MÓDOSÍTOTT FELÜLET Ű KAOLINIT AGYAGÁSVÁNYOK KOMPLEX ANALITIKAI VIZSGÁLATA
Doktori (PhD) értekezés
Készítette:
Vágvölgyi Veronika okleveles vegyészmérnök
Témavezető:
Dr. Horváth Erzsébet egyetemi docens
Készült a Pannon Egyetem
Vegyészmérnöki Tudományok és Anyagtudományok Doktori Iskolája keretében 2008
MÓDOSÍTOTT FELÜLETŰ KAOLINIT AGYAGÁSVÁNYOK KOMPLEX ANALITIKAI VIZSGÁLATA
Értekezés doktori (PhD) fokozat elnyerése érdekében Írta:
Vágvölgyi Veronika
Készült a Pannon Egyetem
Vegyészmérnöki Tudományok és Anyagtudományok Doktori Iskolája keretében
Témavezető: Dr. Horváth Erzsébet
Elfogadásra javaslom (igen / nem)
(aláírás) A jelölt a doktori szigorlaton …... % -ot ért el,
Veszprém, 2007. december 17. ………..
a Bíráló Bizottság elnöke
Az értekezést bírálóként elfogadásra javaslom:
Bíráló neve: …... …... igen /nem
……….
(aláírás) Bíráló neve: …... …... igen /nem
……….
(aláírás)
A jelölt az értekezés nyilvános vitáján …...% - ot ért el
Veszprém, 2008. ……….
a Bíráló Bizottság elnöke
A doktori (PhD) oklevél minősítése…...
………
Az EDT elnöke
TARTALOMJEGYZÉK
KIVONAT...3
ABSTRACT ...4
ABSTRAKT...5
1. SZAKIRODALMI ÖSSZEFOGLALÓ ...6
1.1. A kaolinit szerkezetének felépítése, helye az agyagásványok rendszerében...6
1.1.1 Az agyagásványok...6
1.1.1.1 A kétdimenziós atomköteg...7
1.1.1.2 Az oktaéderes réteg...8
1.1.2 A rétegkomplexumok egymáshoz való csatlakozása ...9
1.1.3 A kaolinit szerkezetében lévő különböző OH-csoportok ...11
1.1.4 Szerkezeti deformációk ...12
1.1.5 Az agyagásványok szerkezeti polimorfizmusa, politipizmusa ...14
1.1.6 A kaolinit kristályszerkezeti rendezettségéből adódó eltérések...16
1.2. A kaolinit analitikai vizsgálata...18
1.2.1 Röntgendiffraktometria alkalmazása...18
1.2.2 Termikus analízis alkalmazása...19
1.2.3 Rezgési spektroszkópia alkalmazása...20
1.2.4 Mágneses magrezonancia spektroszkópia alkalmazása ...22
1.2.5 Elektronmikroszkópia alkalmazása...23
1.2.6 Morfológiai vizsgálatok (fajlagos felület, pórusméret) ...23
1.2.7 Felületi sav-bázis tulajdonságok vizsgálata...25
1.2.7.1 A tesztmolekulák megválasztásának kritériumai ...26
1.2.7.2 Néhány gyakran alkalmazott tesztmolekula...28
1.3. Az interkaláció ...30
1.3.1 Az interkaláció kinetikája ...31
1.3.2 Az interkaláció jelentősége ...33
1.3.2.1 Az interkaláció, mint vizsgálati módszer...33
1.3.2.2 Az interkaláció, mint eljárás...33
1.4. A mechanokémiai aktiválás...34
1.4.1. A mechanokémiai aktiválás reakciói...35
1.4.2. A mechanokémiai aktiválás során végbemenő változások...36
2. KÍSÉRLETI RÉSZ...39
CÉLKITŰZÉS ...39
2.1. Alkalmazott módszerek, anyagok, eljárások és készülékek ...40
2.2. Hidrazin-hidráttal interkalált szegi kaolinit vizsgálata...44
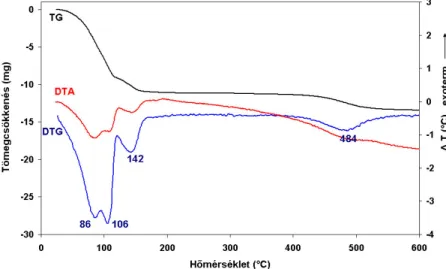
2.2.1. Kísérleti eredmények és értékelésük ...44
2.2.1.1. Termoanalitikai eredmények...44
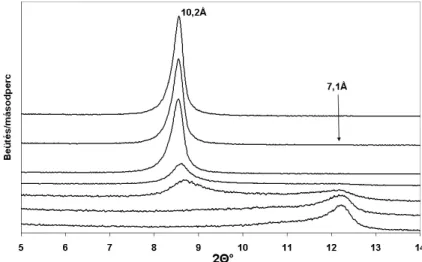
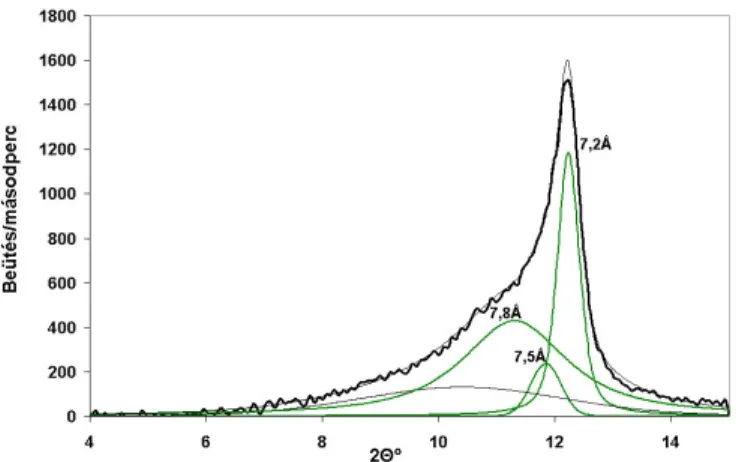
2.2.1.2. Röntgendiffraktometriai eredmények ...47
2.2.1.3. DRIFT spektroszkópiai eredmények ...50
2.3. Mechanokémiailag aktivált valamint formamiddal interkalált szegi kaolinit vizsgálata ...57
2.3.1. Kísérleti eredmények és értékelésük ...57
2.3.1.1. Röntgendiffraktometriai eredmények ...57
2.3.1.2. Termogravimetriai-tömegspektrometriai (TG-MS) vizsgálatok ...59
2.3.1.3. DRIFT spektroszkópiai eredmények ...61
2.4. Mechanokémiailag aktivált valamint formamiddal interkalált és termikusan deinterkalált szegi és zettlitzi kaolinit minták vizsgálata ...66
2.4.1. Kísérleti eredmények és értékelésük ...66
2.4.1.1. Termoanalitikai eredmények...66
2.4.1.2. Fajlagos felület mérési eredmények (BET, BJH)...69
2.4.1.3. Ammónia adszorpciós vizsgálatok ...71
2.4.1.4. DRIFT spektroszkópiai eredmények ...75
3. ÖSSZEFOGLALÁS...77
3.1. Hidrazin-hidráttal interkalált szegi kaolinit vizsgálata...77
3.2. Mechanokémiailag aktivált valamint formamiddal interkalált szegi kaolinit vizsgálata ...78
3.3. Mechanokémiailag aktivált valamint formamiddal interkalált és termikusan deinterkalált szegi és zettlitzi kaolinit vizsgálata...79
4. IRODALOMJEGYZÉK...81
A DOKTORI ÉRTEKEZÉS TÉZISEI...91
THESES ...94
TUDOMÁNYOS KÖZLEMÉNYEK JEGYZÉKE...97
KIVONAT
Széleskörű ipari felhasználása miatt a kaolinit analitikai vizsgálata nagyfokú érdeklődésre tarthat számot. Alkalmazása nagyban függ szerkezeti, illetve felületi tulajdonságaitól és ezen keresztül reaktivitásától. Ezek a tulajdonságok különböző felületmódosító eljárásokkal megváltoztathatók.
