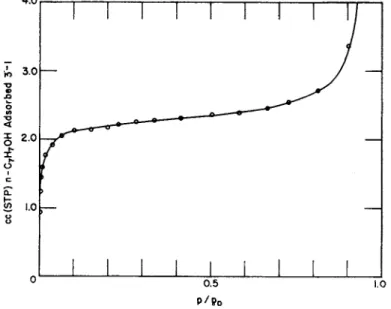
Figure 5 exhibits t h e adsorption isotherm of η-propyl alcohol on a
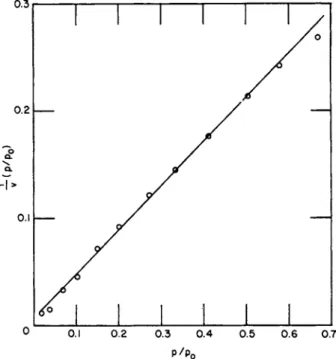
sample of titanium dioxide in t h e form of anatase a t 25°C. Figure 6 shows t h e linear form of t h e Langmuir equation. If 25°A.2 is used as the effective cross-sectional area of t h e η-propyl alcohol molecule t h e area is found t o be 13.6 m .2g ._ 1, which is in good agreement with t h e 13.8 m .2g ._ 1 obtained by other methods for this sample. For t h e com-
FIG. 6. The data of Fig. 5 represented by the linear Langmuir equation.
bination of this gas and solid, the Langmuir isotherm is the only method of obtaining t h e area; all other adsorption theory methods fail.
5. T H E O R Y O F B R U N A U E R , E M M E T T , A N D T E L L E R
5.1. Introduction
The most general method for the determination of the areas of the surfaces of solids is t h a t of Brunauer, E m m e t t , and Teller (18). This theory yields substantially t h e same results as an empirical method developed by Brunauer and E m m e t t (17, 24, 25, 26, 27, 28). Only t h e theoretical approach is discussed. T h e development of Brunauer, E m m e t t , and Teller is a theory of adsorption which a t t e m p t s to explain t h e adsorption characteristics where only Van der Waals' forces are the means of interaction between the solid and gas. T h e results of the theory are useful as a method of area determination because one of the
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 279
parameters in t h e resulting equations is vm, t h e volume of gas required to form a monomolecular layer on t h e surface of t h e solid.
T h e fundamental postulate of t h e theory of Brunauer, E m m e t t , and Teller is t h a t t h e adsorption of gases on a free surface is polymolecular.
Because of t h e complexity of t h e general problem of polymolecular adsorption, a completely general solution is not possible. M a n y equa
tions can be derived which depend upon t h e assumption concerning the energy of interaction of t h e adsorbed molecules with t h e molecules of t h e solid, t h e interaction of t h e adsorbed molecules with themselves, the packing of t h e molecules in t h e various layers, t h e n u m b e r of layers actually formed, etc. T h e fewer t h e assumptions made, t h e more undetermined constants appear in t h e final result.
T h e simplest equation contains two parameters a n d is obtained on t h e assumption t h a t t h e adsorbed molecules do not interact with each other, t h a t t h e net energy of adsorption of t h e second a n d higher layers is zero a n d t h a t an infinite n u m b e r of layers can be adsorbed on t h e surface. T h e equation is
Ρ = C J^1 L + _ L (23)
V(P0 — V) VmC Po VmC
By the proper choice of t h e values of the parameters t h e equation qualita
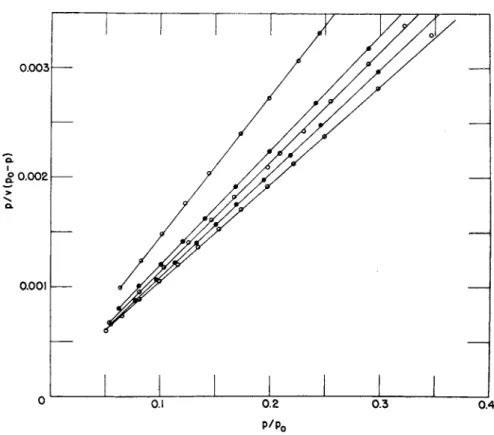
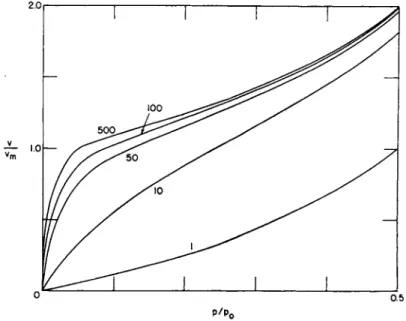
tively reproduces all t h e known isotherms a n d in nearly all cases, better t h a n 99 % q u a n t i t a t i v e agreement can be obtained between t h e observed values a n d eq. (23), in t h e relative pressure region of 0.05 t o 0.30. T h e exact region in which agreement is obtained depends upon t h e system under observation.
