Targeted mutagenesis of multiple chromosomal regions in microbes
Ba´lint Cso¨rg Å
1,2, Akos Nyerges
3,4and Csaba Pa´l
3Directedevolutionallowstheeffectiveengineeringofproteins, biosyntheticpathways,andcellularfunctions.Traditional plasmid-basedmethodsgenerallysubjectoneoroccasionally multiplegenes-of-interesttomutagenesis,requiretime- consumingmanualinterventions,andthegenesthatare subjectedtomutagenesisareoutsideoftheirnativegenomic context.Othermethodsmutagenizethewholegenome unselectivelywhichmaydistorttheoutcome.Recent recombineering-andCRISPR-basedtechnologiesradically changethisfieldbyallowingexceedinglyhighmutationratesat multiple,predefinedlociintheirnativegenomiccontext.Inthis review,wefocusonrecenttechnologiesthatpotentiallyallow acceleratedtunablemutagenesisatmultiplegenomiclociin thenativegenomiccontextofthesetargetsequences.These technologieswillbecomparedbyfourmaincriteria,including thescaleofmutagenesis,portabilitytomultiplemicrobial species,off-targetmutagenesis,andcost-effectiveness.
Finally,wediscusshowthesetechnicaladvancesopennew avenuesinbasicresearchandbiotechnology.
Addresses
1DepartmentofMicrobiologyandImmunology,UniversityofCalifornia, SanFrancisco,94143,SanFrancisco,CA,USA
2GenomeBiologyUnit,EuropeanMolecularBiologyLaboratory,69117, Heidelberg,Germany
3SyntheticandSystemsBiologyUnit,BiologicalResearchCentre,6726, Szeged,Hungary
4DepartmentofGenetics,HarvardMedicalSchool,02115,Boston,MA, USA
Correspondingauthors:
Cso¨rgÅ,Ba´lint(Balint.Csorgo@ucsf.edu),Pa´l,Csaba(cpal@brc.hu)
CurrentOpinioninMicrobiology2020,57:22–30
ThisreviewcomesfromathemedissueonMicrobialsystems biology
EditedbyAthanasios(Nassos)TypasandGene-WeiLi ForacompleteoverviewseetheIssueandtheEditorial Availableonline26thJune2020
https://doi.org/10.1016/j.mib.2020.05.010
1369-5274/ã2020TheAuthors.PublishedbyElsevierLtd.Thisisan openaccessarticleundertheCCBY-NC-NDlicense(http://creative- commons.org/licenses/by-nc-nd/4.0/).
Introduction
On a sufficiently long timescale with a large enough populationsize,biologicalevolutioncanproducemyriad intricate solutions to various selective pressures. Over time, thebest performinggenetic variants are continu- ously selected resulting in highly specialized gene
products with optimal properties. Humans have long sought to speed up and controlthis process to produce whole organisms or specific biomolecules with desired traits[1].Withtheadventandcontinuingadvancementof molecular biologicaltechniques, efforts to directevolu- tion have greatly increased in specificity, capable of targeting single genes withinorganisms [2,3]. The con- currentdevelopmentofhighlyefficient methodsforthe screeningofgenevariantlibraries[4,5]hasallowedforthe isolation of a range of enzymes with improved or completelynovelfunctions[6].
The most comprehensive approach for achieving these objectives requires saturation mutagenesis, that is, the ability to generate and screen all possible amino acid variantsandtheircombinationsatasmanypositionsofa proteinaspossible.Althoughavarietyoftechniqueshave longexistedcapableofgeneratinggenevariantlibraries towardsthisgoal,recentyearshaveseenthedevelopment ofmorerefinedmutagenesistechnologieswithincreased targetingprecision,increasedrangesofattainable muta- tionrates,anddecreasedbiasesinmutationalspectra.We brieflysummarize thesemostrecentadvances andtheir related applications, focusing on tools developed and employed in microbial systems. These technologies canbebroadly categorizedbased onwhetherthemuta- genizedtargetDNAisonanextrachromosomalelement orongenomicDNA.Whiletheformerismoreamenable toawidevarietyofhighlyprecisestrategies,whichallow fortruesaturation,thelatterallowsprobingtheeffectsof mutationsin theirtrulynativecontextsbycouplingthe mutationgenerationandvariantselection steps.Finally, wehighlightrecentapproachesthatareovercomingthese limitationsof mutagenesisofuser-definedchromosomal segmentsandoffernewpossibilitiesforbothfundamen- tal evolutionary biology questions, as well as industrial applications.
Extrachromosomalmutagenesis
Extrachromosomalmutagenesismethodshavetheinher- entabilityoffocusingthegenerationofgeneticvariantsto a specified segment of DNA, allowing for saturation studies of selected regions of interest. However, these libraries are generated separately from the functional selection process and require labor-intensive cloning and transformation steps that often present a limit to thefinalnumberofvariantsthatarescreened.Themost long-standingsuchmethodhasbeenerror-pronePCR[7], whichmakesuseofthelowfidelityofDNApolymerases under certain conditions. This approach has long been
used for numerous protein engineering applications to attain novel variants for example, with new catalytic activities [8], improved stability [9], or novel binding capabilities[10].Drawbacksoferror-pronePCRmethods includerelativelylowperbasemutationratesandinher- ently biasedmutational spectramakingit impossibleto achievesaturation[11].Improvementstoovercomethese limitations have been made in techniques such as
‘sequencesaturationmutagenesis’whereauniversalbase isinsertedthroughoutthetargetsequence[12]andalsoin
‘casting error-prone PCR’, where target sequences are dividedupintosmallerfragments[13],makingforhigher levelsof mutationalcoverage.
