HALOECETSAVAK ÉS FÉM-KELÁT KOMPLEXEK ANALITIKAI ELVÁLASZTÁSA
NAGYHATÉKONYSÁGÚ IONKROMATOGRÁFIÁVAL
Doktori (PhD) értekezés
Készítette
Tófalvi Renáta
okleveles vegyész,
német-kémia szakos középiskolai tanár
Témavezető
Dr. Hajós Péter
egyetemi docens
Készült a Pannon Egyetem
Kémiai és Környezettudományi Doktori Iskolája keretében
Pannon Egyetem Mérnöki Kar
Környezetmérnöki Intézet Analitikai Kémia Intézeti Tanszék
2012.
HALOECETSAVAK ÉS FÉM-KELÁT KOMPLEXEK ANALITIKAI ELVÁLASZTÁSA NAGYHATÉKONYSÁGÚ IONKROMATOGRÁFIÁVAL
Értekezés doktori (PhD) fokozat elnyerése érdekében Írta:
Tófalvi Renáta
Készült a Pannon Egyetem Kémiai és Környezettudományi Doktori Iskolája keretében Témavezető: Dr. Hajós Péter
Elfogadásra javaslom (igen / nem)
(aláírás)**
A jelölt a doktori szigorlaton ...%-ot ért el,
Az értekezést bírálóként elfogadásra javaslom:
Bíráló neve: Dr. Idei Miklós igen /nem
……….
(aláírás) Bíráló neve: Dr. Kertész Vilmos igen /nem
……….
(aláírás) ***Bíráló neve: …... …...) igen /nem
……….
(aláírás) A jelölt az értekezés nyilvános vitáján …...%-ot ért el.
Veszprém, ……….
a Bíráló Bizottság elnöke A doktori (PhD) oklevél minősítése…...
………
Az EDHT elnöke
Megjegyzés: a * közötti részt az egyéni felkészülők, a ** közötti részt a képzésben résztvevők használják, *** esetleges
Kivonat
A szerző a dolgozat első felében az ivóvíz fertőtlenítése során keletkező haloecetsavak (HAA) analitikai elválasztásának kapacitás gradiens optimalizálását végezte el. Az elválasztásokhoz Cryptand A1 típusú makrociklusos állófázist (n-decil-2.2.2.) tartalmazó analitikai oszlopot használt, amelynek ioncsere-kapacitása az elúció során változtatható. Új, nagyhatékonyságú ionkromatográfiás módszert fejlesztett ki kilenc klórozott-brómozott haloecetsav elválasztására. A gradiens módszert elméleti és kísérleti megfontolások alapján optimalizálta. Optimális módszernek a 10mM NaOH-10mM LiOH lépcső gradiens bizonyult, amelyben az eluensváltás az analízis harmadik percében történt. Az optimalizált módszer lehetővé teszi a kilenc haloecetsav és hét lehetséges interferáló szervetlen anion elválasztását kevesebb, mint 25 perc retenciós idő alatt megfelelő felbontással. A módszert alkalmazták csapvíz analízisére, amely során az érzékenység növelése érdekében előzetes dúsítást végeztek fordított fázisú szubsztráton (LiChrolut EN).
A dolgozat második részében az átmenetifémek anioncserés elválasztását tanulmányozta.
A nyommennyiségű fémkomplexek és ligandumaik azonosítása, ill. gyors analízise egyre inkább az analitikusok és a környezetkutatók érdeklődésének középpontjában áll. Ha az oldat a nagy töltésű komplexképző aniont, pl. etilén-diamin-tetraecetsav (EDTA), transz-1,2- diamin-ciklohexán-tetraecetsav (DCTA), feleslegben tartalmazza, akkor a fémionok többsége anionos komplexként van jelen, ezért anioncserével elválaszthatók és vezetőképességi detektálás alkalmazásával a komponensek követhetők. Különböző koncentrációjú és pH értékű karbonát eluenst használt ioncserélő rendszerben, figyelembe véve a mintában jelenlévő komplexképződési mellékreakciókat. A módszer előnyös körülménye, hogy mind a fémkomplexek stabilitása, mind a komponensek elúciója szempontjából ugyanaz a lúgos pH- tartomány a kedvező. Mivel a poliaminokarbonsavak a környezetre potenciális veszélyt jelentenek, fontos, hogy hatékony analitikai technikát fejlesszünk ki nemcsak a fémkomplexek, hanem a feleslegben jelenlévő ligandumok meghatározására is. Ennek a munkának a célja volt az EDTA és DCTA ligandummal komplexált fémionok, ligandumok és szerves anionok szimultán kromatográfiás elválasztásának és azonosításának optimalizálása szuppresszált anioncsere kromatográfiában. Az eluens pH értékeinek és koncentrációinak széles tartományára meghatározta a komplex ionok kapacitási tényezőit és az elválasztások paramétereit. A csúcsok azonosítását ICP-AS és FTIR-ATR vizsgálat is igazolta.
Abstract
„Analytical Separation of Haloacetic Acids and Metal-chelate Complexes by Highperformance Ionchromatography”
In the first part of the thesis the capacity gradient optimize of analytical separation of haloacetic acids was performed. A macrocycle-based adjustable capacity anion-exchange IonPac Cryptand A1 separator column (n-decil-2.2.2.) was used to the separations. A new high performance ion chromatographic method has been developed for the separation of the nine chlorinated–brominated haloacetic acids (HAAs) that are the disinfection by-products of chlorination of drinking water. A gradient method based on theoretical and experimental considerations has been optimized in which 10mM NaOH–10mM LiOH step gradient was performed at the third minute of the analysis. The optimized method allowed us to separate the nine HAAs and seven possibly interfering inorganic anions in less than 25 min retention times with acceptable resolution. Application of this method to the analysis of haloacetic acids in real tap water samples is illustrated. To increase sensitivity, a preconcentration step on a reversed phase substrate (LiChrolut EN) has been coupled.
In the second part of the work the anion exchange separation of transition metals was studied. The trace analysis of metal-complexes and their ligands has long been an area of interest for analytical chemists and environmental researchers. Complexing eluents have been used to improve the selectivity of the chromatographic separation of metal ions. When basic solution contains an excess of a strong complexing anion of high charge such as ethylenediaminetetraacetate (EDTA) and trans-1,2-diamine-cyclohexane-tetraacetate (DCTA) ion, most metal ions will occur as anionic complexes and can be separated by anion exchange using suppressed conductivity detection. The compounds can be followed using this detection system. This study incorporates an ion-exchange system with a carbonate eluent at various concentrations and pH levels. A theoretical framework is developed when complexation effects are present in the sample. An advantageous condition of the method is that the same basic pH-range is favourable to the stability of the metal complexes and also to elution of the components. Because polyaminocarboxylic acids are a potential risk to the environment, it is important also to develop an effective analytical technique for their determination. The aim of this work was the optimization of a simultaneous chromatographic separation and identification of metal ions precomplexed by the ligand EDTA or DCTA and non-metallic species by suppressed anion-exchange chromatography. The capacity factors of complex ions
and the conditions of separations were determined for wide ranges of pH values and eluent concentrations. The identification of the peaks was verified using ICP-AS and FTIR-ATR method, too.
Kurzfassung
„Analytische Trennung der Haloessigsäuren und Metallkomplexen mit Hochleistungender Ionchromatographie”
Im ersten Teil der Arbeit wurde die Optimierung der Kapazität-Gradienttrennung der Haloessigsäuren (HAA) durchgeführt, die während der Desinfektion des Trinkwassers entstehen. Zu den Trennungen wurde eine IonPac Cryptand A1 analytische Kolonne benutzt, die eine makrozyklische stationäre Phase enthält, und die Kapazität der Säule kann geändert werden. Eine neue analytische Methode mit hoher Leistung wurde für die Trennung der neun chlorierten und bromierten Haloessigsäuren entwickelt. Die Gradient-Methode wurde durch theoretische und experimentelle Berücksichtigungen optimiert. Das 10mM NaOH-10mM LiOH Stufegradient erwies sich als optimale Methode, in der die Eluentänderung in dem dritten Minuten der Analyse passiert. Die optimierte Methode ermöglichte die Trennung der neun Haloessigsäuren und der sieben möglichen interferierenden anorganischen Anionen in weniger als 25 Minuten mit geeigneter Auflösung. Diese Methode wurde für die Analyse des Leitungswassers anwendet. Für die Erhöhung der Sensitivität wurde Pre-Anreicherung in Substrat mit reversen Phase (LiChrolut EN) durchgeführt.
