K F K I - 7 5 - 5
J, K O L L Á R G. S O L T
S I M P L E M O D E L F O R T H E T O T A L E N E R G Y OF N O B L E M E T A L S
‘H u n g a ria n ‘A cad em y o f°S c ie n c e s
CENTRAL RESEARCH
INSTITUTE FOR PHYSICS
BUDAPEST
2017
KFKI-75-5
SIMPLE MODEL FOR THE TOTAL ENERGY OF NOBLE METALS
J. Kollár and G. Solt
Theoretical Solid State Physics Department Central Research Institute for Physics
H-1525 Budapest 114, POB 49 Hungary
Submitted to Journal of the Physics and Chemistry of Solids
ISBN 963 371 004 9
ABSTRACT
An expression for the total metallic energy is constructed as the sum of an average s-electron energy and of a d-energy term that has a mini
mum near the equilibrium lattice spacing. The model predicts reasonable cohesive energies for copper and silver. The calculation of the elastic con
stants reveals that, in the case of gold, the contribution from d-electrons to the total energy can not be represented by a sum of pairwise forces be
tween the ions. The sensitivity of the results to the choice of the prefixed atomic d-orbitals is tested.
АННОТАЦИЯ
Полная энергия для благородного металла пишется как сумма двух член: средняя энергия s-электронов и d -энергия, имеющая минимум близко к равновесному кристаллическому объему. Энергии связи получаются разумно для Си и Ад. Вычисление упругих постоянных указывает, что в .случае Au вклад из d -электронов в полную энергию не может быть преставлен как сумма паровых сил между ионами. Чувствительность результатов на изменение использованных атом
ных d -функций исследуется.
KIVONAT
A nemesfémek teljes energiáját az átlagos s-elektron-energia és egy d-energia összegeként értelmezzük; ez utóbbi minimummal rendelkezik az ész
lelt rácsállandó közelében. A modell elfogadható kötési energiát szolgáltat réz és ezüst esetén. A rugalmas állandók számításából kitűnik, hogy arany esetében a d-energia nem értelmezhető az ionok közötti párjellegü erők ősz- szegeként. Megvizsgáljuk az eredmények érzékenységét különböző rögzített atomi pályák felhasználása esetén.
The total metallic energy of noble metals have frequently been described by parametric expressions consisting of two terms: a nearly free s-electron energy and a repulsive pairwise interaction from the d-electrons^. Though the co
hesive energy and the elastic constants could more or less be reproduced in this way, nonphysical values of the model parameters /like imaginary core radius/ show that the
part played by the d-electrons in the binding is more complex than mere repulsion. In fact, the result of a
detailed calculation of the volume dependent total energy 2 of copper has shown that the nature of the d-type energy contribution basically differs from a simple monotonic
repulsion, since both the value of, and the pressure contri
bution from, the d term proved to be slightly negative at the equilibrium atomic radius, becoming repulsive only for smaller lattice spacings.
In this paper a new analytical expression is proposed to describe the d-electron contribution to the volume dependent
energy of noble metals that reproduces this complex behaviour Besides, a theoretical estimate of the band structure energy
of the s-electrons is given, in order to check its effect on both the elastic and the volume dependent properties.
The total metallic energy is written as2
E - t b -
Ej,
/1/where is the average s-electron energy and E cj_ is a
2
d-type contribution. An expression with the required
2 3
properties * for the d-energy can be constructed using the overlap integral О
Ej* -O.MJ v few 1 - и-- /2/
к Л К
where is the nearest neighbour distance in the lattice and the "effective" overlap integral is defined through the radial d-function "R(jL by
OO
j = J'Rji ( (lr - B w l ) r ' - A v -
o
Since the overlap energy integral in a tight binding scheme is essentially proportional to S / V . * . * . » ^ varies with the lattice spacing roughly like the width of the d band.
The energy of an s-electron as the function of the atomic radius "iRa can be written as
^5 = Eo( Ьл ( Ra ) +■ (R i \ ) f~ £ BS ] 'V'c ) ^^
where the first term contains the kinetic, exchange and correlation energy of the homogeneous electron liquid,
(Г - 2 . 2 1
ö 1
-Ra. +■ trCO xrr (K,V ) /4/
£ И is the Madelung energy - -Fr— /in rydbergs/ and Ra.
is the "band structure" energy arising from the scattering of the s-electrons in the periodic lattice.
The factor ? \0 accounts^ for the stretching of the
- 3 -
free в-band; it is the common "orthogonalization hole”
factor
= ( j - i-'c /r ,,)3 ;
/ 5 /
with the ionic radius ~X~C determined from the inner atomic 2 s-functions.