AmoretargetedPCR-basedapproachthatallowsfortrue saturation ofselectedpositionsbutgeneratesvariantsof much shorter sequences, is site saturation mutagenesis (SSM),whichutilizessyntheticoligonucleotidescarrying one or more degenerate codon (such as NNK). To increase efficiency of this approach, prior identification of keyresidues ofthegivengene-productthroughphy- logeneticanalysisofhomologousproteinsisusuallyper- formed, andregionsdeemedimportant forfunctionality arethentargeted.Numerousvariationsofthistechnique exist,themostcommonbeingQuikChangemutagenesis [14] where overlapping oligonucleotides carrying the degenerate codons are used to amplify the target sequence from a plasmid. Recent variations of SSM include nicking mutagenesis[15]andmutagenesis with reversibly terminated deoxyinosine triphosphates [16], both of which allowed for comprehensive saturation librariesoftheactive sitesof thebla genein Escherichia coli encoding TEM-1 b; as well as a two-step PCR strategy applicable to difficult-to-randomize genes[17].
SSMhasnumerousapplications,includingtheengineer- ing of protein binding and selectivity [18], increased enzymaticactivity[19],orenhancedtherapeuticefficacy [20].Mostrecently,SSMwasutilizedforthemutagenesis of phage tail fiber residues, limiting bacterial phage resistance,therebyincreasingefficiencyofphagetherapy [21].
Recent advances in DNA synthesis capabilities have allowedthemassivelyparallelinvitrogenerationofgene variantlibrariesbyhigh-throughput oligosynthesis[22].
Althoughsucholigosarelimitedinsize(350nucleotide maximal length), this has enabled complete saturation mutagenesis of short genes, including a tRNA gene in yeast [23]. Recently, tiling of multiple(19 in this case) sucholigonucleotidelibrariesallowedforthegeneration of the complete first-order fitness landscape of a much largeradeno-associatedviruscapsidgene[24].Thehigh costofDNAsynthesishasbeenanobstacleingenerating largegenevariantlibrariesinthisfashion,howevertech- niques such as DropSynth, an emulsion-based DNA synthesis method [25] hold promise in making this approach moreattainable.
Severalmethodologiesalsoexistformutatingepisomal target genes in a continuous manner, which has the advantage of not requiring prior in vitro synthesis of variants of PCR-based or synthesis-based approaches.
Many of these techniques utilize error-prone (EP) variants of DNA polymerases for replicating plasmid DNA leading to mutagenesis of the target sequence.
Thiswasoriginallyachievedbytransformingthevector into a mutator host strain with EP DNA polymerase enzymes and defective/deleted mismatch-repair sys- tems such as E. coli strain XL1-Red [26]. However, the systematically high mutation rates of suchstrains eventuallyleadtodeleteriouseffectsinthecell,slow- inggrowthandmakingthecellsdifficulttotransform.A more targetedapproachutilizesanEP DNA polymer- aseI(PolI)enzymetomutagenizethecargoofaPolI- dependentplasmid[27].Amorerefinedversionofthis principle was developed recently in yeast termed OrthoRep,wherethe replicationofaplasmidcarrying thetargetedDNAsequenceisfullydependentuponan engineeredorthologousEPDNApolymerasethatoth- erwise does not replicate the host genome or other plasmids [28]. This system allowed the generation ofadetailedfitnesslandscapeofthemalarialdihydro- folate reductase against the anti-malarial drug pyri- methamine. An entirely different approach utilizes bacteriophage andtheirlifecycles asvessels formuta- genizing a target gene. In phage-assisted continuous evolution (PACE), propagation of the M13 bacterio- phage relies onbacterial production ofthe pIII infec- tivityprotein,whichinturnisdependentonfunctional libraryvariantsencodedwithinthephage.Inthisfash- ion, genes of interest can be encoded on M13 and continuously mutagenized to rapidly generate satura- tion libraries.PACE wasrecently employedto evolve Bacillusthuringiensisd-endotoxinvariantsabletotarget previously resistant insect pests [29], to generate a variety of proteins with improved soluble expression [30], and to evolve Cas9 variants with altered PAM specificity andhigher precision[31].
Genomic strategies
Targeting chromosomalDNA sequencesformutagen- esis has the advantages of eliminating labor-intensive cloningandPCRstepsandcouplingthevariantgener- ation and selection steps, all while maintaining the native genetic context of the target. The first approachesaimingtogeneratechromosomalgenevari- ant librariesutilized various DNA damaging forces or compounds affecting the entire chromosome of an organism. A range of both physical (e.g. ultraviolet irradiation, gamma rays) and chemical (e.g. ethyl methanesulfonate, nitrous acid) mutagens induce mutationsatrandomsequencesthroughoutthegenome [32]andcan beutilized forgenome-widegene inacti- vation screens.