Im zweiten Teil der Arbeit wurde die anionenaustauschende Trennung der Übergangselementen studiert, weil die schnelle Analyse der in Spuren vorkommenden Metallkomplexen immer mehr im Mittelpunkt des Interesses der Analytiker und Umweltforscher steht. Wenn die Lösung die komplexbildenden Anionen mit größer Ladung z.B. Äthylendiamintetraessigsäure (EDTA), Trans-1,2-Diaminocyclohexantetraessigsäure (DCTA) im Überschuss beinhaltet, vorhanden die meisten Metallionen als anionische Komplexe, deshalb können sie mit der anionenaustauschenden Trennung mit der Anwendung der suppressierten Leitfähigkeit-Detektierung separieren werden. Die Karbonat-Eluenten mit unterschiedlichen Konzentrationen und pH-Werten wurden im ionenaustauschenden System genutzt, die komplexbildenden Nebenreaktionen in der Probe wurden berücksichtigt. Der vorteilhafte Umstand der Methode ist es, dass das gleiche pH-Bereich sowohl für die Stabilität der Komplexe als auch für die Elution der Komponenten güngstig ist. Weil die Polyaminocarbonsäuren potenzielle Gefahr für die Umwelt bedeuten, ist es wichtig, eine wirksame Technik für die Analyse nicht nur der Metallkomplexe, sondern auch der im Überschuss wesenden Ligands entwickelt zu werden. Die Retention wird durch mehrere Faktoren bei der Separation der Anionen beeinflusst: komplexbildende Reaktionen,
ionenaustauschende Gleichgewichte und Protolyse können auch während der Separation abhängig von pH vorkommen. Das Ziel dieser Arbeit ist die Optimierung der simultanen chromatographischen Trennung und Identifizierung der mit EDTA und DCTA Ligands komplexierten Metallionen, Ligands und der organischen Anionen in suppressierten anionenaustauschenden Chromatographie. Die Kapazitätfaktoren der Komplexionen und die Parameters der Separationen wurden im breiten Bereich der pH-Werten und Konzentrationen des Eluents bestimmt. Die Identifizierung der Peaks werden auch durch ICP-AS und FTIR- ATR Messungen rechtfertigt.
Tartalomjegyzék
Kivonat ... 4
Abstract ... 5
Kurzfassung... 7
Tartalomjegyzék... 9
Bevezetés... 11
1. Irodalmi összefoglaló ... 13
1.1. A folyadékkromatográfia típusai... 13
1.2. A kromatográfia alapfogalmai ... 14
1.3. Ionkromatográfia ... 16
1.3.1. Az ioncsere-kromatográfia elve ... 18
1.3.2. Az ionkromatográfiában használt eluensek és mintakomponensek ... 19
1.3.3. Az ionkromatográfia eszközei... 20
1.4. Az ionkromatográfiában alkalmazható vezetőképességi detektálás ... 22
1.5. Az ionkromatográfiában használt állófázisok fizikai-kémiai szerkezete ... 23
1.5.1. A makrociklusos állófázist tartalmazó oszlop működési elve ... 24
1.6. Az ionkromatográfia alkalmazásai... 26
1.7. A retencióra ható tényezők... 28
1.7.1. A pH szerepe ... 28
1.7.2. Az eluens koncentrációjának hatása... 28
1.7.3. Az állófázis szerepe... 29
1.8. A haloecetsavak jelentősége és analitikai elválasztási lehetőségei ... 30
1.9. Az átmenetifémek és a poliaminokarbonsavak környezeti jelentősége ... 34
1.10. A fém-kelátok kialakulása, jellemzői... 36
1.11. A fém-kelátok elválasztásának mechanizmusa többkomponensű karbonát eluens alkalmazásával ... 39
1.12. A fémkomplexek elválasztásának egyéb (HPLC) módszerei ... 42
1.13. Célkitűzés ... 44
2. Kísérleti rész... 45
2.1. Felhasznált eszközök, anyagok ... 45
2.1.1. Haloecetsavak elválasztása kriptand bázisú oszlopon ... 45
2.1.2. Átmenetifém kelátok elválasztása pellikuláris töltetű oszlopon ... 46
2.2. A haloecetsavak elválasztása ... 47
2.2.1. A haloecetsavak izokratikus elválasztása... 48
2.2.2. A haloecetsavak optimalizált gradiens elúciója ... 59
2.2.3 Analitikai jellemzők ... 65
2.2.4 Az elválasztások alapján tett következtetések... 66
2.3. A fém-kelátok anioncsere-kromatográfiás elválasztása és retenciós jellemzői ... 68
2.3.1. A kromatográfiás rendszer kémiai jellemzői ... 68
2.3.2. A fém-kelát komplexek retenciójának vizsgálata ... 71
2.3.3. Komponens csúcsok azonosítása ... 75
2.3.3.1. Komponens csúcsok azonosítása a retencióprofil alapján ... 75
2.3.3.2. Komponens csúcsok azonosítása a fémion és a ligandum mólarányának változtatása alapján ... 78
2.3.4. A komponenscsúcs-azonosítás helyességének igazolása csatolt analitikai módszerekkel... 85
2.3.4.1. A fémtartalom igazolása ICP atomspektroszkópiás mérésekkel... 85
2.3.4.2. A szabad ligandumok igazolása infravörös spektroszkópiás mérésekkel ... 87
2.3.5. A módszer érzékenységének és linearitásának vizsgálata... 88
2.3.6. Az elválasztások alapján tett következtetések... 90
3. Összefoglalás... 92
4. Függelék ... 94
5. A szerző tudományos munkássága... 97
5.1. Publikációk... 97
5.2. Konferencia előadások ... 97
6. Tézispontok ... 100
6. Theses... 104
7. Az eredmények hasznosítása... 108
Irodalomjegyzék... 109
Köszönetnyilvánítás ... 114
Bevezetés
A természetben kis, esetleg nyomnyi mennyiségben jelenlévő káros hatású anyagok és különböző konfigurációinak, komplexeinek felismerése, mennyiségi kimutatása egyre fokozódó mértékben foglalkoztatja mind a szakértőket, mind a lakosságot. A vizekben előforduló halogénezett szerves anyagok, főleg a haloecetsavak a termékek széles variációit mutatják, amelyeknek biológiai aktivitása, környezetszennyező tulajdonságai is különbözőek.
A haloecetsavakhoz hasonlóan lényeges a természetes vizekben előforduló fémek és poliaminokarbonsavak nyomon követése, mivel vízoldható komplexeket képeznek, miáltal bekerülhetnek az ivóvízcikluson át az emberi szervezetbe is.
Az elmúlt évszázad második felében az elválasztástudományok minden területén jelentős fejlődés volt tapasztalható. A kémiai elválasztások változatossága és bonyolultsága, sebessége, hatékonysága növekedett, a szelektivitás, a felbontás jelentősen javult. A kromatográfia történetében a legjelentősebb áttörésre az intenzív folyadékkromatográfia bevezetésekor került sor a hetvenes években, amellyel az addigi módszerekhez képest sokkal gyorsabban, pontosabban, reprodukálhatóan és szelektíven lehet igen bonyolult elválasztási feladatokat megoldani [1, 2, 3, 4, 5]. Napjainkban a humán-genommal kapcsolatos kutatások, a bioanalitikai módszerek fejlesztése egyre nagyobb követelményeket támasztanak az elválasztástudomány alapkutatásaival szemben. A környezeti kémiai hatások, a természetes és ivóvizek kémiai összetételének fluktuáló változása nagy szelektivitású, gyors működésű vizsgálatokat igényelnek.
A haloecetsavak (HAA) az ivóvíz klóros fertőtlenítésekor keletkező melléktermékek fontos csoportját alkotják. Az utóbbi években egyre inkább felismerik ennek a vegyületcsoportnak az emberi egészségre gyakorolt káros hatásait. A dolgozat első részében bemutatom azt az új, nagyhatékonyságú ionkromatográfiás módszert, amelyet kilenc klórozott, ill. brómozott haloecetsav elválasztására fejlesztettünk ki, a mérésekhez makrociklusos alapú, szabályozható kapacitású anioncserélő elválasztó oszlopot használva.
A fémkomplexek analízise régóta az analitikusok és a környezettel foglalkozó kémikusok érdeklődésének középpontjában áll. A fémek és ligandumaik biológiai és toxikus tulajdonságai bonyolult, sokkomponensű folyamatban alakulnak ki. A dolgozat második felében bemutatandó módszer lehetővé teszi a fém kationok, komplexek és a szervetlen, ill.
szerves anionok szimultán felismerését, elválasztását anioncsere kromatográfiával. A módszer nagy töltésű, erős kelátképző anion használatán alapul. A poliaminokarbonsavak, köztük az
EDTA és a DCTA, a komplexképzés révén mobilizálhatják az átmeneti- és nehézfémeket.