T h e application of the theory is sufficiently successful so t h a t it is generally accepted. Recently, Cassie (19), with implausible arguments, derived an equation of exactly t h e same form b y t h e use of statistical mechanics. Hill (41) obtained t h e same result. More recently, Hill (42) has shown t h a t usable results can be obtained even when t h e restric
tion t h a t the adsorbed molecules in the first layer do not interact is removed. T h e statistical thermodynamic t r e a t m e n t s of Cassie and Hill demonstrate beyond question t h e fundamental soundness of the Brunauer, E m m e t t , Teller theory. T h e sole question is, how seriously do t h e assumptions made for mathematical reasons affect t h e value of vm, which is used to calculate t h e area. This question is answerable only by experiment, a n d the experiments indicate t h a t t h e values for vm are substantially correct.
5.2. Theory
Two approaches are possible in the t r e a t m e n t of multimolecular adsorption: (1) a kinetic t r e a t m e n t similar to t h a t used b y Langmuir
in his derivation of monomolecular adsorption and (2) a statistical approach. T h e original kinetic approach of Brunauer, E m m e t t , a n d Teller is reproduced.
If a volume of gas is adsorbed onto t h e surface of a solid, s0 c m .2 of t h e solid will be bare, Si c m .2 will adsorb one layer, s2 c m .2 two layers, Si c m .2 i layers, etc. T h e exact n u m b e r of molecules in each layer will depend on t h e energy of t h e molecule in its particular position. If t h e energy of t h e molecule could be calculated or measured as a function of its position in t h e various layers, t h e Boltzmann distribution function could be used t o determine t h e n u m b e r of molecules t h a t are present in a n y layer when t h e total n u m b e r of adsorbed molecules is known. T h e total area of t h e solid is
(24)
and t h e total number of adsorbed molecules is
t = oo
v = VQ ^ isi (25)
where vo is the volume of gas required to cover t h e surface with a mono- molecular layer of gas.
From eqs. (24) and (25)
l — o o
1 · . 4=°— (26)
i- 0
E q u a t i o n (26) is a completely general expression, a n d is true regard
less of a n y condition or restraint t h a t m a y be placed upon t h e system.
For example, if only η layers can be adsorbed then all Si greater t h a n η are zero. T h e equation, however, is useless until t h e ratio of t h e s u m m a tions can be evaluated. I t is with respect t o t h e evaluation of t h e sums t h a t the various approximations are introduced into t h e theory.
W h e n equilibrium is established between t h e gas on t h e surface of t h e solid a n d t h e vapor, t h e n u m b e r of molecules in each layer m u s t remain constant. However, molecules are always leaving t h e surface, and molecules from t h e gas phase are always entering t h e surface region.
If a molecule evaporates from t h e iih layer, another molecule must condense in t h e ι layer. If t h e same assumptions are m a d e as in t h e
DETERMINATION OF AREA OF THE SURFACES OF SOLIDS 281
dipSo = 61S1I ' RT Et
= 62S2I RT Ει
= 63S3I RT (27)
dipsi-i = biSil RT Ei_
where a» and bi are constants whose form is given in t h e previous section, a n d E, E2} Ez, etc. are t h e energies of adsorption of t h e molecules in t h e respective layers.
If t h e following additional assumptions are m a d e :
E2 = Ez = · · · Ei = · · · = EL (28)
62 _ bz __ bi _ _ a% as α» ^ it becomes possible to express s» in t e r m s of so.
where V = IT peRT
Οι
and SI = cx^o
where c = - χ
and χ = -A=-V_ EL
9eR T
E q u a t i o n (26) becomes
%— 00 cso ^ ix*
i = \
%= CO
So{l + c) ^ xi
» = 1
B o t h of t h e above summations are well known
l = 00
Σ
ι = 00
Xi = 1 - χ
1 = 1
% = 00
ι
IX (1 - *)·«= 1
(29)
2/So (30)
£1
(31)
(32)
(33) derivation of t h e Langmuir equation (see Section 4) then t h e following equations can be w r i t t e n :
Ex