A kutatás célja módosított felületű kaolinit agyagásvány szintézise valamint a felületmódosító eljárások paramétereinek hatásvizsgálata az ásvány külső és belső felületére illetve reaktivitására. A felület módosítása mechanokémiai aktiválással, interkalációval valamint termikus kezeléssel történt. Mechanokémiai aktiválás, azaz száraz őrlés során nemcsak a szemcsék morfológiája változik meg, hanem a felületen lévő aktív centrumok sav-bázis tulajdonságai is módosulnak. Interkaláció segítségével az ásvány rétegei közé vendégmolekulák építhetők be, melynek során nanoszerkezetű komplex keletkezik. Ezzel az eljárással egyben a rétegek belső felületének reaktivitása is vizsgálható. A keletkező komplex termikus kezelése termoanalitikai berendezésben (termomérlegben) történt, ami lehetővé teszi, hogy a kezelés során lejátszódó folyamatokat in situ kövessük nyomon.
A kísérletek kétféle előfordulásból származó kaolinit mintára irányultak. A kétféle ásvány főként hidratáltsági fokában és szerkezeti rendezettségében tért el egymástól.
A módosított felület vizsgálata termoanalitikai, tömegspektrometriai, diffúz reflexiós Fourier-transzformációs infravörös spektroszkópiai valamint röntgendiffraktometriai technikákkal valósult meg. A felületanalitikai vizsgálatok morfológiai tanulmánnyal egészültek ki, illetve kísérletek készültek a felület aktív centrumai sav-bázis tulajdonságainak meghatározására.
Az alkalmazott analitikai módszerek együttes használatával lehetővé vált a felületmódosító eljárások segítségével kialakított újfajta tulajdonságokkal bíró ásványfelület komplex analitikai vizsgálata.
A kutatás eredményei a jövőben hozzájárulhatnak egy tervezhető felületi tulajdonságokkal rendelkező, természetes alapú, környezetbarát ipari adszorbens illetve adalék fejlesztéséhez.
ABSTRACT
Complex analytical investigation of kaolinite clay minerals with modified surface
The industrial utilization of kaolinite is closely related to its reactivity and surface properties. The physical and chemical nature of the active clay surface depends strongly on the parameters of surface modification by several methods.
The aim of the research is to study the effects of the mechanochemical activation, intercalation and thermal treatment on the surface of kaolinite. The modified surface was investigated by the following instrumental techniques: thermal analysis (thermogravimetry, TGA; derivative thermogravimetry, DTG, controlled-rate thermal analysis, CRTA) coupled with mass spectrometry (TG-MS), X-ray diffraction (XRD) and diffuse reflectance Fourier-transform infrared spectrometry (DRIFT). The investigations was complemented with surface acidity investigations by adsorption of ammonia as the probe molecule.
With a systematic study of the effect of the treatment parameters on the surface properties of the clay, natural, environmentally friendly adsorbents with tailored surface properties can potentially be produced.
ABSTRAKT
Komplexe analytische Untersuchung von Mineralien mit modifizierter Oberfläche
Die Anwendung von Kaolinit hängt von seinen strukturellen und oberflächigen Eigenschaften und dadurch auch von seiner Reaktivität ab.
Ziel der Forschung ist die Synthese des Minerals Kaolinit mit modifizierter Oberfläche und die Wirkungsanalyse der Parameter der Oberfläche-modifizierenden Verfahren vor allem auf die äussere und innere Oberfläche des Kristalls und auf die Reaktivität. Die Oberfläche wurde durch mechanochemisches Aktivieren, Interkalation und termisches Verfahren modifiziert. Durch mechanochemisches Aktivieren, trockenes Mahlen wird nicht nur die Morphologie der Körner, sondern auch die Säure-Base-Eigenschaften der aktiven Oberflächenzentren verändert. Durch Interkalation können zwischen die Mineralschichten Gastmoleküle eingebaut werden und dadurch entsteht ein nanostruktureller Komplex.
Durch die verwendeten analytischen Methoden wurde die komplexe analytische Untersuchung der mit Hilfe von Oberfläche-modifizierenden Verfahren ausgestalteten Mineraloberfläche und ihrer neuen Eigenschaften ermöglicht.
1. SZAKIRODALMI ÖSSZEFOGLALÓ
1.1. A kaolinit szerkezetének felépítése, helye az agyagásványok rendszerében 1.1.1 Az agyagásványok
A kaolinit a szilikát ásványok csoportjába tartozó agyagásvány, azon belül pedig a réteges (kétdimenziós) szerkezetű szilikát ásványok közé sorolható [1].
Az agyagásványok kémiailag olyan hidroszilikátok, melyek felépítésében Si4+, Al3+, Cr3+, Fe3+, Fe2+, Mg2+, Li+, Ti4+, Ni2+, Cu2+, Na+, Ca2+, K+ kationok és O2-, F-, OH- anionok vesznek részt. A kationok és anionok fontosabb adatai az 1.1. táblázatban olvashatók. A felsorolt kationok és anionok közül a kaolinitban Si4+, Al3+ kation és O2- illetve OH- anion fordul elő.
A kationok az ionsugár aránytól függő koordinációs képességük szerint négyes, hatos, nyolcas, tízes koordinációban foglalnak helyet. A hatos vagy kisebb koordinációjú kationok (ilyen a Si4+ és az Al3+ kation is) leggyakrabban négyes (tetraéderes) vagy hatos (oktaéderes) koordinációt alakítanak ki attól függően, hogy a kation/anion ionsugár arány 0,35-nél kisebb vagy nagyobb.
1.1. táblázat: Az agyagásványokban szereplő kationok és anionok fontosabb adatai [2]
AIV – aktív négyes koordinációs számú kationok AVI – aktív hatos koordinációs számú kationok
I – inaktív hatnál nagyobb koordinációs számú kationok [3, 4]
Iontípus Ionok Koordinációs
szám Ionsugár (Å)
Ion térfogat
(Å3)
Sugár arány oxigénnel
Ionizációs potenciál
(V)
Si4+ 4 0,40 0,27 0,29 44,95
AIV
Al3+ 4 0,49 0,49 0,35 29,31
Al3+ 6 0,51 0,56 0,36 28,31
Cr3+ 6 0,63 1,05 0,45 32,10
Fe3+ 6 0,64 1,10 0,46
Mg2+ 6 0,66 1,20 0,47 14,97
Li+ 6 0,68 1,32 0,49 5,29
Ti4+ 6 0,68 1,32 0,49 44,66
Ni2+ 6 0,69 1,35 0,49 18,13
Cu2+ 6 0,72 1,56 0,51 3,35
Fe2+ 6 0,74 1,70 0,53 16,24
Na+ 6 0,97 3,82 0,69 5,138
AVI
Ca2+ 6 0,99 4,09 0,71 11,82
Na+ 8 1,01 4,32 0,72 5,138
Ca2+ 8 1,03 4,58 0,74 11,82
K+ 10 1,42 12,0 1,01 4,339
Kationok
I
K+ 12 1,45 12,8 1,04 4,339
O2- 1,40 11,49 137,48
F- 1,36 10,50 184,00
Anionok
OH-
A szilikátok szerkezeti felépítése során a következőket kell figyelembe venni:
1. A Si-atomok a tetraéderes sp3 hibridszinteken lévő elektronok útján kovalens kötésben négy oxigénnel vannak körülvéve (majdnem tetraéderesen).
2. A keletkező (SiO4)4- -csoportot szilikátionnak is nevezhetjük, amely önállóan illetve egymáshoz kapcsolódva csoportokban fordulhat elő. A kapcsolódás mindig csúcsokon keresztül történik, mivel így lesz a Si-Si távolság a legnagyobb. Él menti csatlakozás csak kivételes esetben fordulhat elő, mivel így nagyobb lenne a kation-kation taszítás (1.1. ábra).
3. Az alumínium a Si-atomot helyettesítheti a szilikátokban, de a helyettesítés ritkán haladja meg az 50%-ot. Ezen kívül elvétve előfordulhat még Fe atom helyettesítés is a Si-atom helyén.