The term accounts for the fact that the average potential energy (s|Vls^ of the s-electrons differs from that of a homogeneous charge density in the pure Coulomb field of a unit charge. For simple metals, where the ion- electron potential becomes coulombic outside a certain core radius <. ~Ra one has . In the case of noble metals, however, due to the incomplete screening by d-electrons, the potential differs from - е г/ г " even in the outer regions of the atomic sphere, thus
where |sj is the actual s-electron state /e.g. an OPW state/, is the sum of the Coulomb and exchange interactions of the s-electron with the lo d-electrons. If one neglects
and calculates the Madelung energy in the spherical approximation, the formulae /3/ - /6/ become equivalent to
the expression for the s-energy used in Ref• 2.
The first two terms on the right-hand side of /6/ can be brought to a form that can be compared to (s iVls) for a simple metal, by introducing the effective charge Zeff felt, on the average, by the s-electron inside the atomic sphere,
- 4 -
U t v i 4 ) s ( s i - ' - v I * л -Е ь л =
/7/Further, assuming the e-density constant outside , and zero inside, one gets /in ry units/
( S I V b ) = Z etf (- F | - j /а /
which is the well known expression for the bottom of the s-band for an s-p metal. The presence of d-electrons makes Zeff depend on "R^ , which has physical consequences even though Zeff is nearly stationary at the observed atomic volumes. /The form /8/ with a nearly stationary Zef.^ was also found by using actual OF// s-functions 2 ./ That Z e££
must have a maximum where the d orbitals begin to overlap appreciably is easily understood. For large Ra , Zeff tends to unity, while for smaller lattice spacings the screening by d-electrons becomes more complete due to the OPW-shaped в-functions and the readjustment of the d-density on compres
sion.
In calculating Z renormalized atomic d-functions wore used^. One notices that the model has only two adjustable parameters a_ and b, and all the other parameters (Vc / ÍW )
needed in the calculation are independently determined atomic data.
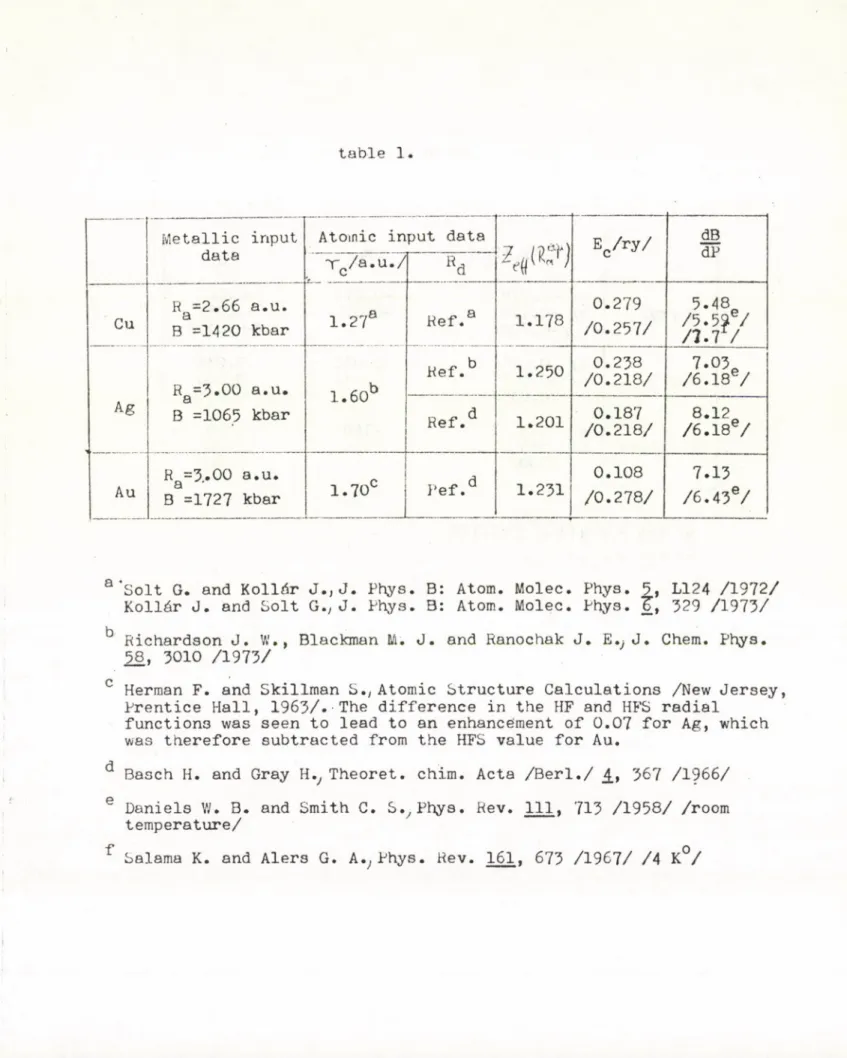
The atomic and metallic input data and the derived Ze;f;£.