Chemicalmutagenesisprotocolsareconceptuallysimple and broadly applicable,but hey have associated health hazards,andtheassociatedmutationalspectraaregener- allybiased.Inasimilarvein,amutagenesisplasmid(MP) approachhasbeendeveloped,whereselecteddominant mutatorgenesareexpressedfromavectorinthebacteria ofinterest, leadingtoasystematicincreasein mutation rateswithlessbiasinthemutationalspectrathanphysical and chemical approaches [33]. Overall, mutator strains andchemicalmutagenesisdonotrequirespecificationof thegenomicregionsrelevantfortheselectedphenotype:
theyincreaseoverallbacterialgenomicmutationrate.As aconsequence,theycannotbefocusedtospecificregions forin-depthsaturationstudies,andresultin deleterious off-targeteffects.
Alternatively, synthetic constructs can be genomically integratedtoachievetargetedmutagenesisinacontinu- ousfashion.Inoneapproach dubbedinvivocontinuous evolution(ICE),retroelementsareconstructedinyeastto encodeatargeted geneofinterestwhich undergoesEP reverse-transcription and genomic integration in a con- tinuous fashion [34]. In another approach, an array of specific sites can be genomically integrated next to a selectedregionofinteresttowhichaglycosylaseenzyme is recruited that is capable of mutagenizing a 20kb genomicregion[35].Theseapproachessolvetheproblem of localizing mutagenesis within the genome, however they require extensive prior construct development
resulting in considerable modifications to the native contexts.
Targetedmutagenesisofmultiplegenomicloci
Recentyearshaveseenthedevelopmentofanumberof diversestrategiesthatallaimtocombinethehighpreci- sion of extrachromosomal mutagenesis with genomic targeting for the saturation mutagenesis of specified genomic sequences within their native contexts. Two keytechnologieshaveenabledtheseadvances:thedevel- opmentandoptimizationofsingle-strandedoligonucleo- tide-basedrecombineeringmethods[36],andtheadvent of CRISPR-Cas genomeengineering technologies[37].
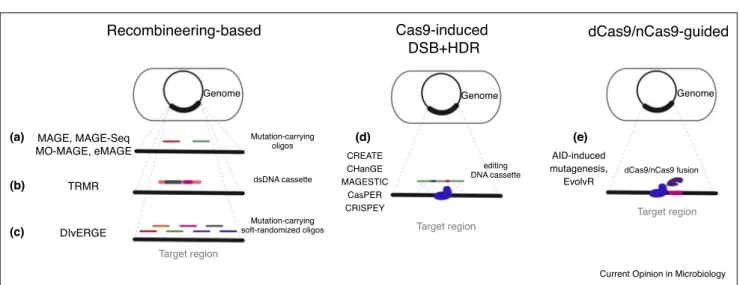
Theseapproachesallowfor unprecedented precision in the targeted modification of microbial genomes, and, throughvariousstrategies,canbeadaptedtomutagenize distinctchromosomalregions(Figure1 andTable1).
Recombineering-basedapproachesrelyontheannealingof syntheticsingle-strandedoligonucleotidesto thelagging strands at open replication forks. This process requires specific single-stranded DNA annealing proteins (e.g.
the phagel’sRed Beta proteinfor E. coli [38]or other RecTvariants[39])toworkatahighefficiencyinagiven organism.Inalandmarkpaper,recombineeringwasdevel- opedtointroducemultiplemutationsacrossthegenomein aprocesscalledmultiplexautomatedgenomeengineering (MAGE) [40]. 20 separate oligonucleotides containing degenerateribosome bindingsite(RBS)sequenceswere
Figure1
(a)
(b)
(c)
(d) (e)
Recombineering-based Cas9-induced DSB+HDR
dCas9/nCas9-guided
Genome Genome Genome
MAGE, MAGE-Seq MO-MAGE, eMAGE
TRMR
DIvERGE
CREATE CHanGE MAGESTIC
CasPER CRISPEY
AID-induced mutagenesis, EvolvR Mutation-carrying
oligos
dsDNA cassette
Mutation-carrying soft-randomized oligos
editing
DNA cassette dCas9/nCas9 fusion
Target region Target region
Target region
Current Opinion in Microbiology
Schematicrepresentationofmicrobialgenomeeditingmethodscapableoftargetedsaturationgenome-mutagenesis.Recombineering-based approachesrelyonsingle-strandedDNAoligonucleotide-strandedordouble-strandedDNAcassette-mediatedhomologousrecombination.(a) MAGE,MAGE-Seq,MO-MAGE,andeMAGEutilizesingle-strandedDNAoligonucleotidesthatcarryuser-definedmutationsandincorporatethose intothegenomictarget.(b)TMMRachievesthesameoutcomebyrecombiningselectabledsDNAcassettes,thatcarrythedesiredmodification, intothetarget.(c)DIvERGEusespartiallyoverlappingDNAoligonucleotidesthatcarryrandomlydistributedrandompoint-mutationsalongtheir entirelengthtoperformmutagenesisatthetargetregion.(d)MethodsrelyingonCas9-induceddouble-strandedbreaksplushomologous recombination(Cas9-inducedDSB+HDR)exploitthelethaleffectofCRISPR-Cas9-inducedDSBstoselecttheintegrationofaneditingDNA cassettethatiscarryingthemodification-of-interest.(e)CatalyticallyimpairedCas9-(dead(d)-ornicking(n)-)guidedmethodsexploitCas9’sability tosequence-specificallyrecognizethetargetsequenceandbringittotheproximityofaCas9-fusedmutatorenzymeandthusintroducedesired mutations.Seesection‘Targetedmutagenesisofmultiplegenomicloci’foranextendeddescriptionofeachmethod.