1.1. ábra. A (SiO4)4- -csoport csúcsmenti és élmenti kapcsolódása
A szilikátok nagyobb részében határozatlan számú (SiO4)4- tetraéder kapcsolódik össze atomköteggé a következőképpen:
• egydimenziós atomköteg, (SiO3)n2- vagy (Si4O11)n6-
• kétdimenziós atomköteg, (Si2O5)n2-
• háromdimenziós atomköteg, (SiO2)n
1.1.1.1 A kétdimenziós atomköteg
A kaolinit esetén kétdimenziós atomkötegekről beszélhetünk. Ez a kétdimenziós atomköteg olyan tetraéderes elrendeződést mutat, amelyben egyik lapjukkal a síkon álló tetraéderek e síkban lévő három bázisoxigén-atomjuk (OB) által szomszédos tetraéderekkel kapcsolódnak össze, míg a negyedik, a csúcson lévő oxigén atom (OA) szabadon marad (1.2. ábra). A réteg kémiai összetétele így: [(Si2O5)2-]n. A tetraéderek csatlakozása olyan, hogy a súlypontokban lévő Si-atomok és az oxigénatomok egyúttal szabályos hatszög csúcspontjain helyezkednek el. Ez a kapcsolódási mód a tetraéderek szabályos hatszöges elrendeződése. A valóságban persze a tetraéderek ideális elrendeződésétől a konkrét ásványok szerkezetében különféle eltéréseket észlelhetünk.
1.2. ábra. A tetraéderek szabályos hatszöges elrendeződése
A (Si2O5)n2- -rétegben negatív töltésfelesleg van, mert a tetraéder csúcs-oxigénatomja csupán egy vegyértékkel kapcsolódik a Si-atomhoz. E töltésfelesleg kiegyenlítésére a tetraéderes Si-O réteghez további kétdimenziós atomkötegek csatlakoznak. Legegyszerűbb esetben a vegyértékfelesleg kiegyenlítésére elég csupán egy oktaéderes réteg csatlakozása.
Ezáltal az agyagásványok, és így a kaolinit szerkezeti felépítésének tárgyalása során az oktaéderes atomköteg a következő fontos szerkezeti építő egység.
1.1.1.2 Az oktaéderes réteg
Az oktaéder központjában alapesetben Mg2+ vagy Al3+ foglal helyet, míg a csúcspontokon (OH)- illetve O2- ionok helyezkednek el. Az alapesettől eltérően előfordulhat Fe3+ helyettesítés is az oktaéder központjában. Ha az oktaéderpozíciókban két vegyértékű ionok (pl. Mg2+) foglalnak helyet, akkor az oktaéderréteg elvben megegyezik a brucit ásvány egy rétegével. Ezért ekkor brucit-szerű rétegről, vagy trioktaéderes rétegről beszélünk, mivel ilyenkor fél elemi cellánként mindhárom kationhelyzet be van töltve.
Ha viszont Al3+ ion foglal helyet az oktaéder központjában, akkor az így kialakuló réteg a gibbsit ásvány szerkezetében lévő rétegnek felel meg, amit így gibbsit-szerű rétegnek vagy dioktaéderes rétegnek nevezünk. Ez esetben fél elemi cellában két oktaéderes kationpozíció van betöltve. A dioktaéderes réteg részlete az 1.3. ábrán látható [5].
A fentiekben leírt tetraéderes és oktaéderes rétegek az agyagásványokban különféle rétegkomplexumokká kapcsolódnak össze, amelyek az ide tartozó ásványok, így a kaolinit ásványcsoport jellegzetes szerkezeti egységei.
1.3. ábra. Az oktaéderek ideális elrendeződése [5]
1.1.2 A rétegkomplexumok egymáshoz való csatlakozása
A rétegkomplexumok különbözőképpen kapcsolódva egymáshoz, különböző ásványtípusokat alakítanak ki, melyeket az 1.2. táblázatban foglaltam össze [6]. A rétegkomplexumok a következő szerkezeteket alakíthatják ki:
Legegyszerűbb esetben egy tetraéderes réteg (T) egy oktaéderes réteggel (O) kapcsolódik, vagyis két rétegből álló rétegkomplexum (TO), vagy összetett kettős réteg keletkezik. Ezek az (1:1) típusú agyagásványok. Fontos körülmény, hogy ezekben a kettős réteg fő vegyértékei kiegyenlítettek, tehát a rétegkomplexum elvileg semleges. A rétegkomplexumokat H-híd kötések csatolják egymáshoz.
A (TO) rétegkomplexumok kb. 0,71 nm-enként ismétlődnek a c-tengely irányában, s ezért a 0,71 nm körüli (001) reflexió jellemző rájuk, amely felismerésüket is lehetővé teszi. Attól függően, hogy di- vagy trioktaéderes réteget tartalmaznak, két fő csoportra oszlanak: kaolinokra és szerpentinekre.
A rétegek kapcsolódásának egy másik típusába két, egymással szembeforduló tetraéderes réteg fog közre egy oktaéderes réteget, s így három rétegből álló rétegkomplexum (TOT), vagy összetett hármas réteg keletkezik. Ezek az ásványok a (2:1) típusú ásványok.
Ezekben - ellentétben az (1:1) típusúakkal – a pozitív és negatív töltések nincsenek okvetlenül kiegyenlítve, s ebből a szempontból három fontos csoportot különböztetünk meg:
- A (2:1) rétegkomplexumok semlegesek (TOT), s így van der Waals erők tartják össze őket. Ide tartozik pl. a pirofillit és a talk. A rétegkomplexumok 0,95 nm távolságra követik egymást.
- Változó nagyságú negatív töltésfelesleg van a (2:1) rétegkomplexumban, s ezeket víz (Ai) és cserélhető kationok (C) kapcsolják össze egymással (TOTAiC). Ilyen ásványok a szmektitek, vermikulitok. Mivel ezek rétegközi terében változó mennyiségű víz adszorbeálódik, a (00l) reflexióhoz tartozó d-érték is változó, ezért a szmektit ásványok feltűnő sajátsága a rétegközi duzzadás (expandáló ásványok).
- Elemi cellánként határozott nagyságú negatív töltésfelesleg van a (2:1) rétegkomplexumban, ezért a rétegközi térben határozott helyzetben inaktív kation (I) foglal helyet a rétegkomplexumok között (TOTI). Ide tartoznak a csillámszerkezetből levezethető agyagásványok. A rétegkomplexumok ismétlődési távolsága ebben az esetben kb. 1 nm.
Önálló csatlakozási típusnak tekintjük azt az esetet, amikor a kiegyenlítetlen (2:1) rétegkomplexumok között inaktív kation helyett egy második oktaéderréteg fordul elő (TOTO). Ezek voltaképpen a [2:(1+1)] vagy (2:2) típusú ásványok, amelyek a klorit- csoport tagjait tartalmazzák. A négyes réteg periódusa és jellegzetes d(001) értéke kb.
1,4 nm.
1.2. táblázat: A filloszilikátok csoportosítása a rétegkomlexumok kapcsolódása alapján [6]
Típus Csoport Alcsoport Példa
dioktaéderes kaolinitek kaolinit, halloysit 1:1 trioktaéderes szerpentinek antigorit, lizardit
dioktaéderes pirofillit
trioktaéderes talk
dioktaéderes montmorillonoit
trioktaéderes szmektitek szaponit
dioktaéderes
trioktaéderes vermikulitok
dioktaéderes muszkovit, paragonit
trioktaéderes valódi csillámok
flogopit, annit
dioktaéderes margarit
trioktaéderes merev csillámok clintonit
dioktaéderes donbassit
di-, trioktaéderes sudoit
2:1
trioktaéderes
kloritok
chamosit, klinoklor
Mindezek alapján elmondhatjuk, hogy a kaolinit ásványcsoport dioktaéderes kétdimenziós atomköteget tartalmazó, (1:1) típusú rétegkomplexumokkal rendelkező szilikát.
A kaolinit ásványcsoport tagjait, így a kaolinitet is tehát tetraéderes és oktaéderes kettősrétegek alkotják. A kettősrétegek, vagy rétegkomplexumok ideális esetben úgy helyezkednek el egymás felett, hogy a kitöltetlen hatos gyűrűk illeszkednek egymáshoz. Ez a szerkezet persze a későbbiekben ismertetett erőhatások miatt bizonyos mértékben deformálódhat. A két réteg egyfajta kapcsolódási módját az 1.4. ábra szemlélteti. A réteg poláros, mert egyik oldalát oxigén-ionok, a másik oldalát pedig OH-ionok határolják, és minden ásványban a c-tengely irányában ezen rétegek azonos orientációban helyezkednek el. Így a réteg és egyúttal az ásvány kristálykémiai képlete: Si2O5(OH)4Al2, amiből kitűnik, hogy a szerkezetben nincsenek vízmolekulák, csak OH-csoportok.