are shown in table 1. The parameters a and b_ were deter
mined by fitting to the observed values of the atomic radius and bulk modulus. The resulting d-energies for Cu and Ag displaying the characteristic minimum near the equi
librium volume are seen in fig. 1. From the results for
the cohesive energy and the pressure derivative of the bulk modulus, shown in table 1., one finds acceptable agreement v/ith experiment for copper and silver. As
for gold, the much larger disagreement for the energy indi
cates the breakdown of the assumption of weakly overlapping atomic-like d-functions. /Gold has, in fact, a much broader d-band than copper and silver./
To illustrate the sensitivity of the results to varying the input atomic parameters /d-functions, / table 1. shows two sets of numbers obtained by using different analytical atomic d-functions. We notice that the largest
change occurs, through the variation of Zeff * in the value of , while ( рд and 13 д proved to be rather insensitive.
The difference in the E:0 values belonging to different sets of HP orbitals shows the absolute limit of accuracy by using prefixed atomic orbitals.
The role of the different contributions to the total energy from 3- and d-electron interactions Í3 analysed in table 2. in the case of Cu. One sees, first, that the most important contribution to the energy сотез from E c t- 1 4 +- * Second, both the s and d energy constituents are close to a stationary value at ]sc=2..b6 that is shown by the small pressure values /in units of the equilibrium bulk
modulus/. Hence one realizes again ’ that accounting only for the s-energy would already lead tc quite a good estimate for the equilibrium volume and cohesive energy, while E c^
becomes essential in determining B> . The term was
- 6 -
£
estimated by using an empty core potential with the charge
^eff* cal°u la‘t;e the elastic constants С,С* the variations of and with shear deformations are needed. As to
E s
, the contributions to the shear moduli from the electrostatic and structural terms are calculated as for an alkali metal. Although the usual assumption concerning the transformation properties of is that it can be written as a sum of pairwise central interactions between the ions, its substantial role in determining C^ /table 3./suggests that any deviation from this behaviour affects the final result considerably. With this in mind should one look at table 3. which shows the results with the assumption that is the sum of pair potentials. We notice first, that the contributions from are rather small. Further, one sees that the results generally improve with taking into account the"Rc^ dependence of /the agreement for Cu is the best when we use the d-energy found in ref. 2./ The disagreement
is striking in the case of gold, both for
C
andC*
• Thatthis discrepancy is probably not caused by the particular choice of the d-energy is shown by the last row in table 3. In fact, by assuming besides the volume dependent energy
7 terms only pairwise interactions, c ti- s h o u l d be equal to B v- I V y , where the index \J refers to the purely volume dependent terms in the energy; this holds indepen
dently of the form of the pair forces. Comparing the cal
culated values of с,г— с.цц with the observed deviation from the Cauchy relation for Cu, Ag, Au one can say, first, that the thboretical values are close to each other in all
three cases. Second, the observed c l2_-c44 for gold is
- 7 -
much too large compared to the calculated value, showing that here the actual d-electron energy contributes signi
ficantly to the deviation from the Cauchy relation. The results for Ag and Cu are less conclusive in this respect.
One can see, incidentally, that the rearrangement of the d-charge on compression affects the calculated values of rather seriously, because of the variation of %e f f
Non-zero values in column EB0 appear since this term contains other than pairwise interactions as well.7
In conclusion, the energy expressions /1/, /2/, / 3 / involving a d-energy that is near to a minimum in equilibrium, seem to be appropriate to describe the volume dependence of the total energy for Cu and Ag. In calculating the second derivatives of the energy, the readjustment of d-electrons leading to the variation of the effective charge is essential. Describing the role of the d-electróns in the binding by means of pair forces is certainly wrong for gold, while it can mot be ruled out for Ag and particularly for Cu.
8
References
1 S. S. Jaswal and L. A. Girifalco, J. Phys.Chem. Solids 28, 457 /1967/
K. Hsieh and P. Bolsaitis»
J.Phys. Chem.Solids 22» 183S /1972/
2 J. Kollár and G. Solt,
J.Phys.Chem. Solids 22» H 2 1 /1974/
3 G. Solt And J. Kollár,
Solid State Commun. 12» 957 /1974/
4 Watson R.E., Ehrenreich II. and Hodges L., Phys.Rev.Lett. 2 ± , 829 /197о/
5 J. Kollár and G. Solt,
J.Phys.Chem.Solids 22» 651 /1972/
6 G. Solt and J. Kollár,
Solid State Commun. /to be published/
7 E. G. Brovman and Yu.M.Kagan, Zhetf 22, 1635 /1969/
- 9 -
•Figure Caption
fig. 1. The variation of the d-energy with the atomic radius, as given by equ. 2.