simultaneouslytargetedtovariousgenesallinvolvedinthe biosynthesisoflycopene,leadingtoafivefoldincreasein theproductionofthisindustriallyrelevantisoprenoidcom- poundin onlythreedays.Inamethodtermedtrackable multiplexrecombineering,theMAGEapproachwasfur- therrefinedtoincludebarcodeswithineacholigonucleo- tidetoallowformassivelyparallelmutagenesisofmultiple genomicregionsandthesubsequentidentificationofmod- ifiedsequencesthatresultedin improvedphenotypesof interest[41].Thisapproachallowedforthemutagenesisof
the RBSs of close to allgenesin E. colito modify their expression levels allowing improved growth in various environments [42].MAGE canbeemployedinahighly focusedmanner, assynthesizingalibraryofoligonucleo- tideseachcarryingadegeneratecodonofthesamegene, allowedforthesaturationcodonmutagenesisoftheessen- tialgeneinfAinE.coli[43].Measuringthefitnessofeach individualvariant,combinedwithamplicondeepsequenc- ing,enabledthein-depthanalysisoftheeffectsofcodon usageacrossanentiregene.
Table1
Efficacyandcostsoftargetedmutagenesismethods Basisof
technology
Method Targetingwindow,efficiency Applicablespecies Off-targeteffects Cost Recombineering-
based
MAGE[40], MAGE-seq[43]
Upto30nucleotidesusinga singleoligoorhundredsof nucleotides(e.g.219of essentialgeneinfA)inparallel usingmultipleoligos(1per 1 2saturatedcodons)
OptimizedforE.coli andavailablein multiple
Gammaproteobacteria
HighbecauseMMR deficientstrainrequiredfor highefficiency,buttheuse ofinducibledominant- negativeMMRvariant (pORTMAGE)can eliminateoff-targeteffects
Cost-effective, howevereach oligonucleotide canonly mutagenizea targetupto30bp TRMR[41] Thousandsofnucleotidesin
parallelusingmultipleoligos
OptimizedforE.coli Low Highcost,dueto theneccesityof high-throughput DNAsynthesis MO-MAGE[44] Thousandsofnucleotidesin
parallelusingmultipleoligos
OptimizedforE.coli High,MMRdeficientstrain requiredforhighefficiency
Highcost,dueto theneccesityof high-throughput DNAsynthesis EukaryoticMAGE[48] Hundredsofnucleotidesin
parallelusingmultipleoligos, constrainedbyrequirement forreplicationfork,rarerin eukaryotes
OptimizedforS.
cerevisiae
High,MMRdeficientstrain requiredforhighefficiency
Cost-effective, howevereach oligonucleotide canonly mutagenizea targetupto30bp DIvERGE[47] Thousandsofnucleotidesin
parallelusingmultipleoligos
OptimizedforE.coli, applicabletoarange ofEnterobacteriacae
Undetectableduetousage ofinducibledominant- negativeMMRvariant
Cost-effective, each
oligonucleotide canmutagenizea targetupto72bp Cas9-induced
DSB,HDR
CREATE[53] Thousandsofnucleotidesin parallelusingmultiplerepair cassettes
OptimizedforE.coli, applicabletoS.
cerevisiae
Notexaminedin-depth, expectedtobelow
Highcost,dueto theneccesityof high-throughput DNAsynthesis CRISPRlibrary[55],
CHAnGE[56], MAGESTIC[57], CRISPRvariant libraries[58],CasPER [59],CRISPEY[60]
Thousandsofnucleotidesin parallelusingmultiplerepair cassettes
OptimizedforS.
cerevisiae
Low,Cas9-mediated targetingshowedhigh specificity
Highcost,dueto thenecessityof high-throughput DNAsynthesis
dCas9/nCas9- guided
AID-induced mutagenesis[61–63]
Maximumof2paralleltargets demonstrated,mutagenesis limitedto100nucleotides surroundingPAM- constrainedtargetsite,high biasinmutationalspectra
E.coli,S.cerevisiae, humancells
Potentiallyhigh Moderate,dueto thenecessityof plasmid construction before mutagenesis EvolvR[64] Targetingof2paralleltargets
demonstrated,mutagenesis limitedto50 350nucleotides invicinityofPAM-constrained targetsitewithdeclining mutagenesiswithincreased distance
OptimizedforE.coli High,low-fidelityDNA polymeraseraises backgroundmutationrate over100-fold
Moderate,dueto thenecessityof plasmid construction before mutagenesis
In order to scale up the mutagenizing capabilities of MAGE to allow for potential saturation of extended genomic targets and enhanced multiplexability, micro- array-oligonucleotide (MO)-MAGE was developed, wherethemutation-inducingoligosaresynthesizedfrom microarraychips,allowing for parallelsynthesisof large (>55000)libraries[44].Alternatively,theintroductionof exogenous oligos to generate variants may be circum- ventedthrough aretroelement-basedapproach wherea mutagenicT7 RNA polymerase enzyme generatesvar- iants of a sequence encoded on a retroelement in a continuousmanner[45].Aspecializedreversetranscrip- tase ultimately generates variants of single-stranded DNA which then edits the target sequence through ssDNA-recombineering.