1.4. ábra. A kettős rétegkomplexumok kapcsolódása
1.1.3 A kaolinit szerkezetében lévő különböző OH-csoportok [7]
A kettősrétegek egymáshoz a bennük lévő OH-csoportok által kialakított H-híd kötéssel kapcsolódnak. A kaolinitben háromféle OH-csoportot különböztetünk meg (1.5. ábra):
1) „külső” OH-csoportnak nevezik a mikrokristályok felületén található hidroxil- csoportokat, amelyek egyrészt a töret mentén, másrészt az oktaéderes (legkülső) réteg felületén helyezkednek el.
2) A „belső felületi” OH-csoportok a rétegkomplexum oktaéderes részének felületén találhatók. Mivel a kaolinit sok ilyen rétegből áll, ezért nevezte el a szakirodalom
„belső felületi OH-csoportnak”.
3) A „belső” OH-csoportok: az oktaéderes és tetraéderes rétegek közös síkjában helyezkednek el, és dipólusuk egy üres oktaéderes pozíció felé mutat. Vagyis a belső OH-csoportok H-atomjai az Al-atomok alatti oxigénhez kapcsolódnak, és a kaolinit rétegek közötti tér irányába mutatnak. Ezek az OH-csoportok tulajdonképpen a (001)- es síkkal párhuzamosan állnak és a kaolinit szerkezetben a dioktaéderes rétegben lévő üreg felé mutatnak. A belső OH-csoportok elhelyezkedéséről még nem alakult ki általánosan elfogadott nézet. Valószínűleg ezen csoportok irányultsága függ attól, hogy az Al-O-H-kötés hajlított vagy lineáris [8].
1.5. ábra. A kaolinit elemi cellájában lévő atomok és OH-csoportok
1.1.4 Szerkezeti deformációk
Mivel az oktaéderes és tetraéderes rétegek paraméterei nem egyeznek meg pontosan egymással, ezért e rétegek kapcsolódása során deformáció áll elő. Az agyagásványok nem tekinthetők minden további nélkül tömör anion illeszkedésű szerkezeteknek, melyeknek hézagaiban megfelelő nagyságú kationok passzív értelemben vett neutralitásra törekedve foglalnak helyet. Ehelyett a valóságos helyzetnek jobban megfelel, ha az ásványt a kötőerők – legkisebb belső energia elérésére irányuló – dinamikus egyensúlyának fogjuk fel. E kötőerők vegyesek és különböző kényszerhatás alatt állnak, mégis – a véglegesség igénye nélkül – legcélszerűbben komplex ionosnak foghatók fel.
Ennek megfelelően az agyagásványok szerkezetét a következő szabályszerűségek jellemzik:
• A kötőerők nagyobb része – a Si-O és O-H kötéstől eltekintve – nem irányított.
• A kötéstávolságok általában fordítva arányosak az elektrosztatikus kötéserősséggel [9-11]. Bár ezt a szabályszerűséget általában érvényesülni látjuk a kristályszerkezetben, de mégsem kivétel nélkül. Különleges sztérikus hatások
ugyanis rövid kötéstávolságokra vezethetnek olyan esetekben is, amikor a kötéserősség elenyészően csekély.
• A kötésszögek állandóbbak, mint a kötéstávolságok, s ezen belül az O-T-O szög állandóbb, mint a T-O-T szög. (A T-O-T szög magyarázata az 1.6. ábrán látható).
• Az anionok kölcsönös taszító hatása gyorsan növekszik a közöttük lévő távolság csökkenésével.
• Még jelentősebb a kationok kölcsönös taszító hatása, különösen több értékű ionok esetén, amelyeknek nagyságrendje elérheti a legerősebb kötőerőét is. Ezért a csúcsokon érintkező oktaéderek háromértékű kationjainak taszító hatása a rétegszilikátokban fellépő erők egyik legfontosabbika.
• A szilikátrétegben a kötésfelesleg helye és a rétegközi kation nem lehet túlságosan távol egymástól, mert nagy szabad energia alakulna ki.
A felsorolt szabályszerűségek arra vezetnek, hogy a tetraéderes rétegben a nem pertubált tetraéderek T-O kötéstávolsága megfelel a T ionsugara alapján várható értéknek, ami azonban csökkenhet, ha valamely O-ionnak nem minden vegyértékét köti le csatlakozó kation.
A SiO4-tetraéder O-T-O szöge a Si-O kötés részben kovalens jellege ellenére is gyakran eltér az ideális 109°28’-től (99-109°), vagyis a SiO4-tetraéder többnyire deformált.
A T-O-T szög még nagyobb ingadozásnak van kitéve, mert a tetraéderek csúcsokon történő érintkezése és a kis Si/O sugárarány együttes hatása a Si atomokat jól árnyékolja egymástól.
A tetraéderek ideális és deformált kapcsolódását az 1.6. ábra szemlélteti.
ω
ω ω
a) b) c)
1.6. ábra.
a) Tetraéderek trigonális szimmetria szerinti kapcsolódása, ω = 120° esetén b) Tetraéderek hexagonális szimmetria szerinti kapcsolódása, ω = 180° esetén c) Tetraéderek ditrigonális szimmetria szerinti kapcsolódása, 180° > ω > 120°
ω: T-O-T szög
Az oktaéder paramétereit három különféle erő egyensúlya határozza meg: a kation- anion vonzás, a kation-kation taszítás és az anion-anion taszítás, melyek együtt rendszerint az oktaéder deformációjához vezetnek. Ebben a fő szerepet a kation-kation taszító hatása játssza, mert az élek menti érintkezés miatt ezek nincsenek eléggé árnyékolva, továbbá az oktaéderes kationok változó nagysága, töltése és a kation-anion kötés nem irányított jellege is nagymértékben hozzájárul az oktaéder esetről esetre változó deformációjához.
Az oktaéderek alsó és felső oxigéntriádja például az élek mentén szomszédos két oxigén távolságának csökkenésével, egymással ellentétes irányban elfordul. Ezen viszonyokat értelmező úgynevezett „kation-betöltetlenségi szabálynak” [12] legfontosabb következménye az, hogy
a) dioktaéderes szerkezetben erős tendencia alakul ki az üres kation-pozíció körüli szabályos hexagonális elrendeződésre,
b) az oktaéderes réteg paramétereit a kation-anion kötéserősség és a lehetséges O-O távolság szabja meg,
c) centrumában különféle kationokat tartalmazó, egymással élek mentén kapcsolódó oktaéderek nagyjából hasonló minimális éltávolságot megközelítve rövidülnek meg.
1.1.5 Az agyagásványok szerkezeti polimorfizmusa, politipizmusa
Az agyagásványok körében gyakran lépnek fel a kétdimenziós kristályokra jellemző szerkezeti variációk. Ezek amiatt alakulhatnak ki, mivel a szerkezeti „rétegek”, különösen a semleges rétegkomplexumok, amelyek igen gyenge erőkkel kapcsolódnak egymáshoz, elég magas saját szimmetriájuk következtében a szomszédos rétegekhez változó nagyságú transzlációs vagy rotációs elmozdulások közbejöttével kapcsolódhatnak. Ezáltal egy meghatározott összetételű agyagásvány polimorf módosulatai jönnek létre; mivel azonban a szerkezet alapegységei változatlanok – a tetszőleges eltérést magában foglaló polimorfizmus helyett – ilyen esetben célszerűbb a politipizmus fogalom használata [13].
A rétegkomplexumok transzlációja vagy rotációja természetesen módosítja a teljes szerkezet szimmetriáját is. Ez az agyagásványok körében gyakran észlelhető jelenség homopolitipizmusnak fogható fel, mert az elmozdulásban részt vevő rétegek összetétel és szerkezet tekintetében azonosak. Mivel a politipizmus befolyással van az agyagásvány jelzésére, elnevezésére, illetve azok tulajdonságaira, viselkedésükre, ezért szükség van a
1. az ismétlődő identikus távolságban lévő szerkezeti egységek, rétegkomplexumok száma,
2. a teljes szerkezet szimmetriája,
3. a szerkezet „szabálytalanságának” mértéke.
A kaolinit ásványcsoport homopolitípusait az 1.3. táblázatában foglaltam össze. A homopolitípusok rétegismétlődését pedig az 1.7. ábra szemlélteti.