Ezáltal a poliaminokarbonsavak potenciális veszélyt jelentenek a környezetre.
1. Irodalmi összefoglaló
1.1. A folyadékkromatográfia típusai
A folyadékkromatográfiás áramló rendszerekben a különféle kémiai komponensek áthaladva az elválasztó oszlopon a mozgófázissal együtt a detektorba jutnak. A detektor az elválasztás kezdete óta eltelt idő függvényében regisztrálja a koncentrációjukat. Ideális esetben minden komponens Gauss-görbe alakú csúcs formájában lép ki az oszlopból. Minden csúcs az illető anyagra jellemző idő után hagyja el az oszlopot. A minta injektálása és a csúcsmaximum megjelenése között eltelt idő a retenciós idő, jele tR, amely a kémiai komponens szerkezetére jellemző, minőségi információ. Minél nagyobb két anyag retenciós ideje közti különbség, annál könnyebben elválaszthatók egymástól. Minél keskenyebbek a csúcsok, azaz minél kisebb a sávszélesség, annál hatékonyabb az elválasztás.
A kromatográfiás elválasztás a minta, valamint az álló- és mozgófázis molekulái között kialakuló specifikus kölcsönhatások eredményeként jön létre [1]. A különböző állófázisok szelektív kölcsönhatások kiválasztását teszik lehetővé és többféle elválasztás valósítható meg.
A normál fázisú kromatográfiában az állófázis poláris, mozgófázisként apoláris fázist használnak. Ez a rendszer jól használható poláris komponensek elválasztásához, mert azok számára kedvezőbbek a körülmények az állófázisban. A két fázis fel is cserélhető, ekkor a kevésbé poláris fázis lesz az álló-, a polárisabb pedig a mozgófázis. Ezzel az utóbbi, ún.
fordított fázisú (RP) rendszerrel elsősorban apoláris komponenseket tudunk elválasztani.
Manapság ez a domináns LC módszer.
A folyadékkromatográfiás elválasztások során az illékonyság vagy a hőstabilitás nem jelent problémát, tehát ideális módszer a biokémiai szempontból érdekes makromolekulák és ionos anyagok, a bomlékony, természetes eredetű termékek, és egyéb, változatos eredetű és összetételű, nagy molekulatömegű és / vagy kevésbé stabilis vegyületek, pl. fehérjék, nukleinsavak, aminosavak, gyógyszerek, vitaminok, poliszacharidok, poláris lipidek, szteroidok, színezékek, stb. analitikai elválasztására, ill. széleskörű vízanalitikai problémák megoldására [2].
1.2. A kromatográfia alapfogalmai
Egy komponens visszatartásának mértéke jellemző az adott vegyületre, így annak minőségi azonosítására is szolgál. A legegyszerűbb módszer a retenció, azaz visszatartás számszerűsítésére az injektálás kezdete és az adott komponens detektorban történő megjelenése közti idő mérése, amit retenciós időnek (tR) hívunk [6]. A retenciós idő arányos a komponens visszatartásával és fordítottan arányos az eluens térfogatáramával.
A VR retenciós térfogat az az eluenstérfogat, amely az X komponens csúcs- középpontjának eluálásához szükséges. Ez a minta injektálása és a csúcsmaximum detektorban való megjelenése között átáramló folyadék térfogata [7]. A retenciós térfogat két részből tevődik össze:
- Holttérfogat (V0): az az eluens térfogat, amely az idő alatt áramlik keresztül az oszlopon, amíg a mintakomponens a mozgófázisban tartózkodik, értéke minden mintakomponensre nézve állandó.
- Korrigált retenciós térfogat: az a mozgófázis térfogat, amely az idő alatt áramlik keresztül az oszlopon, amíg a mintakomponens az állófázison tartózkodik. Értéke minden mintakomponensre más.
A retenciót univerzálisan jellemzi a retenciós tényező (k’), amely a komponens álló- és mozgófázisbeli mennyiségének hányadosa és megadható a retenciós idő és a holtidő segítségével [6, 8]:
0
' 0
t t t n
k n R
m
s = −
= /1.1./
ahol ns a vizsgált komponens állófázison, nm a mozgófázisban lévő móljainak száma, t0 a holtidő. Adott oszlop, eluens, elválasztási hőmérséklet és X komponens esetén, kis mintamennyiséget alkalmazva k’ állandó, ezáltal lehetővé teszi az adott mintakomponens minőségi azonosítását.
Az elválasztó oszlopban a komponensek vándorlási sebessége azok álló- és mozgófázis közötti egyensúlyi megoszlásától függ.
dx
dv M DM
= 1
/1.2./
ahol dx az állófázis infinitezimálisan kis mennyisége, amelyen a megoszló anyag dv ml fluid fázis bejuttatásának hatására előrehalad, DM a megoszlási hányados. Az elválasztandó
komponensek oldódnak a mozgófázisban, ill. megkötődnek az állófázison. Ezt a folyamatot a megoszlási hányadossal jellemezzük.
D M
M = (M)
[ ] /1.3./
ahol (M) a minta állófázisbeli, [M] pedig a mozgófázisbeli koncentrációja. DM értéke a két fázis kémiai tulajdonságaitól függ, így általában egytől különböző értékeket vesz fel. DM
nagy, ha nagy az ion affinitása az állófázishoz, és kicsi, ha az inkább a mozgó fázisban tartózkodik. A retenciós viselkedés leírására a gyakorlatban alkalmasabb a már ismertetett retenciós tényező, k’:
0 0 0
' t
t t V D V
k = M álló = R − /1.4./
Az elválasztás hatékonysága a komponens retenciójától és kromatográfiás csúcsának szélességétől függ. Egy mintakomponens retenciója a molekula és az állófázis közötti kölcsönhatás erősségére, valamint az állófázis fajlagos felületére utal. A komponens sávjának szélesedése ellenben alapvetően kinetika által szabályozott, és az állófázis részecskéinek átmérőjétől, porozitásától, pórusméretétől és az oszlop méretétől függ. A kromatográfiás oszlop hatékonyságára jellemző fontos mérőszám az elméleti tányérszám, amely a kromatográfia tányérelméletéből eredeztethető [9]. Értéke a következő összefüggés segítségével számítható:
2
16 tR
N w
=
/1.5./
ahol tR a retenciós idő és w a csúcs bázisszélessége.
Minél nagyobb egy elválasztó rendszer hatékonysága, annál keskenyebb csúcsokat kaphatunk adott retenciós idő esetén, azaz annál több komponens kromatográfiás csúcsát tudjuk megkülönböztetni egy adott retenciósidő-tartományban.
Két különböző komponens szelektivitásán azok retenciós tényezőinek vagy korrigált retenciós időinek hányadosát értjük [2, 10]. A szelektivitás egy kromatográfiás rendszer elválasztási erejét mutatja meg adott komponensekre vonatkozóan. Értéke nagyobb 1-nél, mivel mindig a nagyobb retenciójú komponens retenciós tényezőjét osztjuk a kisebbével:
' '
B B
A A
k t
k t
α = = /1.6./
Ez a paraméter független az oszlop hatékonyságától, értéke csak az elválasztott komponensek természetétől, az eluens fajtájától és összetételétől, valamint az állófázis tulajdonságaitól függ.
Ha két komponens szelektivitása 1, akkor az adott kromatográfiás rendszerben nincs mód azok elválasztására.
A komponensek kielégítő mértékű elválasztásához megfelelő felbontást kell biztosítani.
Két szomszédos A és B sáv felbontása definíciószerűen a két sáv középpontja közti távolság és az átlagos sávszélesség hányadosával egyenlő [6]:
( )
2 RA RB
S
wA wB
t t
R t t
= −
− /1.7./
ahol tRA és tRB az A és B sáv tR értéke, twA és twB pedig a sávok alaplapi szélessége ugyanabban az időegységben, mint a tR értékek. Ha RS = 1, akkor a két sáv jól elválik egymástól, maximum 2 %-ban fednek át. Nagyobb felbontás érték jobb, kisebb kevésbé hatékony elválasztást jelent.
1.1. ábra. Az elválasztás mechanizmusa a kromatográfiában
1.3. Ionkromatográfia
Az ionkromatográfiát (IC) mint új módszert 1975-ben vezette be Small, Stevens és Baumann. Az ionkromatográfia a folyadékkromatográfia egy kiemelt részterülete. E módszernek három típusát ismerjük:
- Ioncsere-kromatográfia (HPIC: High Performance Ion-Exchange Chromatography): Ez az elválasztási eljárás az állófázison kötött ionos csoportok és a mozgófázis közti ioncsere- folyamaton alapul. Az állófázis polisztirol-gyanta, amelyet divinil-benzollal kopolimerizáltak.