A key drawback of MAGE-based recombineering approaches is the requirement of a mismatch repair (MMR)-deficient host forhigh efficiency mutagenesis.
Thisleadstoahighbackgroundmutationrate,leadingto severaloff-targetmutations,potentiallyconfoundingthe phenotypic effects of saturation mutagenesis of the targeted region. One solution to this obstacle is the utilization of counter-selection markers such as the tetA-sacB system [46] or a system employing ccdB [47]. Through a two-step recombination process, the counter-selectablemarkersareintegratedatthegenomic site of interest, which is subsequently targeted using mutagenizing oligos. Counter-selection allows enrich- mentofcellswhichhaveincorporatedthemutagenizing oligosallwithouttherequirementofMMRinactivation.
Alternatively, a simplified approach dubbed portable, plasmid-based MAGE (pORTMAGE) was developed, whichutilizesinducibleexpressionofadominantnega- tive MMR protein allele to achieve high efficiency recombineering while eliminating off-target effects [48]. Building on this advance, it became possible to specificallytargetextendedgenomicregionsforsatura- tion mutagenesis without any detectable off-target effects.This wasachieved inamethod calleddirected evolutionwithrandomgenomicmutations(DIvERGE), which utilizes pools of oligonucleotides synthesized using a soft-randomization protocol (where the alternative nucleotides are spiked in at low (0.5–2%) amounts) at each nucleotide position [49]. Such a synthesisapproachsignificantlyreduces theoligonucle- otidecosts ofothermethodssuch asMO-MAGE. The tiling of such 90mer oligos allows for the coverage of entire chromosomal genes for saturation mutagenesis.
DIvERGE simultaneously targets multiple, user- defined regions, up to 10s of kilobases in total, and has broad, controllable mutagenesis spectra for each nucleotide position [49]. Importantly, DIvERGE is applicable to a range of bacterial host species without the needfor prior genomic modification and off-target mutagenesis rate is expected to be very low [49].
DIvERGE was utilized to perform simultaneous
combinatorial saturation mutagenesis of the 4 genes (a total of 9.5kb) encoding the target proteins of the antibioticsciprofloxacinandgepotidacin[39,49],while saturation mutagenesis ofthe target gene for the drug trimethoprim resulted in combinations of 5 mutations showinga>3900-foldincreaseindrugresistance[49].
Overall,recombineering-basedapproachesnowallowfor the most extensive and controllable mutagenesis of multiple chromosomal regions in microbes, opening entirely new possibilities for future applications (see futureperspectives).
Despitetheseunmatchedcapabilities,recombineering- based approaches do have some inherent limitations.
Recombineering relies on active replication forks withinthetargetcell,meaningtheslowerdivisiontime of eukaryotes makes the approach less efficient [50].
Also, ssDNA annealing proteins are not universal in theirefficienciesindiversebacterialorganisms,mean- ingspecificsystemshavetobeoptimizedfordifferent species[39,51–53,68].Finally,itgenerallyreliesonthe invitrosynthesisofoligonucleotidestogeneratediver- sity. The advent of CRISPR-Cas-based gene editing technologies has offered solutions to some of these limitations. Double-stranded breaks of chromosomal DNA greatly enhance the recombination frequency of introduced homologous templates. Repurposed CRISPR-based systems (generally employing Cas9) canspecificallycleaveagenomicsequence ofinterest, leading to a vast improvement in the frequency of edited microbial cells [54]. Combining this capability with large-scale oligonucleotide synthesis led to the development of CRISPR-enabled trackable genome engineering (CREATE), which utilizes pools of 104– 106barcodedoligostoachievegenomicmutagenesisat chromosomalsitesinbacteria[55].Anumberofsimilar approaches were recently developed in yeast [56,57,58,59,60,61], demonstrating the expanded potential of CRISPR-based targeted mutagenesis approachesineukaryotes(see Box1forspecificappli- cationsofthesetechnologies).