1.3. táblázat: A kaolinit ásványcsoport homopolitipizmusa Ásvány Homopolitípusai A homopolitípusok neve
kaolinit-T kaolinit
kaolinit-2M dickit
Kaolinit
kaolinit-2M (6R0) nakrit
1.7. ábra. A kaolinit ásványcsoport politipizmusa
1.1.6 A kaolinit kristályszerkezeti rendezettségéből adódó eltérések A kaolinitd (fire-clay) szerkezete
A kaolinithoz kémiai összetétel tekintetében igen hasonló ásványok gyakran olyan röntgendiffraktogrammot adnak, amelyen a kaolinit sok vonala gyengül vagy eltűnik, mások diffúzzá válnak. E diffrakciós jelenségek a kristályszerkezet szabályosságának változó mértékű csökkenésére, rendezetlenségére vezethetők vissza.
Ilyen fajta eltérést az úgynevezett fire-clay ásványnak is nevezett kaolinitd produkál, ahol az indexben lévő kis d az angol disordered (rendezetlen) szóból ered. Az (1:1) típusú ásványok rendezetlensége tulajdonképpen két különféle tényezőből fakad: az egyes rétegek belső rendezetlenségéből és a rétegek halmozódásának szabálytalanságából. A rendezettség illetve rendezetlenség mértéke mesterséges úton megváltoztatható, pl.
jókristályos kaolinitből hosszabb ideig tartó nedves őrlés vagy nagy nyomás vagy γ- besugárzás vagy interkaláció és deinterkaláció után kaolinitd-t kapunk. Másrészt azt is tapasztalhatjuk, hogy a rendezetlenség hő hatására csökken, pl. a szegi kaolinitd
meglehetősen diffúz (001) vonala 150°C-ig történő hevítés vagy hidrotermális kezelés hatására fokozatosan élesebbé válik.
A halloysit, metahalloysit és dehidrált halloysit
A halloysit Al2O3:SiO2=1:2 arány tekintetében a kaolinittal megegyező ásvány, kémiailag attól csupán a hidratáltsági fokában különbözik. Víztartalmának egy részét természetes körülmények között is gyakran elveszíti, így metastabilis metahalloysittá alakul. Végül 200°C-ra való hevítés során minden molekuláris víz eltávozik, és formálisan a kaolinittal megegyező összetételű dehidrált halloysit keletkezik.
Röntgenfelvételen az 1,01 nm-es reflexió a hidráthalloysit (001) indexű vonala, amely 110°C-os hevítés hatására eltűnik, és átadja a helyét a dehidrált halloysit 0,72 nm-es reflexiójának, amely megegyezik a kaolinit (001) síkhálótávolságával. A hidrát és dehidrált forma közti bázistávolság-különbség: 1,01 - 0,72 = 0,29 nm, ami ez egy vízmolekula- rétgenek megfelelő távolság. Hendricks szerint ez a vízmennyiség egy vízmolekula-réteg alakjában két kaolinitréteg között helyezkedik el [14]. Elektronmikroszkópi morfológiai tanulmányozások során megfigyelhető, hogy a halloysitok - az egyéb kaolinit ásványok táblás alakjával ellentétben – belül üres, felcsavarodott csöveket alkotnak. Ennek magyarázatát abban a méretkülönbségben kereshetjük, amely a Si2O5 és gibbsitréteg b- paraméterei között fennáll. A kaolinréteg O és OH oldala közötti hosszúságkülönbség a
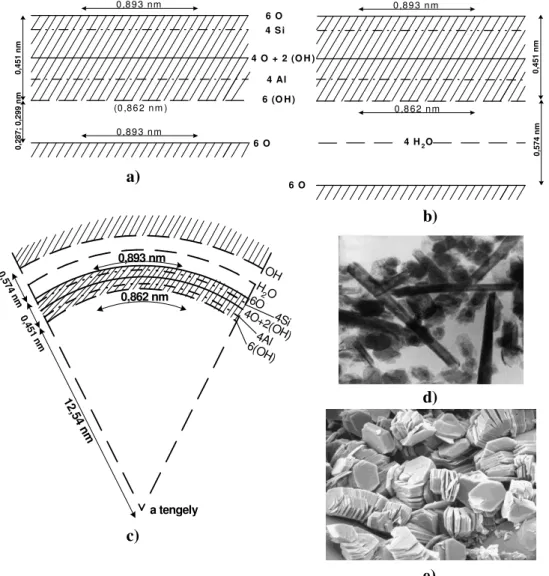
szerkezet fenntartásában a szomszédos rétegek (O-OH-kötés) is részt vesznek. Ha azonban – mint a halloysit szerkezetében – az a- és b-tengelyek szerint rendezetlenség miatt a rétegek közötti együttműködés csökken, érvényre jut az a kompressziós hatás, amelyet a gibbsit-szerű réteg a szilikátrétegre gyakorol. Minthogy a halloysitban a szomszédos rétegek 0,574 nm-re vannak egymástól, és a vízmolekula-réteg is leárnyékolja a szemben lévő rétegkomplexum oxigénhálózatának az OH-hálózatra gyakorolt „szétfeszítő” hatását, a gibbsit-szerű rétegben a hidroxil-csoportok a normál távolságot igyekeznek felvenni, amely – ha a függőleges kötéstávolságot változatlannak képzeljük – csakis a réteg meggörbülése útján lehetséges. Az 1.8. ábrán a fent leírt kaolinit- illetve halloysitréteg szerkezet vázlata, valamint a kaolinit táblás és a halloysit felcsavarodott morfológiájáról készült elektronmikroszkópos felvételek láthatók.
1.8. ábra. A kaolinit és a halloysit rétegszerkezeti és morfológiai összehasonlítása
a) kaolinit, b) halloysit-réteg, c) felcsavarodott, belül üres halloysithenger d)
e) b)
0,8 9 3 nm
(0 ,86 2 n m ) 0,89 3 n m
0,451 nm0,287; 0,299 nm
6 O 4 S i
4 O + 2 (O H ) 4 Al 6 (O H )
6 O
0,89 3 n m
0 ,8 62 n m
0,451 nm0,574 nm
4 H2O
6 O
0,893 nm
0,862 nm
OH H
2O 6O 4O+6(O4Al2(OH)4SiH) 0,574 nm
0,451 n m
12,54 nm
a tengely
c) a)
1.2. A kaolinit analitikai vizsgálata
Az agyagásványok analitikai vizsgálata során attól függően, hogy milyen információra van szükségünk többféle módszert alkalmazhatunk:
Röntgendiffraktometria Termikus analízis
Rezgési spektroszkópia (IR, Raman)
Szilárd fázisú mágneses magrezonancia spektroszkópia (NMR) Elektronmikroszkópia (SEM, TEM)
BET-felület, pórusméret vizsgálat
Sav-bázis tulajdonságvizsgálatok (tesztmolekulák adszorpció) 1.2.1 Röntgendiffraktometria alkalmazása
A röntgendiffraktometria szabályos, kristályos anyagok és így az agyagásványok vizsgálatára is alkalmazható. A kaolinit vizsgálata szempontjából a diffraktogramból többféle információhoz juthatunk:
− ásványazonosítás reflexiók intenzitásának és pozíciójának megállapításával,
− szerkezeti rendezettség meghatározása bizonyos reflexiók arányainak vizsgálatával,
− a kettősrétegek közötti bázislaptávolság meghatározása a (001)-es reflexió helyének és intenzitásának vizsgálatával (interkaláció hatásfokának nyomon követése).
Szerkezeti rendezettség meghatározása röntgendiffraktogramból
A rendezettség diffraktogramból történő meghatározására többféle módszer terjedt el [15]:
1) Hinckley index (HI) [16]. Az egyik legszélesebb körben alkalmazott mutató, amely az 1ī0 és 11ī reflexiók az alapvonaltól mért magasságainak valamint a 20-23 2Θ° közötti tartományra illesztett alapvonaltól mért 1ī0 reflexió magasságának aránya. Értéke 1,5 körül rendezett, míg a 0,5 alatti HI rendezetlen kaolinit struktúrára utal.
2) QF [17]: Szintén nagyon széles körben alkalmazott kristályossági mutató, amely az 11ī és 02ī reflexiók területének aránya egy olyan négyszög területéhez képest, aminek élei az 11ī reflexió magassága valamint a 11ī és 02ī reflexiók közötti távolság. Értéke 0,26 (rendezett) és 0,60 (rendezetlen) között változik.