Az ioncsere-kromatográfia felhasználható szervetlen és szerves anionok és kationok elválasztására. Az anion-kromatográfiában a funkciós csoport többnyire kvaterner ammóniumion, a kation-kromatográfiában pedig szulfonátcsoport. Az ioncsere- kromatográfiát nevezik a gyakorlatban ionkromatográfiának [3]. Az atomenergia-ipar kifejlődésében fontos része volt az ioncserés kromatográfiának a hasadási termékek és a ritkaföldfémek elválasztása révén. Ioncsere kromatográfiával határozzák meg a fehérjék összetételét is. Az aminosavszekvencia-analízis az 50-es évek végén szelektív HPIC elválasztásokkal vált lehetővé.
- Ionkizárásos kromatográfia (HPICE: High Performance Ion Chromatography Exclusion):
Az elválasztás mechanizmusát a Donnan-kizárás, a sztérikus kizárás és az adszorpció határozza meg. Az elválasztáshoz nagy kapacitású teljesen szulfonált kationcserélőt használnak polisztirol-divinil-benzol bázison. Az ionkizárásos kromatográfia elsősorban gyenge szerves savak teljesen disszociált erős ásványi savaktól való elválasztására alkalmas, valamint aminosavak, karbonát és borát meghatározására. Ezt a módszert alkalmazzák az élelmiszeriparban, borászatban és tartósítóiparban, valamint ily módon végzik a vér analízisét is. Az ionkizárásos kromatográfia elsősorban alifás és aromás savak (ecetsav, citromsav, ftálsav, laktát, piruvát) elválasztására alkalmas. Segítségével a di- vagy trikarboxilsavak is elemezhetők. Lényeges szempont az elválasztandó anyagok kiválasztásánál, hogy a pK jól megkülönböztethető legyen az erős ásványi savak pK-jától.
- Ionpár-kromatográfia (MPIC: Mobile Phase Ion Chromatography): A domináló mechanizmus az adszorpció. Az MPIC segítségével elválaszthatók a fémkomplexek, valamint a felületaktív anionok és kationok. Az eluenshez a szervetlen és szerves moderátorok mellett ionpár-reagenst is kell adni. Az állófázis semleges makropórusú polisztirol / divinil-benzol gyanta, amely apoláris. Ionpár-kromatográfiával neutrális és ionos komponensek egyidejűleg elválaszthatók egymástól ionpárképzés révén. A pH és a párképző típusa nagymértékben befolyásolja a párképzést.
Az ionkromatográfia kitűnik a korábbi analitikai módszerek közül szelektivitása és a detektálás érzékenysége révén. A módszer sokoldalúságát felhasználási lehetőségeinek nagy száma igazolja: vízvizsgálatok, félvezetőipar, galvánelemek gyártása, gyógyászat, mezőgazdaság, erőművek kémiája, gyógyszerészet, papíripar, bányászat és fémfeldolgozás, élelmiszeripar, környezetvédelem [4]. Különös jelentőségre tett szert az ionkromatográfia a
vízben található anionok mennyiségi analízisének területén [5]. A különféle ionok rövid időn belül, szimultán meghatározhatók.
Az ionkromatográfia céljaira intenzív, reprodukálható ioncserélő gyantát fejlesztettek ki.
Az injektálási térfogatot 10-100 µl-re csökkentették. A legjelentősebb újítás a szuppresszált elektrokémiai detektálás volt, amely lehetővé tette a kromatográfiás jelek érzékeny feldolgozását. A vezetőképességen alapuló detektálás bevezetésével egyre nagyobb alkalmazási teret nyert az ionkromatográfia.
1.3.1. Az ioncsere-kromatográfia elve
Elúciós oszlopkromatográfiás elválasztás esetén kis mennyiségű mintát juttatunk az elválasztó oszlopra, amelyben az ioncserélő tulajdonságú állófázis található. Az oszlop irányába áramoltatjuk a mozgófázist, amely az ellenionokat tartalmazza. Az ioncserélő gyantán töltéssel rendelkező funkciós csoportok találhatók, amelyek kívülről elektromosan semlegesek az ellenionok miatt. Az elúció folyamán a funkciós csoport ellenionja kicserélődik a mintaionra, ezáltal a gyanta visszatartja az illető iont. A minta komponensei az állófázishoz való különböző affinitásuk alapján választhatók el egymástól.
Anioncsere esetén a következő reakció játszódik le a gyantán:
– x y
– y
x E A R -A E
yR − +x ↔x + y /1.8./
Kationcsere esetén:
+
+ ↔ +
+xKy xR -K yEx E
-
yRx y /1.9./
ahol A és K a minta anion, ill. kation, E az eluensben levő ion, R az ioncserélő töltéssel rendelkező funkciós csoportja. Az anioncserélő kromatográfiában a vizsgált Ay−-ion verseng a mozgófázisban levő Ex−-ionnal az ioncserélő gyantán levő R kationos csoportért.
Kationcserés kromatográfia esetén a minta Ky+-kationjai versengenek az eluens Ex+- ionjaival az ioncserélő gyanta Ranionos csoportjaiért. Az elválasztásnak az az alapja, hogy az így kialakuló ionpár-kölcsönhatások különböző erősségűek. A gyantával gyenge kölcsönhatásba lépő ionoknak kicsi a retenciója, ezért ezek az ionok a kromatogram elején jelennek meg. A gyantával erős kölcsönhatásba lépő ionok retenciója nagyobb, ezért csak később eluálódnak (1.1. ábra). Az ioncserélő gyantán néhány mikron vastagságban vannak az aktív csoportok. A réteg vastagságának növekedésével nő az oszlop kapacitása, és ezáltal a retenció.
1.3.2. Az ionkromatográfiában használt eluensek és mintakomponensek Az eluens kiválasztása döntően az elvégzendő elemzés fajtájától függ. Az eluens állófázishoz való affinitása hasonló kell legyen a mintaion affinitásához. A minta molekuláinak retenciója erősen függ a mozgófázisban levő ion típusától, mert a különböző ellenionok különböző mértékben lépnek kölcsönhatásba a gyantafázissal. Az eluens koncentrációja a legfontosabb retenciót meghatározó paraméterek közé tartozik, növelése gyorsítja a retenciót. A mozgófázis növekvő sókoncentrációjával csökken a komponensek retenciója, mivel a minta ionjai így kevésbé tudnak versenyre kelni az ioncserélő funkciós csoportjaiért. A szelektivitás pH-függő, de ennek mértékét nehéz előre megállapítani. Az eluens összetételének változtatásával az elválasztás szelektivitása is változtatható [2]. Az eluens átfolyási sebessége és a retenciós idő között fordított arányosság van.
Ha az eluensnek nagy a vezetőképessége, akkor részt kell vennie a szuppresszorban egy reakcióban, amely során csekély vezetőképességű anyag keletkezik. Erre a reakcióra nincs szükség, ha az eluensnek olyan kicsi a vezetőképessége, hogy az amúgy is érzékeny vezetőképességi detektálást tesz lehetővé [3].
Ioncsere-kromatográfiával savak és bázisok egyaránt elválaszthatók. A HA gyenge sav disszociál:
A–
H
HA↔ ++ /1.10./
A disszociációfok az oldat pH-jának változtatásával befolyásolható. A legtöbb ioncserés elválasztást vizes közegben végzik, mert a víz oldó és ionizáló képessége nagyon jó.
Az 1.1. táblázat bemutatja, hogy adott mintaion csoportokat milyen kísérleti körülmények mellett célszerű elválasztani, detektálni.
1.1. táblázat: Főbb ionkromatográfiás módszerek
Minta / Irodalom Eluens Detektor Elválasztó
oszlop Szervetlen anion
(Cl–,Br–,NO−3,SO24−) [3]
Na2CO3
elektromos
vezetőképességi anioncserélő Szerves anion
(formiát, acetát, laktát, oxalát) [18]
NaOH elektromos
vezetőképességi anioncserélő Szervetlen kation
(alkáli, alkáliföldfém) [10]
aminosav + HNO3 elektromos vezetőképességi
felületi kationcserélő Szerves kationok
aminszármazékok, (metil, etil, amin) [11]
aminosav + HNO3
elektromos vezetőképességi
felületi kationcserélő
1.3.3. Az ionkromatográfia eszközei
A mozgófázist az egész kromatográfiás rendszerben egy kettősdugattyús pumpa mozgatja, amely állandó áramlási sebességet biztosít. A mintát mikrofecskendővel visszük a rendszerbe.
A kromatográfiás készülékben található mintabemérő hurok (loop) állandó térfogatának köszönhetően igen kis térfogatú minták is reprodukálhatóan injektálhatók, miközben igen kis sávszélesedéssel kell számolni. A mintabemérő hurok használata egyszerű, kényelmes, olcsó.