Allofthese CRISPR-basedmethods requirethe prior synthesisoflargepoolsofDNAoligonucleotideswhich serveastheeditingtemplatesforgenevariationgener- ation.Fusing variousmutagenizingenzymestoacata- lytically inactive version of Cas9 (dCas9) allows for theirtargetedlocalizationwithinthegenome,allowing for highly specific mutagenesis. One such approach utilizes fusion [62] or recruitment [63] of activation- inducedcytidinedeaminase(AID)todCas9togenerate targeted mutagenesis specified by the single guide RNA (sgRNA) sequences. Using multiple sgRNAs allowed for tiling of longer mutagenized sequences and was used to identify drug resistance mutations againstvariouscancertherapeuticsinmammaliancells [62,63].AsimilarapproachfusedAIDtozinc-fingerand
transcriptionactivator-like effectorproteinstoachieve targeted variant generation in E. coli [64]. Finally, a CRISPR-guided Cas9 nickase was recently used to guide an engineered EP nick-translating DNA poly- merasetospecificgenomictargetsites,raisingmutation rate by 3–4 orders of magnitude compared to back- ground levels [65]. This system, termed EvolvR is capable of generating all single substitutions in a 60- nucleotide window after 16hours in 1ml of saturated culture.Notwithstandingcertainlimitationsofexisting CRISPR-guided targeted genomic mutagenesis tools suchasbiasesinmutationalspectra,potentialoff-target effects,limitedtargetingwindowsize,andanincreased backgroundmutationrateinthecaseofEvolvR,these technologies hold great promise in potential applica- tions goingforward.
Future perspectivesforin vivo chromosomal saturationmutagenesis
The technologies currently allowing for the most con- trolled and complete mutagenesis of chromosomal sequences of interest (such as DIvERGE[49],CRE- ATE [55], and EvolvR [65]) will open new doors in whatispossibleindirectedevolution.Broadly,examples of these future applications include: (1) Targeted
mutagenesis along the full length of multiple genes withinagenome.Thiswillallowtheengineeringofnovel cellular functions involving multiple proteins, such as evolving novelmetabolic functionsfrom complexpath- ways. (2) Metabolic engineering in previously under- utilizedspecies.Theseabovetechniquescanbeadapted to a range of bacteria, including those with untapped metabolic potential resulting in optimization of novel industriallyrelevantpathways.(3)Saturationmutagene- sisofmultiplegenes,allowing thedirectedevolutionof multiproteincomplexes.Improvementofcomplextraits often requires co-evolution of interacting amino acids coded atdistinct loci,whosemutationsprovide noben- efits individually.(4)Forecasting thedynamicsofresis- tance evolution to novel antimicrobial drugs. System- wide mutagenesis affecting gene expression levels will aidinidentifyingprimarydrugtargetsandmechanismsof action.Oncetheseareidentified,saturationmutagenesis of the encoding genes will allow detailed fitness land- scapesinthepresenceofagivendrug.(5)Optimizationof invitrosynthesizedDNAconstructs.Invitroconstructed DNAelementsencodingforexample,biosyntheticpath- ways,geneticcircuits,orentiregenomicsegmentsoften lack clear design principles thus leading to suboptimal performance. High-throughputvariantgeneration ofthe constructs will lead to rapid optimization. Finally, (6) fundamental evolutionary biology questions, such as the conservation of epistatic effects between related speciesorthephenotypiceffectsofvaryingcodonusage indifferentspeciescouldbestudiedingreatlyenhanced detail.
In summary, the last several years have seen great stridesintheabilitytogenerategeneticvariantlibraries capable of saturation of selected sequences. Many of these techniques can complement each other and dependingonthestudiedorganism,thelevelofspeci- ficity,targetingwindowsize,andlevelofsaturation,the ideal strategy can be chosen for a range of diverse applications.
Conflictofinterest statement
Theauthorsdeclarecompetingfinancial interest.B.C.,A.N., andC.P.arelistedasinventorsonpatentapplicationrelated to DIvERGE (PCT/EP2017/082574 (WO2018108987) MutagenizingIntracellularNucleicAcids).
Acknowledgements
B.C.issupportedbytheEo¨tvo¨sNationalScholarshipofHungaryanda MarieSkłodowska-CurieActionsIndividualGlobalFellowship(number 844093)oftheHorizon2020ResearchProgramoftheEuropean Commission.A.N.wassupportedbyaLong-TermFellowship(number ALTF160-2019)fromEMBO.C.PissupportedbytheEuropeanResearch CouncilH2020-ERC-2014-CoG648364–ResistanceEvolution;‘Ce´lzott Lendu¨let’ProgrammeoftheHungarianAcademyofSciencesLP-2017–10/
2017;‘E´ lvonal’KKP126506,andGINOP-2.3.2–15–2016–00014 (EVOMER).