3) IK [18]: Ugyanabban a zónában mérik, mint a HI-t és a QF-et. Kiszámítani a 020 és a 1ī0 csúcsok alapvonaltól mért magasságainak arányából lehet. Értéke 0,7 alatt rendezett,
4) R2 [19]: ez a módszer csak véletlenszerű hibastruktúra kimutatására érzékeny. Értéke az és az 131 csúcsok intenzitásából és a két csúcs közötti „völgy” magasságából számolandó, amely 0,7 alatt rendezetlen szerkezetre utal, míg 1,2 körül már rendezett szerkezetről beszélhetünk.
5) H&B [20]: ez a típusú index talajban lévő kaolinok kristályossági fokának megállapítására alkalmas igazán. Számítása a 020 csúcs magasságának valamint az és 003 reflexiókhoz húzott alapvonal magasságának arányán alapul.
6) FWHM [21]: ez a mutató a 001 és 002 reflexiók félértékszélességéből következtet a rendezettségre. Értéke 0,4 fölött rendezetlen, míg 0,3 alatt rendezett szerkezetre utal.
7) A szakirodalomban a fenti módszerek kombinálására is van példa [22].
1.2.2 Termikus analízis alkalmazása
Termikus analízis során a hő hatására lejátszódó átalakulási folyamatok vizsgálhatók.
Ásványok vizsgálatánál a termikus analitikai módszerek közül a termogravimetriát és a differenciáltermoanalízist alkalmazzák elterjedten.
a) A termogravimetria (TGA) a minta tömegének változását regisztrálja a hőmérséklet függvényében.
b) A differenciáltermoanalízissel (DTA) a mintában hő hatására bekövetkező, mérhető entalpiaváltozással járó folyamatok tanulmányozhatók. A minta és az inert anyag hőmérsékletének különbségét a minta vagy az inert anyag, vagy a kemencetér hőmérsékletének függvényében regisztráljuk.
c) A szimultán termikus analízis során egyidejűleg regisztrálható a minta tömegének változása (TG), a tömegváltozás sebessége (DTG), az entalpiaváltozással arányos (DTA) görbe és a minta hőmérséklete (T).
d) A termikus analízissel kinyerhető információ kiegészíthető csatolt technikákkal, pl.
tömegspektrométerrel (TG-MS) vagy IR spektrométerrel (TG-FTIR) kombinált termomérleg alkalmazásával.
Kontrollált sebességű termoanalitika
A hőbomlási folyamatok sebességét a hő- és anyagtranszport folyamatok lassúsága alapvetően befolyásolja. Ennek következtében a gyakorlatban alkalmazott fűtési sebesség (5-10°C/perc) mellett a bomlásfolyamatok jelentős mértékben átlapolhatnak, és az egyes bomlásreakciók hőmérsékleti tartománya is jelentősen függ a kísérleti körülményektől (minta mennyisége, tömörítettsége, mintatartó geometriája, kemence szellőzésének
mértéke, stb.). A Paulik-testvérek által kidolgozott ún. Q-TG technika lehetőséget biztosít arra, hogy lassú, állandó bomlási sebesség alkalmazásával a bomlásfolyamatok reprodukálható módon kváziegyensúlyi körülmények között játszódjanak le [23].
1.2.3 Rezgési spektroszkópia alkalmazása
A rétegszilikátok infravörös rezgési spektruma a hidroxil-csoportok (O-H), a szilikát anion (Si-O), az oktaéderes kation (Al-O) és a rétegközi kationok rezgési sávjaiból tevődnek össze [24]. Ezek közül a kaolinit esetében az első hárommal kell számolni. A rétegszilikátokra jellemző rezgési tartományokat a 1.4. táblázatban foglaltam össze.
1.4. táblázat: A rétegszilikátok jellemző IR rezgési sávjainak spektrális pozíciója
Spektrális tartomány (cm-1) Rezgés forrása
3400-3750 OH vegyértékrezgés
600-950 OH libráció
700-1200 Si-O vegyértékrezgés (gyengén átfed más szerkezeti rezgésekkel) 150-600 Si-O deformációs rezgés (erősen átfed a rácsrezgésekkel)
Az OH-vegyértékrezgési tartomány kitüntetett figyelmet kap a kutatások során, mivel a belső felületi változások megfigyelésének egyik legjobb módja az ebben a tartományban történő változások nyomon követése. Ledoux és White [25] hidrazinnal történő interkalációt vizsgálva kimutatta, hogy a 3697, 3669 és 3652 cm-1-nél jelentkező sávok OH rezgések. A 3620 cm-1-es sáv pedig a belső OH-csoport rezgéseként azonosítható [26]. Az újabb szakirodalom 3685 cm-1-nél egy ötödik OH vegyértékrezgést is említ a Raman spektrumban, mely nagyobb rendezettséggel bíró kaolinitek esetén az IR spektrumban is megjelenik [27].
Az OH-csoportok térbeli orientációjának magyarázatában kezdetben nem volt egyetértés. Ellentmondás alakult ki a röntgendiffrakcióval és az infravörös spektroszkópiai technikával kapott eredmények között. Jacobs szerint [28] a belső OH csoport orientációja, valamint a 3669 és 3652 cm-1-es rezgésekhez rendelhető csoportok közel párhuzamosak a rétegekkel, míg a 3697 cm-1-es sávhoz rendelhető OH-csoport inkább merőlegesen áll. A röntgendiffrakciós vizsgálatok alapján mindhárom OH csoport merőlegesen állna, mert így válik lehetővé, hogy az OH-csoportok protonjai kölcsönhatásba tudjanak lépni a szomszédos réteg oxigénjével. Ledoux és White [25]valamint Wada [29] szerint, azonban a három nagyobb frekvenciájú rezgés különböző OH csoportokhoz tartozik. Az ellentmondás feloldására a következő magyarázat született [30, 31]: az elemi cellában a c tengellyel 14-17°-ot bezáró három OH-csoport van feltételezett háromszoros
szimmetriával. Azonban háromszoros szimmetria mellett csak két sáv jelenne meg: egy intenzívebb, a rétegekre merőleges rezgés (a három OH-csoport fázisban mozgó vegyértékrezgése 3697 cm-1-nél), valamint egy gyengébb, a rétegekkel párhuzamos rezgés (a három OH-csoport nem fázisban mozgó degenerált rezgése). Mivel a háromszoros szimmetria nem teljesen pontos, ezért az utóbbi felhasad két sávra: 3652 és 3669 cm-1-re.
Hidratált halloysit esetén a 3400-3500 cm-1-es tartományban az adszorbeált víz széles sávja is jelentkezik és a 3696 cm-1-es sáv gyengülése figyelhető meg, ami más interkalált komplexnél is észlelhető [32-35]. Ezek mellett a halloysit spektrumában 3570 cm-1-nél lehet azonosítani H-híd kötésben lévő OH-csoport rezgést [36].
Az OH-csoportok rezgései az alacsonyabb hullámszámoknál is megfigyelhetők: a belső felületi OH-csoport deformációs rezgése 936 cm-1-nél, míg a belső OH-csoportra jellemző rezgés 915 cm-1-nél jelentkezik [29].
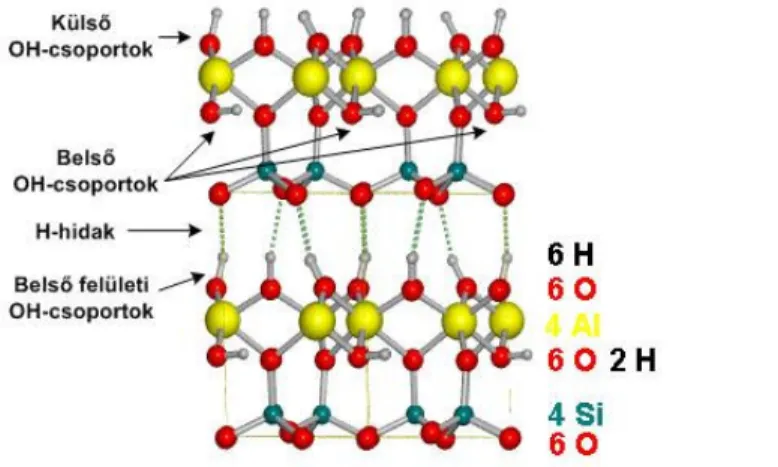
A szilikát anion rezgései az alacsonyabb hullámszám tartományban azonosíthatók (1.9.