A kromatográf legfontosabb része az elválasztó oszlop. A megfelelő állófázis és a megfelelő kromatográfiás feltételek kiválasztása meghatározza az analízis minőségét. Az előkolonna (guard) azt a célt szolgálja, hogy megvédje a szennyeződésektől az analitikai oszlopot, amelyben a töltéssel rendelkező funkciós csoportokat tartalmazó ioncserélő gyanta található.
A detektor a vizsgált minta minőségi és mennyiségi kimutatására szolgál. Az ionkromatográfiában leggyakrabban elektromos vezetőképességi detektorokat használnak. A vezetőképességet váltóáramú ellenállásmérésre vezetjük vissza. A detektorban annál nagyobb a jel, minél nagyobb különbség van a minta és az eluens vezetőképessége között, azaz minél nagyobb a jel-zaj viszony. A detektor elé helyezett szuppresszornak az a szerepe, hogy az ionok elúciójához használt elektrolit vezetőképességét kémiailag csökkentse. Tehát a szuppresszor elnyomja az eluens vezetőképességét, ezáltal növeli a jel-zaj viszonyt és ezzel összefüggésben a detektor érzékenységét is. Lúg eluens (pl. NaOH) esetén szükséges a szuppresszálás, mivel a lúgnak nagy a vezetése. Ekkor a szuppresszorban ioncserélt víz keletkezik, amely gyakorlatilag nem vezet, így a detektálás nagyon érzékeny lesz.
R-H+ + NaOH → R-Na+ + H2O /1.11./
Ebben az esetben a szuppresszor kationcserélő kapilláris cső, amelyben az eluens Na+- ionjai H+-ionokra cserélődnek a reakció során. Az ioncserélő membrán regenerálása elektrokémiai úton vagy kénsavval történik, amelynek protonjai biztosítják, hogy H+ -ionok legyenek a membrán felületén.
A kromatográfiás jeleket számítógép rögzíti. Minőségi eredményeket kapunk a csúcs helye alapján és mennyiségi eredményeket a csúcsterület kiértékelésével. Ehhez a digitális integrátorok nyújtanak segítséget.
1.4. Az ionkromatográfiában alkalmazható vezet ő képességi detektálás
Az ionos komponensek elektromos vezetőképességük alapján detektálhatók. A vezetőképességi detektálás, amely egy időben viszonylag mellőzött volt, ma az ionok kromatográfiás analízisében a legfőbb detektálási módszerek egyike. Két fontos előnye van:
minden ion elektromos vezető, ezért ez a detektálási mód általánosan használható, a másik szempont, hogy viszonylag egyszerű szerkezetű és működésű. A vezetőképességi detektor részei: a cella, amely tartalmazza a mikro-Pt-elektródokat, egy kimenetijel-mérő és a szükséges elektronika, amely méri a vezetőképességet és módosítja az érzékenység szabályozását. A detektor érzékenysége előre megbecsülhető, ha ismerjük a vezetőképességi adatokat. A detektor a koncentrációval arányos jelet ad [2]. A cella holttérfogata kisebb, mint 2 µl. Az oldatban változik az ionok mozgékonysága az oldat hőmérsékletével, ezért azt gondosan termosztálni kell [5].
Az analitikai eljárás érzékenysége azt a jelváltozást jelenti, amely a vizsgált minta egy alkotója koncentráció- vagy tömegegységnyi megváltoztatásának hatására bekövetkezik. Ez tulajdonképpen az analitikai mérőgörbe meredeksége. Definíció szerint:
érzékenység= ∆
∆ x
C /1.12./
ahol ∆x a műszerrel megállapított mérőszám (kimenő jel) megváltozása ∆C koncentrációváltozás közben. Az analitikai rendszer érzékenysége a részrendszerek érzékenységének szorzata [14].
T. Okada és T. Kuwamoto szuppresszált rendszerben egyezést talált a mért és a fajlagos vezetőképesség alapján számított érzékenység között [15]. Másrészt Gjerde és munkatársai arról számoltak be, hogy az anionok detektálhatók olyan vezetőképességi detektorral, amely közvetlenül kapcsolódik az elválasztóoszlophoz, amelyben kis vezetőképességű eluens van [16]. Viszont ennek az oszloprendszernek általában kisebb az érzékenysége, mint annak, amelyik szuppresszáló rendszerrel van ellátva.
1.5. Az ionkromatográfiában használt állófázisok fizikai-kémiai szerkezete
Az ionkromatográfiában elsősorban szerves polimer, pl. sztirol-divinil-benzol alapú állófázisok terjedtek el, de szervetlen alapú tölteteket tartalmazó oszlopok is használatosak [17]. Az anion-kromatográfiában gyakran alkalmaznak latex alapú anioncserélőt, amely szerves polimer alapú és pellikuláris szerkezetű, felépítése az 1.3. ábrán látható.
1.3. ábra. Latex alapú anioncserélő állófázis felépítése
A latex alapú anioncserélő 5-25 µm átmérőjű felületileg szulfonált sztirol-divinil-benzol kopolimerből és az erre felvitt, elektrosztatikusan vagy van der Waals kölcsönhatással rögzített teljesen aminált pórusos anioncserélő gyöngyökből áll, melyek polivinil-kloridból vagy polimetakrilátból készülnek Az utóbbi ún. latex részecskék átmérője kb. 0,1 µm.
Mindezek alapján a latex alapú állófázis három régióra különíthető el:
• egy inert és mechanikailag ellenálló hordozóra
• egy vékony szulfonsav borításra a hordozó felületén
• egy külső aminált latex rétegre, mely az anioncserélő csoportokat hordozza [3]
Egy ilyen ioncserélő pásztázó elektron mikroszkóppal készített felvétele az 1.4. ábrán látható. A latex réteg önmagában véve nagy ioncsere kapacitással rendelkezik, a kisméretű gyöngyök mégis kis ioncsere kapacitású állófázist eredményeznek. A latex alapú állófázisok
kémiailag nagyon stabilak, még 4 mol/l koncentrációjú NaOH sem képes megbontani a szulfonált felület és a latex részecske közti kötést.
1.4. ábra. Latex alapú állófázis pásztázó elektronmikroszkópos felvétele [3]
Ezen anioncserélők pellikuláris struktúrája felelős a nagy kromatográfiás hatásfokért. A latex-agglomerált anioncserélők szelektivitása megváltoztatható a kvaterner ammónium-bázis kémiai természetének változtatásával. Mivel a latex anyagot külön lépésben szintetizálják, lehetővé válik az elválasztó oszlop szelektivitásának optimalizálása speciális analitikai problémára vagy a latex gyöngyökhöz kötött funkciós csoportok, vagy a térhálósság mértékének változtatásával.
1.5.1. A makrociklusos állófázist tartalmazó oszlop működési elve
Anionok elválasztására töltéssel nem rendelkező makrociklikus vegyületek is alkalmasak.
Kifejlesztettek egy kriptand-bázisú anioncserélőt (IonPac Cryptand A1) [16, 18], amelyben a kapacitás, és kisebb mértékben a szelektivitás egyszerűen szabályozható a mozgófázis megválasztásával.
A makrociklusos ligandumos anioncserélő anioncsere képessége a fém kationok komplexképzésének köszönhető a makrocikluson belül. Amint egy fém kation komplexálódik, kialakul egy pozitív töltésű funkciós csoport. A makrociklus kation-megkötő képességének köszönhetően a pozitív töltésű fém-kriptand komplexek anioncserélő helyekként viselkednek. Az 1.5. ábrán látható az n-decil-2,2,2-kriptand molekula, amely az alkilcsoportján keresztül kovalensen köthető a sztirol-divinil-benzol gyantára.
1.5. ábra. Makrociklikus állófázis (n-decil-2.2.2-kriptand molekula)
A makrociklikus vegyületek, mint a koronaéterek, kriptandok, kalixarének, jellemző tulajdonsága, hogy képesek fémionokat szelektíven megkötni. Ionkromatográfiás állófázisként szóba jöhetnek egyrészt kationok elválasztása esetén, ahol a makrociklikus vegyületek említett tulajdonságának köszönhetően az eltérő átmérőjű fémek eltérő mértékben szenvednek visszatartást az oszlopban, így azok ligandcserés mechanizmussal elválaszthatók egymástól, másrészt alkáli hidroxid (LiOH, NaOH, KOH) eluenst használva pozitív töltésű anioncserélő helyek alakulnak ki az oszlopban, mivel a mozgófázis fémionja komplexet képez a makrociklikus vegyülettel, amelyen a mintaionok ioncserés mechanizmussal elválaszthatók egymástól. A koronaéterek felhasználhatók a szelektivitás szabályozásában a kationelválasztások során, azonban csak korlátozottan használhatók anioncserélőként [19]. A kriptandokat, annak köszönhetően, hogy sokkal erősebben képesek bizonyos kationokat képezni, messzemenően alkalmasnak találták az anioncserélő elválasztásokhoz.