Box1CurrentapplicationsofCRISPR-basedmutagenizing technologies
CRISPR-enabledtrackablegenomeengineering(CREATE)combines thegenomeeditingcapabilitiesofCRISPR-Cas9withlarge-scale DNAoligosynthesistoachievetargetedchromosomalmutagenesis inbacteria[55].Thisapproachwasusedtosaturateallcodonsofthe folAdrug-targetgeneinE.coli,andidentifyallresistanceconferring individualmutations.CREATEcanbeusedinmultiplex,andwas usedtotarget50000genomicsitestoselectforvariantswith improvedtolerancetotemperatureandtotheindustrialsolvents furfuralandacetate[55].CREATEcanalsobeperformediteratively, generatingcombinationsofthousandsofmutationstoachieve60- foldimprovementintheproductionoftheindustriallyimportant chemical3-hydroxypropionicacid[66].Thetechniquehasalsobeen usedfortheparallelmutagenesisof19genesinvolvedinlysine metabolisminE.coli,identifyingdeterminantscapableofincreasing productionofthemetabolite[67].Buildinguponthebasicprinciples laiddownbyCREATE,severalmethodshavebeenrecentlydevel- opedtoexpandthesecapabilitiestoeukaryotesaswell.These approacheshaveallowedthegenomicintegrationoflargelibrariesof variantsandhaveenabledawidevarietyofapplications,including:
determiningthefunctionalconsequencesofpremature-termination codonsatvariouslocationswithinallannotatedessentialgenes [57],thesaturationmutagenesisofa29aminoacidregionofthe Siz1proteinforincreasedtolerancetothegrowth-inhibitorfurfural [58],thesaturationeditingoftheessentialgeneSEC14andidenti- ficationofaminoacidscriticalforchemicalinhibitionoflipidsignaling [59],thegenerationofasetoftilingdeletionmutantsforcharac- terizationoftheSGS1DNAhelicaseenzyme[60],thegenerationand screeningofcombinationsofmutationsintwokeyenzymesofthe mevalonatepathwayresultinginimprovedisoprenoidproduction [61],andstudyingthefitnessconsequencesof16006natural geneticvariantsthrougharetroelement-basedapproachtogenerate variation[62].
References andrecommendedreading
Papersofparticularinterest,publishedwithintheperiodofreview, havebeenhighlightedas:
ofspecialinterest ofoutstandinginterest
1. DoebleyJF,GautBS,SmithBD:Themoleculargeneticsofcrop domestication.Cell2006,127:1309-1321.
2. RomeroPA,ArnoldFH:Exploringproteinfitnesslandscapesby directedevolution.NatRevMolCellBiol2009,10:866-876.
3. SimonAJ,d’OelsnitzS,EllingtonAD:Syntheticevolution.Nat Biotechnol2019,37:730-743.
4. BershteinS,TawfikDS:Advancesinlaboratoryevolutionof enzymes.CurrOpinChemBiol2008,12:151-158.
5. PackerMS,LiuDR:Methodsforthedirectedevolutionof proteins.NatRevGenet2015,16:379-394.
6. TurnerNJ:Directedevolutiondrivesthenextgenerationof biocatalysts.NatChemBiol2009,5:567-573.
7. LeungDW,ChenE,GoeddelDV,LeungDW,GoeddelDV:A methodforrandommutagenesisofadefinedDNAsegment usingamodifiedpolymerasechainreaction.Technique1989, 1:11-15.
8. SeeligB,SzostakJW:Selectionandevolutionofenzymesfrom apartiallyrandomizednon-catalyticscaffold.Nature2007, 448:828-831.
9. BordesF,TarquisL,NicaudJ-M,MartyA:Isolationofa thermostablevariantofLip2lipasefromYarrowialipolyticaby directedevolutionanddeeperinsightintothedenaturation mechanismsinvolved.JBiotechnol2011,156:117-124.
10. KleinstiverBP,PrewMS,TsaiSQ,TopkarVV,NguyenNT, ZhengZ,GonzalesAPW,LiZ,PetersonRT,YehJ-RJetal.:
EngineeredCRISPR-Cas9nucleaseswithalteredPAM specificities.Nature2015,523:481-485.
11. PatrickWM,FirthAE,BlackburnJM:User-friendlyalgorithmsfor estimatingcompletenessanddiversityinrandomizedprotein- encodinglibraries.ProteinEng2003,16:451-457.
12. WongTS,TeeKL,HauerB,SchwanebergU:Sequence saturationmutagenesis(SeSaM):anovelmethodfordirected evolution.NucleicAcidsRes2004,32:e26.
13. YangJ,RuffAJ,ArltM,SchwanebergU:CastingepPCR (cepPCR):asimplerandommutagenesismethodtogenerate highqualitymutantlibraries.BiotechnolBioeng2017, 114:1921-1927.
14. HogrefeHH,ClineJ,YoungbloodGL,AllenRM:Creating randomizedaminoacidlibrarieswiththeQuikChangemulti site-directedmutagenesiskit.BioTechniques2002,33:1158- 11601162,1164–1165.
15. WrenbeckEE,KlesmithJR,StapletonJA,AdeniranA,TyoKEJ, WhiteheadTA:Plasmid-basedone-potsaturation
mutagenesis.NatMethods2016,13:928-930.
16. HallerG,AlvaradoD,McCallK,MitraRD,DobbsMB,GurnettCA:
Massivelyparallelsingle-nucleotidemutagenesisusing reversiblyterminatedinosine.NatMethods2016,13:923-924.
17. LiA,Acevedo-RochaCG,ReetzMT:Boostingtheefficiencyof site-saturationmutagenesisforadifficult-to-randomizegene byatwo-stepPCRstrategy.ApplMicrobiolBiotechnol2018, 102:6095-6103.
18. vanderMeerJ-Y,PoddarH,BaasB-J,MiaoY,RahimiM, KunzendorfA,vanMerkerkR,TepperPG,GeertsemaEM, ThunnissenA-MWHetal.:Usingmutabilitylandscapesofa promiscuoustautomerasetoguidetheengineeringof enantioselectiveMichaelases.NatCommun2016,7:10911.