ábra) néhány OH-csoport rezgéssel és oktaéderes kation rezgési sávjaival átfedve:
1100 cm-1 körül: merőleges Si-O rezgés [29]
970 és 1070 cm-1 között két intenzív sáv: Si-O-Si síkbeli vegyértékrezgése
400-550 cm-1: Si-O deformációs rezgés (erősen átfed az oktaéderes kation rezgésével)
800-950 cm-1: librációs rezgések (OH-csoportét is beleértve)
Az oktaéderes kation rezgései a 600 cm-1 hullámszám alatti tartományban jelentkeznek.
1.9. ábra. A szilikát anion rezgései az alacsonyabb hullámszám tartományban [29]
Az IR spektrumok felvételének technikája
Szilárd anyagok infravörös spektroszkópiai analízisének jól bevált módszere az ún.
pasztilla technika, amikor achát mozsárban 1-2mg mintát kb. 100mg szilárd KBr-dal elkeverünk, majd ezt a homogén keveréket speciális présben nyomás alá helyezünk. A nyomás alatt a KBr megolvad, majd a nyomás megszűnése után átlátszó anyaggá szilárdul.
Az így kapott pasztillát, melyben a mintaszemcsék egyenletesen helyezkednek el, a fényútba helyezve a spektrum regisztrálható.
Olyan anyagok esetén, melyek a pasztillázás során deformálódhatnak, esetünkben pl. a KBr a nyomás hatására bejuthat a kaolinit rétegek közé, a pasztillamódszer nem alkalmazható. Ezért a mintát csak elkeverjük a KBr-dal, de nem préseljük össze. Így azonban nem kapunk infravörös sugárzás számára átjárható mintát. Ekkor alkalmazzuk az ún. reflexiós technikát, melynek során a beeső sugárzás a minta és a KBr szemcséin diffúz módon reflektálódik, és a reflektált sugárzás mintegy 50%-a egy homorú gömbtükörrel összegyűjtve a detektorba jut. Az így előállított spektrum nagyon hasonló az abszorpciós spektrumhoz, és annak megfelelően értelmezhető és értékelhető ki. Ehhez a méréshez egy speciálisan kialakított mintatartó és tükörrendszer szükséges, ami az 1.10. ábrán látható. A fény útját az 1, 2, 3, 4, 5 és 6 jelű tükrök és az S-sel jelölt minta segítségével követhetjük nyomon.
1.10. ábra. A DRIFT adapter felépítése és a fény útja
1.2.4 Mágneses magrezonancia spektroszkópia alkalmazása
A szerkezet ún. rövid távú rendezettségének meghatározására a szilárd fázisú MAS/NMR spektroszkópia alkalmas. Segítségével megállapítható:
− a tetraéderes oxigénekkel közvetlenül csatoló atomok kémiai jellege, valamint a Si
− az atomok közötti távolságok és szögek [39, 40],
− a hidrogén híd kötések erőssége [41].
1.2.5 Elektronmikroszkópia alkalmazása
Transzmissziós (TEM) és pásztázó (SEM) elektronmikroszkópiai technikákkal morfológiai sajátságok állapíthatók meg. Az 1.11. ábrán néhány kaolinit SEM illetve TEM felvétele látható.
1.11. ábra. SEM és TEM felvételek
A) SEM felvétel kaolinitről (www.petrotech-assoc.com/images);
B) TEM felvétel kaolinitről (www.gly.uga.edu/schroeder/geol6550/CM07.html)
C) SEM felvétel hidratált halloysit csövekről (www.minersoc.org/.../halloysite8c.html); D) és E) TEM felvétel csöves és gömbszerű halloysitról (www.gly.uga.edu/schroeder/geol6550/CM07.html)
1.2.6 Morfológiai vizsgálatok (fajlagos felület, pórusméret)
Porózus anyagok felületének valamint pórusszerkezetének jellemzésében a gáz adszorpció fontos szerepet játszik. Általában nitrogén gáz adszorpciós izotermájának felvételéből következtetnek a fajlagos felületre illetve a pórusméretekre. A módszer lényege, hogy mérjük a gáz mennyiségét (térfogatát) az adszorpció előtt és után. A fajlagos felület meghatározásának legelterjedtebb, mára már standard eljárássá vált módszere a Brunauer, Emmett és Teller által kidolgozott ún. BET elmélet [42]. A modell alapja a következő:
- az adszorpciós fázis több molekularétegből áll,
- az első rétegnek kitüntetett szerepe van, mert csak ennek a rétegnek a molekulái érintkeznek az adszorbens felületével. Ezt a réteget „valódi” adszorpciós hő jellemzi, a további rétegek molekulái csak az adszorbátum molekulákkal érintkeznek, és adszorpciós hőjük a rétegszámtól függetlenül azonos az adszorptívum kondenzációs hőjével,
- egyensúly esetén az adott számú réteggel borított felületek nagysága állandó.
A BET elméletből levezetett ún. kétállandós egyenlet:
Ahol: u adszorbátum teljes mennyisége
um teljes felület monomolekuláris borítottságához szükséges adszorbátum mennyisége
x relatív nyomás (p/po)
c gáz/szilárd kölcsönhatás erősségére jellemző paraméter
Kísérleti adatok alapján a bal oldalt x függvényében ábrázolva egyenest kapunk, melynek meredekségéből és ordinátametszetéből um és c meghatározható. A monomolekuláris borításhoz szükséges anyagmennyiség (um) és egy adszorptívum molekula felületigényének ismeretében pedig kiszámítható az adszorbens felülete. A BET egyenlet a 0,05 és 0,35 p/p0 relatív nyomástartományban ad jó eredményt az adszorbens felületére.
A nitrogén izoterma segítségével pórusméret elemzés is végezhető, melyre a Barrett, Joyner és Halenda által kidolgozott BJH elmélet ad lehetőséget [43]. A klasszikus megközelítésnek megfelelően a módosított Kelvin-egyenlet segítségével a pórusnyílás mérete meghatározható a pórust kitöltő folyadék gőznyomásából. Mindehhez feltételezzük, hogy a pórusok jól definiáltak, azaz alakjuk hengeres, faluk párhuzamos, valamint azt hogy a meniszkusz görbülete függ a pórus méretétől.
Porózus anyagok esetén ún. IV. típusú adszorpciós izotermával kell számolni, amely az 1.12. ábrán látható a görbe részeinek magyarázatával valamint a részlépésekhez rendelt fizikai tartalommal [44].
( )
( )
x
u x u c
x c
m u cm
1
1 1
− = + −
1.12. ábra. Adszorpció porózus anyagokon
1.2.7 Felületi sav-bázis tulajdonságok vizsgálata
Szilárd anyagok sav-bázis tulajdonsága tesztmolekulákkal vizsgálható, bár ezek a tulajdonságok a felület és a tesztmolekula kölcsönhatásától is függnek.
Ebből az következik, hogy a savasság nem becsülhető csupán a szilárd anyag vizsgálatával, hanem a szilárd felület és a (bázikus) molekula közötti kölcsönhatás vizsgálata is szükséges.
A tesztmolekula/szorpciós centrum adszorpciós sztöchiometriája meghatározható (i) spektroszkópiásan a szilárd sav funkciós csoportjainak (pl. hidroxil csoportok) változásaiból és a szorbeált molekula karakterisztikus sávjainak növekedéséből vagy (ii) párhuzamos gravimetriás illetve térfogatos mérésekből.
A vizsgálat céljától függően bizonyos kritériumokat követve választhatjuk ki a megfelelő tesztmolekulát [45]. A legfontosabb kritériumokat az 1.5. táblázat foglalja össze.