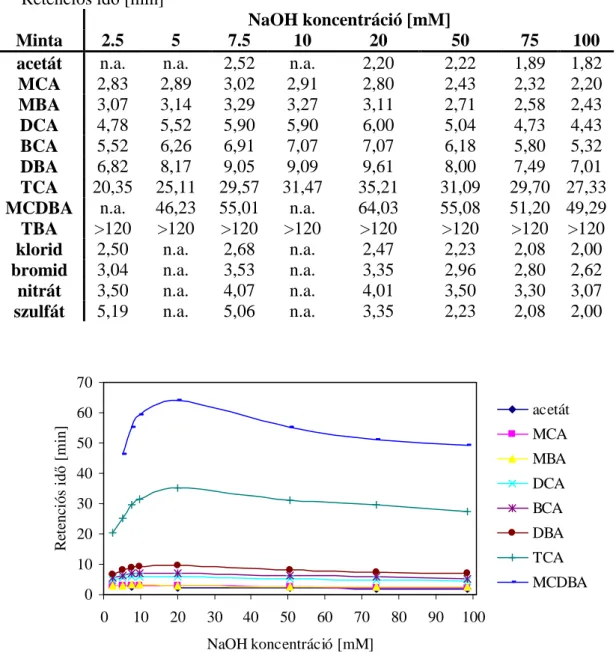
A makrociklikus vegyületek anionkromatográfiás alkalmazásának legfőbb előnye, hogy az alkáli-hidroxid eluens típusának megváltoztatásával befolyásolható az állófázis ioncsere- kapacitása a fémionok eltérő mértékű megkötődésének köszönhetően. Továbbá az elúció közben az eluens típusának váltásával ún. kapacitás gradiens érhető el, mellyel lehetőség nyílik a jelentősen és kevésbé visszatartott komponensek szimultán elválasztására. A mozgófázis ionkoncentrációját állandónak tartva, de kationjának típusát változtatva az oszlop kapacitása változtatható az elúció során. A kapacitás egyenes arányban van a komplexált kation képződési állandójával. Az elválasztást nátrium-hidroxiddal kezdik (mérsékelt kapacitású mód), majd lítium-hidroxid következik (alacsony kapacitású mód). Lítium- hidroxid eluensnél kicsi az anionok retenciója, mivel a lítiumnak nagyon kicsi a kötési állandója, kevesebb, mint 1,0. Amikor nátrium-hidroxid a mozgó fázis, nagyobb kapacitás észlelhető a nátrium nagyobb kötési állandójának köszönhetően (log K = 3,9). Mivel a káliumnak a legnagyobb a relatív kötési állandója (log K = 5,4), az elválasztási idők ennek megfelelően nőnek. A kapacitás-gradiens használatakor nő az elválasztás hatékonysága, az
erősen visszatartott anionok rövidebb idő alatt eluálhatók alacsony eluenskoncentrációkkal, ezáltal a háttérvezetés alacsony marad. Mivel az eluens ionkoncentrációja nem változik, kicsi az alapvonal-torzulás az elúció folyamán. A rendszernek van holttérfogata, így a kapacitás csökkenése csak a holtidő után figyelhető meg. Ezenkívül az oszlopnak is van holttérfogata és az eluensnek időre van szüksége, hogy elérje az oszlop távolabbi részeit. Ezeknek az okoknak köszönhetően minél nagyobb a minták retenciója, annál nagyobb a retenciójuk csökkenése az oszlop kapacitáscsökkenése miatt. Ezidáig a kriptand-bázisú oszlop alkalmazásai szervetlen anionokra korlátozódtak.
A makrociklusos kriptand fázis használata haloecetsavak ionkromatográfiájában számos előnyös tulajdonságot kínál. Két lehetőség van a retenció kontrolljára és szabályozására: az állófázis kapacitása és az eluens erőssége. A makrociklus alapú fázisokon különböző szelektivitások érhetők el, míg a tradicionális fixált ioncserélő helyek és alkanol kvaterner ammónium csoportok használatával a HAA-k elválasztásának szelektivitása ily módon nem változtatható. A kriptand oszlop lehetővé teszi az oszlopkapacitás szelektivitási kontrollját egyszerűen az eluens kationjának változtatásával. A szelektivitás javítására szolgáló megfelelő gradiens használatával az erősen visszatartott polarizálható anionok (tioszulfát, jodid, tiocianát, perklorát) és az egyszerű anionok szimultán elválaszthatók [18]. Ez a technika lehetővé teszi a többértékű anionok elválasztását is, mint pl. az oligoszacharidok, a mozgó fázis viszonylag alacsony koncentrációja mellett. Jó elválasztási profilt figyeltek meg több, mint 25 polifoszfát mintára és komplex mátrixokra is, pl. tömény savakra.
Konvencionális anioncserélő fázisok használatakor a hasonló szelektivitású eredmények elérése időigényes, többszörös gradiens elúciót és költséges eszközöket igénylő folyamat.
1.6. Az ionkromatográfia alkalmazásai
Az ionkromatográfia kitűnik szelektivitásával és érzékenységével a szervetlen és szerves anionok és kationok analízisében [3]. A felhasználási lehetőségek nagy száma bizonyítja ennek a módszernek a sokoldalúságát és fejlesztési irányait.
Különös jelentőséget ért el az ionkromatográfia a vizekben lévő anionok mennyiségi analízisének területén. Így kevesebb mint 10 perc alatt szimultán meghatározhatók a klorid, foszfát, nitrát, nitrit és szulfát ionok, amelyek koncentrációjától alapvetően függ a víz minősége. Az ionkromatográfia előnyei az érzékeny detektálás, a gyors elválasztás [20].
Azoknál a mintáknál, amelyek részlegesen protonálódnak, figyelembe kell venni a különböző mintaoldatok móltörtjeit is.
Mind a karbonsavak, mind a karbonsavak sói széles körben használatosak a vegyiparban és kereskedelmi termékekben [21]. Az ionkromatográfiát sikeresen alkalmazzák karbonsavak analízisére különböző élelmiszermintákban pl. juice, bor, tej rutinanalízisére. A kis móltömegű karbonsavak nagyon fontos szerepet töltenek be, mivel közreműködnek az íz kialakításában és a termék stabilitásában. Az ionkromatográfiát előszeretettel használják a klinikai kémiában, mert gyors analízist tesz lehetővé és minimális mintaelőkészítés szükséges hozzá pl. a vizeletben lévő oxalát meghatározása, humán plazma analízise.
A karbonsavakat általában három különböző folyadékkromatográfiás módszerrel választják el: anioncsere, ion-kizárásos és fordított fázisú kromatográfia. [22] A nagyhatékonyságú szuppresszált ionkromatográfia használata alifás karbonsavak elválasztására vonzó és életképes módszerré vált az utóbbi években [23]. Széleskörűen használják számos elméleti és gyakorlati kérdés megválaszolására, alkalmazza a környezeti kémia, élelmiszerkémia és a gyógyszeripar. Az ionkizárásos technika különösen alkalmas az ioncsere-kromatográfia kiegészítésére, mivel az ezekkel a módszerekkel kapott szelektivitások meglehetősen különbözőek. Az erős szervetlen sav anionok a Donnan elvnek megfelelően egy egyszerű csúcsban záródnak ki a gyantafázisból és a holttérfogatnál eluálódnak. Gyengébb és protonált minták, amelyek főként molekuláris formában léteznek, visszatartódnak az állófázison az ionkizárás és a hidrofób kölcsönhatások kombinációjaként.
A legújabb fejlesztések azt mutatják, hogy a gradiens elúciós ionkromatográfia kiszorítja az ionkizárásos kromatográfiát a szerves és szervetlen minták esetében. A fordított fázisú kromatográfiával sikeresen elválaszthatók a nukleinsavak (Horváth Csaba 1965-1966) [8], a biopolimerek és egyéb, biológiailag fontos molekulák. Nagy hatékonyság és szelektivitás, valamint az egyensúly gyors beállása és rövid analízisidők jellemzők erre a módszerre.
Azonban az alkalmazható pH-tartomány 2 – 8 közöttire korlátozódik a szilikagél alapú RP töltetek esetén.