Table Captions
Table 1. Metallic and atomic input data for the noble metals and the calculated cohesive energies and pressure
I u
derivatives of the bulk modulus • For A g cal
culations were performed with two different sets of atomic d-functions. The experimental values are in brackets.
Table 2. Different energy contributions to the total energy
E
t pressure V and bulk modulus'B
for copper. The numbers in brackets are the results of Ref. 2.Table 3. Different energy contributions to the elastic moduli
C ( C*
and c IL-с нц
for copper and final results for Ag and Au. The numbers in brackets are the results of Ref. 2.table 1
Metallic input data
Atomic in T c/a.u./
put data
Rd
V V
Ес/гу/dB dP
Cu
R =2.66 a.u.
a
В =1420 kbar 1.278 Ref .a 1.178
0.279 /0.257/
5.48o /5.59е/
/7.7 V
Ag
Ra=3.00 a.u.
В =1065 kbar
1.60b
R e f .b 1.250 0.238 /0.218/
7.03- /6.18е/
Ref.d 1.201 0.187 /0.218/
8.12 /6.18е/
Au
Rq=3..00 a.u.
В =1727 kbar 1.70C Pef.d I.23I
0.108 /0.278/
7.13 /6.43е/
a "Solt G. and Kollár J.,J. Phys. B: Atom. Molec. Phys. 5, L124 /1972/
Kollár J. and Solt G., J. Phys. B: Atom. Molec. Phys.
Tt
329 /1973/b Richardson J. W ., Blackman M. J. and Ranochak J. E.; J. Chem. Phys.
58, 3010 /1973/
c Herman F. and Skillman S.; Atomic Structure Calculations /New Jersey, Prentice Hall, 1963/. The difference in the HF and HFS radial
functions was seen to lead to an enhancement of 0.07 for Ag, which was therefore subtracted from the HFS value for Au.
d Basch H. and Gray H T h e o r e t . chim. Acta /Berl./ 4, 567 /1966/
e Daniels W. B. and Smith C. S., Phys. Rev. Ill. 713 /1958/ /room temperature/
f
Salama K. and Alers G. A., Phys. Rev. 161, 673 /1967/ /4 K°/table 2.
E +E,+E..
0 1 M e b g Ed
a/ -0.778 -0.017 -0.065
E /гу/ b/ -0.778 /-0.728/
-0.017 -0.051
/-0.055/
о/ 0.005 -0.056 0.051
P/B b/ -0.005
/0.055/
-0.057 0.040
/-0.055/
a/ 455 -168 1155
В /кЬаг/ b/ 566 -166 1020
/790/ /650/
a/ iV(| = ] =1.178
table 3
r--
Cu Ag Au
E +E,
0 1 EM EBS E^ total/exp/ total/exp/ total/exp/
а/ 0 260 -26 796 1030 780 1250
G/kbar/ b/ 0 260 -26 695 929 /818/
/535/ /795/
692 /453/ 1097 /423/
cVkbar/
а/ 0 29 8 376 413 288 531
b/ 0 29 8 319 356 /256/
/293/ /322/
238 /155/ 429 /145/
t tfb Цй" ^ a/ 159 0 -44 0 115 93 123
/kbar/
b/ 295 /411/
0 -41 0 254 /431/
/411/
214 /509/ 343/1207/
a/£e(f =
)
=const.b / ^ 4 =2e|('Ri4.)
--
Cí7
i
Kiadja a Központi Fizikai Kutató Intézet Felelős kiadó: Siklós Tivadar osztályve
zető
Szakmai lektor: Fazekas Patrik Nyelvi lektor: Fazekas Patrik Példányszám: 295 Törzsszám: 75-063 Készült a KFKI sokszorosító üzemében Budapest, 1975. január hó

![table 2. E +E,+E.. 0 1 M e b g Ed a/ -0.778 -0.017 -0.065 E /гу/ b/ -0.778 /-0.728/ -0.017 -0.051 /-0.055/ о/ 0.005 -0.056 0.051 P/B b/ -0.005 /0.055/ -0.057 0.040 /-0.055/ a/ 455 -168 1155 В /кЬаг/ b/ 566 -166 1020 /790/ /650/ a/ iV(| = ] =1.](https://thumb-eu.123doks.com/thumbv2/9dokorg/823619.41762/16.890.24.880.23.1147/table-e-e-e-ed-гу-в-кьаг.webp)