19. KleinstiverBP,SousaAA,WaltonRT,TakYE,HsuJY,ClementK, WelchMM,HorngJE,Malagon-LopezJ,Scarfo` Ietal.:
EngineeredCRISPR-Cas12avariantswithincreasedactivities andimprovedtargetingrangesforgene,epigeneticandbase editing.NatBiotechnol2019,37:276-282.
20. WhiteheadTA,ChevalierA,SongY,DreyfusC,FleishmanSJ, MattosCD,MyersCA,KamisettyH,BlairP,WilsonIAetal.:
Optimizationofaffinity,specificityandfunctionofdesigned influenzainhibitorsusingdeepsequencing.NatBiotechnol 2012,30:543-548.
21. YehlK,LemireS,YangAC,AndoH,MimeeM,TorresMDT,dela Fuente-NunezC,LuTK:Engineeringphagehost-rangeand suppressingbacterialresistancethroughphagetailfiber mutagenesis.Cell2019,179:459-469.e9.
22. RocklinGJ,ChidyausikuTM,GoreshnikI,FordA,HoulistonS, LemakA,CarterL,RavichandranR,MulliganVK,ChevalierA etal.:Globalanalysisofproteinfoldingusingmassively paralleldesign,synthesis,andtesting.Science2017, 357:168-175.
23. LiC,QianW,MacleanCJ,ZhangJ:Thefitnesslandscapeofa tRNAgene.Science2016,352:837-840.
24. OgdenPJ,KelsicED,SinaiS,ChurchGM:ComprehensiveAAV capsidfitnesslandscaperevealsaviralgeneandenables machine-guideddesign.Science2019,366:1139-1143.
25. PlesaC,SidoreAM,LubockNB,ZhangD,KosuriS:Multiplexed genesynthesisinemulsionsforexploringproteinfunctional landscapes.Science2018,359:343-347.
Thiswork introducesDropSynth,ascalable,low-cost, droplet-based methodtoconstruct1000sofgene-lengthassembliessimultaneously.
TheauthorsappliedDropSynthtosuccessfullybuildmorethan7000syn- thetic,phylogeneticallydiversehomologsoftwoessentialgenesofE.coli andtestedtheirfunctionalityinvivo.
26. GreenerA,CallahanM,JerpsethB:Anefficientrandom mutagenesistechniqueusinganE.colimutatorstrain.Mol Biotechnol1997,7:189-195.
27. CampsM,NaukkarinenJ,JohnsonBP,LoebLA:Targetedgene evolutioninEscherichiacoliusingahighlyerror-proneDNA polymeraseI.PNAS2003,100:9727-9732.
28.
RavikumarA,ArzumanyanGA,ObadiMKA,JavanpourAA,LiuCC:
Scalable,continuousevolutionofgenesatmutationrates abovegenomicerrorthresholds.Cell2018,175:1946-1957.e13.
TheauthorsdescribeOrthoRep,anorthogonal,highlyerror-proneDNA polymerase-plasmid pair inSaccharomyces cerevisiae that stably mutatesplasmid-bornesequences upto100000-foldfasterthanthe host genome. Using OrthoRep, the authors simultaneously evolved antimalarialdrug-resistantdihydrofolatereductasesin90replicates.
29. BadranAH,GuzovVM,HuaiQ,KempMM,VishwanathP,KainW, NanceAM,EvdokimovA,MoshiriF,TurnerKHetal.:Continuous evolutionofBacillusthuringiensistoxinsovercomesinsect resistance.Nature2016,533:58-63.
30. WangT,BadranAH,HuangTP,LiuDR:Continuousdirected evolutionofproteinswithimprovedsolubleexpression.Nat ChemBiol2018,14:972-980.
31. HuJH,MillerSM,GeurtsMH,TangW,ChenL,SunN,ZeinaCM, GaoX,ReesHA,LinZetal.:EvolvedCas9variantswithbroad PAMcompatibilityandhighDNAspecificity.Nature2018, 556:57-63.
32. KodymA,AfzaR:Physicalandchemicalmutagenesis.Methods MolBiol2003,236:189-204.
33. BadranAH,LiuDR:Developmentofpotentinvivomutagenesis plasmidswithbroadmutationalspectra.NatCommun2015, 6:8425.
34. CrookN,AbatemarcoJ,SunJ,WagnerJM,SchmitzA,AlperHS:
Invivocontinuousevolutionofgenesandpathwaysinyeast.
NatCommun2016,7.
35. Finney-ManchesterSP,MaheshriN:Harnessingmutagenic homologousrecombinationfortargetedmutagenesisinvivo byTaGTEAM.NucleicAcidsRes2013,41e99–e99.
36. ThomasonLC,SawitzkeJA,LiX,CostantinoN,CourtDL:
Recombineering:geneticengineeringinbacteriausing homologousrecombination:recombineering.In Current ProtocolsinMolecularBiology.EditedbyAusubelFM,BrentR, KingstonRE,MooreDD,SeidmanJG,SmithJA,StruhlK.John Wiley&Sons,Inc.;2014.1.16.1-1.16.39.