1.7. A retencióra ható tényez ő k
1.7.1. A pH szerepe
Az elválasztás körülményeinek megválasztásakor figyelembe kell venni a minta és az eluens pKa értékét, az oldószer alifás természetét, valamint az eluens pH-ját [19]. Az eluens erőssége egyszerűen befolyásolható a pH változtatásával. Amikor karbonát és bikarbonát anionokat használunk, puffer eluens keletkezik és az elúciós ereje könnyen változtatható a két anion arányának változtatásával. Ahogy változik a mozgó fázis pH-ja, egyidejűleg játszódik le az ioncsere-egyensúly és a protonálódás a kromatográfiás rendszerben. A karbonát eluens, mint többfunkciós eluens három versengő aniont tartalmaz (CO32−, HCO3− és OH−). A megbízható retenciós viselkedés érdekében figyelembe kell venni adott időpontokban az eluens és az oldatkomponensek minden formáját. Az eluens pH csökkenése a retenció növekedéséhez vezet, mert a karbonát eluens túlsúlyban levő formája az egyértékű HCO3− pH<10 esetében.
Mivel a szerves savak, köztük a poliaminokarbonsavak ionizációja függ a pH-tól, az elúciós viselkedésük befolyásolható a mozgó fázis pH-jának változtatásával abban a pH- tartományban, amelyet a pKa-értékeik meghatároznak.
Az elválasztások tervezésekor figyelembe kell venni az adott oszloptöltetnél alkalmazható pH-tartományt. A munkánk során alkalmazott elválasztó oszlopok (AS9-HC és Cryptand A1) minden pH értéken alkalmazhatók, de a szilikagél alapú állófázisok nem használhatók lúgos pH-jú eluensekkel és mintákkal.
1.7.2. Az eluens koncentrációjának hatása
Az eluens koncentrációjának változása jelentős hatást gyakorol a minta retenciós jellemzőire [16]. A növekvő eluenskoncentráció csökkenő kapacitási faktorokhoz (k’) vezet.
Az anionok elúciós sorrendje megfordítható az eluens koncentrációjának növelésével. A /1.13./ egyenletből látszik, hogy az egyensúly befolyásolható az eluens koncentrációjának változtatásával, mivel az ioncsere egyensúlyi folyamat.
A /1.8./ egyenlettel jellemezhető anioncsere folyamat megoszlási hányadosa a következő alakban is felírható:
[ ]
yxx y x AE
M x
K Q D
− −
= x
1
E /1.13./
ahol DM a megoszlási hányados, KA/E az egyensúlyi állandó, Q az effektív oszlopkapacitás,
−
Ex az eluens anionja és Ay− a vizsgált anion. Az egyenletet logaritmizálva a következő összefüggést kapjuk:
[ ]
−− +
=1lg lg lgEx
lg x
y x Q x K y D x
AE M
/1.14./
A /1.14./ egyenlet lineáris kapcsolatot mutat a megoszlási hányados és az eluens koncentrációjának logaritmusai között [24]. Ha ábrázoljuk a retenció mértékének logaritmusát (lgDM) az eluens koncentráció logaritmusának (lg[Ex-]) függvényében, és ha csak tisztán ioncsere folyamat játszódik le, egy (-y/x) meredekségű egyenest kapunk, amely a minta-, illetve eluension töltéseinek hányadosa. A tengelymetszetből KA/E értéke meghatározható.
Többkomponensű rendszerek és a mellékreakciók figyelembevételével bonyolult retenció egyenletek alakíthatók ki, elsősorban az eluens protolízise, a minta protolízise, ill.
komplexálódása révén [19].
1.7.3. Az állófázis szerepe
A minták retenciós viselkedését az anion-kromatográfiában használt állófázisok fő jellemzői is meghatározzák: (1) az állófázisok morfológiai szerkezete (pellikuláris, makroporózus, gél-típusok), (2) a mátrix kémiai szerkezete (polisztirén, polimetakrilát, szilika alapú), (3) a funkciós csoportok kémiai szerkezete (dimetil-etilamin, dimetil-allilamin, trimetilamin) és (4) az ioncsere-kapacitások [23]. Az ionkromatográfiában használt állófázisok többsége pellikuláris polisztirén bázisú. Aktív ioncsere kapacitása a pellikuláris gyantának csak a gyöngy felületén vagy annak közelében van. Az állófázis hidrofobicitása és ioncsere-kapacitása jelentősen befolyásolja a karbonsavak elválasztását, különösen kétértékű és telítetlen anionok esetében.
1.8. A haloecetsavak jelent ő sége és analitikai elválasztási lehet ő ségei
A klóros fertőtlenítés hatékonyan semlegesíti az ivóvízben lévő fertőző mikrobiális szennyező anyagokat [25]. Azonban a fertőtlenítési folyamat során a klór reagálhat azokkal a szerves anyagokkal, amelyek természetes módon vannak jelen a tisztítatlan vízben, és számos szerves Cl-tartalmú melléktermék keletkezik. Ha bromidot is tartalmaz a természetes víz, Br- tartalmú szerves komponensek is keletkeznek. A természetes vizek nyom-mennyiségű oxobróm vegyületei, amelyek a bromidból keletkezhetnek, a közelmúltban különleges és kiemelt szerepet kaptak humán egészségügyi szempontból. Az esetlegesen jelenlevő vegyületek már ng/l koncentrációban is karcinogén hatásúak. A haloecetsavak a fertőtlenítési melléktermékek fontos csoportját képezik. Az utóbbi években egyre inkább felismerik a haloecetsavak emberi egészségre gyakorolt káros hatásait [26, 27, 28]. Ezek a vegyületek toxikusak az emberre, csecsemőfejlődésre, a növényekre, különösen az algákra.
Fordított összefüggés van a HAA-k képződése és a pH növekedése között [35]. A képződési folyamat kinetikájának köszönhetően a hőmérséklet növekedése a nyári hónapok során növekedést okozhat a vízben lévő fertőtlenítési melléktermékek (disinfection byproducts: DBP-k) szintjében. Ráadásul a nyári hónapokban megnövekszik a mikrobiális szaporulat. Az évszakos hatások legyőzése érdekében ilyenkor a klórt nagyobb koncentrációban adagolják a kezelés során, ami a DBP-k nagyobb mennyiségét eredményezi ebben a periódusban. Dojlido és társai tanulmányozták, hogy a HAA tartalmú vízminták kezelés utáni forralása okozza-e a HAA-k bomlását. Arra a következtetésre jutottak, hogy kevesebb, mint 10 perc forralás hatására csökkent a HAA-k mennyisége, a TCA (triklór- ecetsav) 72 %-kal, az MBA (monobróm-ecetsav) 31 %-kal. Azonban később azt találták, hogy a forralás a HAA-kat a versengő trihalometánokká (THM) alakítja. Ez a felismerés lényeges az előzetes dúsítási folyamatok kiválasztásában.
A klór dózisának növekedése a HAA-k szintjének növekedését okozza, szemben a THM- ekkel. Továbbá a triklórozott szerves anyagok mennyisége nagyobb, mint a di- és monoklórozott részecskéké, ha növeljük a klór mennyiségét. A klórozott szerves anyagok mennyisége nagyobb, mint a brómozottaké. A vízkezelési rendszerben hipobrómossav képződik a bróm és a klór reakciójának köszönhetően. A hipobrómossav hozzávetőleg 25- ször gyorsabban reagál, mint a hipoklórossav és DBP-ket képez. A HOBr/HOCl arány fontos
szerepet játszik a THM-ek és HAA-k képződésében. A bromidion eltolja a DBP-k eloszlását a jobban brómozott formába.
Az utóbbi néhány évben az ionkromatográfia alternatív, ígéretes technikaként jelent meg haloecetsavak meghatározására a mikroextrakció [34] mellett. Az ionkromatográfiás elválasztásukhoz alkalmas az egyszerű működésű és költségkímélő vezetőképességi detektálás, míg a mikroextrakcióhoz ESI-MS detektálást használnak. A HAA-kat erősen savas és hidrofil karakterük alkalmassá teszi hatékony ioncserés elválasztásra. [40] (1.2. táblázat). A HAA-k pKa értékei 0,7-től 2,8-ig terjednek, ami azt jelenti, hogy ezek a savak csak protonált formában léteznek erősen savas feltételek mellett, aminek lényeges hatása van az extrakcióra és az előzetes dúsítási technikákra.
1.2. táblázat: A HAA-k és pKa értékeik
Haloecetsav Rövidítés Képlet pKa
monoklór-ecetsav MCA ClCH2COOH 2,86
diklór-ecetsav DCA Cl2CHCOOH 1,29
triklór-ecetsav TCA Cl3CCOOH 0,65
monobróm-ecetsav MBA BrCH2COOH 2,87
dibróm-ecetsav DBA Br2CHCOOH 1,47
tribróm-ecetsav TBA Br3CCOOH 0,66
bróm-klór-ecetsav BCA BrClCHCOOH 1,09
klór-dibróm-ecetsav MCDBA ClBr2CCOOH 1,39
monobróm-diklór-ecetsav MBDCA BrCl2CCOOH n.a.
Az US EPA a következő öt haloecetsav: MCA, DCA, MBA, DBA, és TCA mennyiségére 60 µgl-1 szintet engedélyez, azzal a kitétellel, hogy a DCA lehetőleg egyáltalán ne legyen jelen. Az öt szabályozás alatt álló haloecetsav meghatározására az EPA jelenleg két módszert hagyott jóvá, ezek a standard Method 6251B [29] és az 552,2 [30], amelyek oldószerextrakción, derivatizáción, gázkromatográfia-elektronbefogásos detektálásos (GC- ECD) analízisen alapulnak. Az EPA Method 552,3 [31], amelyet nemrégiben fejlesztettek ki a haloecetsavak meghatározására, teljesítményben felülmúlja az említett módszereket, különösen a nem szabályozott brómozott trihaloecetsavaknál, amelyeknél a detektálási határok 3–8-szor kisebbek. A szabályozásokhoz szükséges alacsony detektálási határok eléréséhez az elektroporlasztásos ionizációs tömegspektrometria (ESI-MS) jó kiegészítője a gázkromatográfiának. Ez a technika előnyös módszer, érzékeny és szelektív. Amennyiben
folyadék-kromatográfiával és előzetes dúsítással kapcsolják össze, az ESI-MS-sel kevesebb, mint 70 ngl-1 detektálási határokat lehet elérni [32]. Gyakran szükség van mintaelőkészítésre is. Az ionpár LC-ESI-MS-t, amelyet szilárd fázisú extrakció (SPE) lépés előz meg, használták vízmintákban a fent említett HAA-k meghatározására [33]. A szelektivitás fejlesztéséhez, amelyre a természetes mátrix interferáló ionjai miatt van szükség, perfluorozott alkil- karboxilátokat használtak. Ezek a reagensek stabil asszociációs komplexeket képeznek a mintákkal, amelyek detektálhatók ESI-MS-sel [34].
Mivel a HAA-k anionként léteznek az ivóvizekben, az IC nyilvánvaló választás az elválasztásukra és detektálásukra, eltekintve az előzetes dúsítási lépésektől, nem igényel további minta-előkezelést [35, 36]. Az utóbbi időben az elektroporlasztásos ionizációs tömegspektrometriát és az induktív csatolású plazma tömegspektrometriát (ICP-MS) sikeresen alkalmazzák a HAA-k detektálására az IC elválasztást követően. Egy sokkal összetettebb és sokkal érzékenyebb IC alapú módszert fejlesztett ki Lopez-Avila és társai [37]. A módszer kombinál egy többlépéses oldószer mikroextrakciós eljárást, egy on-line anion koncentráló oszlopot, amelyen a teljes extrakció mikroextrakcióból történik, és egy 60 perces, 12 szakaszos gradiens elúciós programot. Ez a módszer egy 25 cm-es microbore Dionex AS11 elválasztóoszlopot használ NaOH eluenssel és szuppresszálást egy 2 mm-es anion önregeneráló szuppresszor modul használatával (ASRS, Dionex). A detektálási határok 0,05 és 1,1 µg/l között voltak. Mint látható, néhány ionkromatográfiás módszert már alkalmaztak HAA-k elválasztására [35, 43]. Azonban meg kell jegyezni, hogy sok esetben hidroxid-szelektív oszlopot használtak [39, 44, 45] és a módszerek többségéhez gradiens elúció szükséges (koncentráció, áramlási sebesség, hőmérséklet vagy hőmérséklet kontroll).
NaOH vagy Na2CO3 eluens esetén az volt a tapasztalat, hogy a lényeges haloecetsav-párok felbontása számos esetben nem volt megfelelő. A szelektivitás szempontjából legjobb eredményeket ESI-MS [41, 46, 47] vagy ICP-MS [44] kapcsolással lehet csak elérni, mivel így a szervetlen ionok interferenciája elkerülhető, viszont ezek az eljárások nagyon költségesek. Az utóbbi időben kifejlesztettek egy nagy kapacitású anion-cserélőt, az IonPac AS24-et, kifejezetten haloecetsavak analízisére [47] azzal a céllal, hogy megszüntessék a felbontási problémákat. Ennek az oszlopnak a kémiája, ill. a funkciós csoportjai hasonlók az AS19-AS20 oszlopéhoz. MS/MS technikával kombinálva használták és nagy ionerősségű mátrixokhoz ajánlják mintakezelés nélkül. Ez egy lehetséges jövőbeni irányvonal, habár rutin analízisekhez jelenleg magas költségű, komplex eljárás, valamint az analízis időigényes.
Sarzanini és társai [38] NaOH vagy Na2CO3/NaHCO3 eluensekkel azt találták, hogy az MBA és Cl-, valamint a DCA és NO3− felbontása rossz volt az AS11 oszloppal. Az AS9 oszloppal a fenti minták felbontása némiképp javult. Később Liu és Mou [39] megpróbálták javítani a szuppresszált anioncserés módszer (Na2CO3/NaHCO3 gradiens) detektálási határait egy nagy kapacitású oszlop (AS9-HC) használatával és 500 µl minta direkt injektálásával.
Standard oldatokban a HAA-k detektálási határa 0,4-32 µg/l tartományban volt. Valódi minták analízisében a mátrix anionok problémákat okoznak számos HAA mennyiségi meghatározásában és a minták egy tisztító szakaszt igényelnek Ag töltetű On-Guard kolonna használatával, hogy csökkenjen a klorid, bromid és foszfát mennyisége a minta analízisének érdekében, amelyben meghatározható a nyommennyiségű DCA, BCA, DBA és TCA.
Az ionkromatográfia egyszerűsége HAA-k elválasztási lépéseként (nem igényel minta derivatizálást) kombinálva az ESI-MS detektálás szelektivitásával és érzékenységével, az IC- MS-t potenciálisan ideális alkalmazássá teszi az ivóvízben lévő nyommennyiségű HAA-k meghatározására [40]. Az IC-ESI-MS munkához kis áramlási sebességet kell használni olyan eluensekkel, amelyek csak illékony anyagot tartalmaznak. Ezért a hagyományos standard bore IC oszlopokat nem lehet használni. Roehl és társai [41] javasolnak egy módszert anioncsere kromatográfiát használva (Dionex AS 162 mm belső átmérőjű oszlop) 5-70 mM NaOH gradienssel 0,25 ml/min áramlási sebességnél eluens szuppresszálással. Az eluens szuppresszálás a hidroxid eluens tiszta vízzé való átalakulását eredményezi a tömegspektrométerbe való bevezetést megelőzően. Fontos megjegyezni, hogy a HAA-k nem illékony vegyületek. A szuppresszálás után az eluátum a HAA-k nagyon híg vizes oldatát tartalmazza. Ez az oldat nem teszi lehetővé az oldószer teljes elpárologtatását a elektroporlasztó fúvókánál és alacsony tömegspektrometriai választ eredményez. A probléma megoldásához egy második pumpa szükséges, amely illékony szerves oldószert, pl. metanolt juttat a rendszerbe egy T-illesztésen keresztül az MS előtt, hogy javítsa a minta elpárologtatását és ebből eredően az érzékenységet. A szerves oldószer elválasztás utáni bevezetéséről is igazolták Takino és társai [42], hogy javítja az érzékenységet. Az IC legújabb alkalmazásában HAA-k meghatározására Liu és társai ICP-MS-t használtak nagy érzékenységű és szelektivitású detektálórendszerként. A tanulmányban szuppresszált IC-t használtak hidrofíl anioncserélő oszloppal és NaOH gradienssel, on-line összekapcsolva az ICP-MS-sel. A detektort a klórozott HAA-k 35ClO- ionjainak és a brómozott vegyületek 79Br-- jainak szelektív monitorozására használták (a domináns ionok a plazmában alakultak ki). A detektálási határokat a klórozott savaknál 16 és 24 µg/l között és a brómozott savaknál 0,3 és

![1.4. ábra. Latex alapú állófázis pásztázó elektronmikroszkópos felvétele [3]](https://thumb-eu.123doks.com/thumbv2/9dokorg/874327.47049/24.892.274.644.202.451/ábra-latex-alapú-állófázis-pásztázó-elektronmikroszkópos-felvétele.webp)

![1.7. ábra. Az EDTA fémkomplexeinek stabilitási állandói a pH függvényében [63]](https://thumb-eu.123doks.com/thumbv2/9dokorg/874327.47049/38.892.287.637.381.700/ábra-edta-fémkomplexeinek-stabilitási-állandói-ph-függvényében.webp)