1.5. táblázat: A tesztmolekula kiválasztásának kritériumai [46]
Lewis savas centrum Bronsted savas centrum Szorpciós komplex Elektronpár
donor/akceptor H- híd Ionpár (H-híd)
A komplex kialakulásának detektálása
νB sávpozíció változása νOH sávalak és sávpozíció változása
νOH eltűnése, νB-H
és/vagy δB-H megjelenése A saverősség
meghatározásának módszere
Korreláció a νB és az
adszorpciós hő között νOH eltolódás adott tesztmolekula esetén
A H-hidas ionpár komplex termikus stabilitása Savas centrumok
koncentrációjának
meghatározása νB intenzitásából νOH intenzitásából Az ionpár komplex karakterisztikus sávjának intenzitásából
Spektrális tulajdonságokkal szembeni elvárások
A νB eltolódása jelentős legyen a
félértékszélességhez viszonyítva
A tesztmolekula ne tartalmazzon OH- csoportokat
A karakterisztikus sáv egyértelműen az ionpár komplexhez tartozzon Általában alkalmazott
molekulák Piridin, NH3, acetonitril, benzonitril, CO
Benzol, aceton, piridin, szubsztituált piridinek, aminok, acetonitril
NH3, piridin és származékai jelmagyarázat: νB a B tesztmolekula karakterisztikus rezgése
νOH- a szilárd anyag OH vegyérték rezgése
1.2.7.1 A tesztmolekulák megválasztásának kritériumai
1. kritérium: A tesztmolekula legyen inkább bázikus illetve gyengén savas tulajdonságú.
A molekula a szorpció során elsősorban bázikus (elektronpár donor) csoportjával lép kölcsönhatásba a felülettel, míg savas (elektronpár akceptor) funkciója alig van hatással az anyagra. Ilyen jellegű molekula. pl. az ammónia és a legtöbb amin, ideértve a piridint is. Ammónia alkalmazása során elsődleges kölcsönhatás a savas felületen a nitrogén atom (magányos elektronpárján keresztül) és a Bronsted vagy Lewis savas (elektronpár akceptor) centrum között jön létre. Igaz, nem csak a hidroxil csoport
saverőssége határozza meg a szorpciós komplexben lévő kölcsönhatás erősségét (az ammónia a szorpció során vagy protonálódik vagy nem) [47], vagyis minél komplexebb a savasság meghatározásra használt molekula (pl.
ketonok [48, 49]), annál nagyobb az esély szimultán sav-bázis kölcsönhatás kialakulására, amely megnehezíti a szilárd anyag savassági tulajdonságainak meghatározását.
2. kritérium: Az adszorbeált molekula IR-spektrumában megkülönböztethető legyen a protonos (Bronsted) és aprotikus (Lewis) savas centrum.
Ez akkor alkalmazható, amikor a két fajta módon kötött (Bronsted illetve Lewis) molekula spektrális tulajdonságai jelentősen eltérnek, illetve az adszorpció során karakterisztikus változások mennek végbe a protonos illetve a koordinatív jellegű kötődés eredményeként [50]. Erős bázisok, pl. az ammónia esetén a protonálódás könnyen kimutatható [51], míg gyengébb bázisok (alkoholok, ketonok, víz) esetén ezt nehezebb eldönteni. A koordinatíve telitetlen fém kationokon történő szorpció általában erősebb, mint a hidroxil csoportokon végbemenő [52].
3. kritérium: A tesztmolekulának különbséget kell tenni az ugyanolyan típusú, de eltérő erősségű savas centrumok között.
Általában az erős bázis (pl. piridin) alkalmasabb a kismértékű saverősségbeli különbség kimutatására, mivel nincs protonálódás, míg a gyenge bázis (pl.
benzol) megfelelőbb a nagyobb mértékű különbségek detektálására, de célszerű a sav-bázis kötésben részt vevő orbitálok polarizálhatóságát is figyelembe kell venni [53]. Eszerint az összehasonlítható polarizálhatósággal rendelkező sav-bázis centrumok közötti kölcsönhatások valószínűsíthetők, vagyis inkább kemény-kemény, és lágy-lágy kölcsönhatások alakulnak ki, mintsem kemény-lágy [54-56]. Itt meg kell jegyezni, hogy a molekulák keménységéről felállított rangsor eléggé vitatott [46, 57].
4. kritérium: A tesztmolekula mérete összehasonlítható legyen a későbbi alkalmazás során használandó reaktánsok méretével.
A legkisebb méretű molekulákkal (CO, NH3) a felület összes savas centruma tesztelhető. A méret növelésével egyre jobban érvényesülnek a sztérikus gátlások. Ezt kihasználva a savas centrumok körüli sztérikus gátlás tesztelhető egy növekvő méretű tesztmolekula sorozattal vagy olyan sorozattal, melyben az elektronpár donor (bázis) funkció körül növekszik a sztérikus gátlás.
1.2.7.2 Néhány gyakran alkalmazott tesztmolekula Ammónia
A felületi savasság vizsgálatában az egyik legtöbbet alkalmazott tesztmolekula. Kis méretének köszönhetően minden savas centrum elérhető vele, viszont saverősség vizsgálatára nem igazán alkalmas. Kemény bázisként elvárhatjuk, hogy erős kölcsönhatásba lépjen az olyan savas centrumokkal, mint a hidroxil csoportok protonjai vagy a kis fém kationok. A protonált molekula (ammónium ion) és a koordinatíve kötött ammónia spektroszkópiai úton az NH deformációs és vegyértékrezgésük által megkülönböztethető. Az ammónium ion 1450 és 3300 cm-1-nél, míg a koordinatíve kötött ammónia 1250, 1630 és 3330 cm-1-nél jelenik meg a spektrumban. A legmegbízhatóbb indikátorként az 1450 és az 1630 cm-1-es deformációs sávok használhatók.
Az NH vegyértékrezgés spektrális tartománya viszonylag bonyolult, mert az adszorpció során bekövetkező molekula torzulás szabad és pertubált N-H rezgések összetett sávjaihoz vezet, mely további átlapolásokkal sújtott a deformációs rezgések felhangjai és kombinációs rezgései által [58].
Az ammónia alkalmazásánál további két komplikációval kell számolni. Az egyik a magasabb hőmérsékleten (T>500K) történő kísérletek során fordul elő, amikor az NH3
disszociált formában NH2 és NH csoport formában szeretne szorbeálódni a Lewis savas centrumokon, vagy a meglévő OH csoportokat NH2 csoportokra cseréli [59, 60]. A másik komplikáció a molekuláris víz jelenléte. Ilyenkor ammónium ion keletkezhet például a szilícium-alumínium-oxidokon [61]. Az átlapolás megnehezíti az IR spektrumok kiértékelését.
Alifás aminok
A legtöbbet alkalmazott molekula az n-butil-amin. Oxidok Lewis savas centrumain kötődve 1605 cm-1-nél jelenik meg (-NH2 deformációs) sáv. Protonált butil-amint figyeltek meg szilícium-alumínium-oxidon és zeolitokon [62], mely 1590 és 1510 cm-1-nél jelenik meg az IR spektrumban.
Piridin és származékai
Gáz fázisban a piridin erősebb bázis az ammóniánál, protonálódása könnyebben végbemegy és a protonált forma termikus stabilitása is nagyobb, mint az ammónia esetén
míg az 1445 és 1460 cm-1 közötti sávokat a koordinált módon adszorbeált piridinhez kötik [50]. Míg az 1540 cm-1-es sáv spektrális pozícióját tekintve a szilárd anyag savasságával nem változik, addig a koordinált piridinhez tartozó sáv a nagyobb értékek felé tolódik el a kölcsönhatás erősödésével. A piridin szorpció mennyiségi analíziséről Lavalley és munkatársai számoltak be [64, 65].
Nitrilek
A nitrilek viszonylag gyenge bázisok és a nitril-csoport nitrogénjén keresztül tudnak kapcsolódni a savas centrumokhoz. A kölcsönhatás során a C-N vegyértékrezgés sávja a magasabb hullámszámok felé tolódik el. A hozzáférhető fém kationokhoz történő koordinációkor a kék eltolódás kb. 30-60 cm-1 [66], míg a hidroxil csoporthoz történő koordinációkor 10-30 cm-1-es eltolódás észlelhető [67-69].
Kis mérete miatt számos oxid jellemzésére az acetonitrilt használják [70-77]. Nagyobb nitrilek, melyek kevésbé reaktívak (pl. terc-butironitril) jól alkalmazhatók alumínium-oxid [66], szilícium-dioxid – titán-dioxid és más kevert oxidok [75, 78, 79] vizsgálatában.
Benzol és származékai
A benzolok és a szubsztituált származékaik gyengén bázikus molekulák, melyek π- kötést alakítanak ki a Bronsted és Lewis savas centrumokkal. A kötések erőssége a Bronsted savas hidroxil csoportok OH sáveltolódása alapján, illetve a CH vegyértékrezgéshez és a gyűrűrezgéshez tartozó sávok változásának nyomon követésével vizsgálható [80, 81]. Amorf SiO2/Al2O3 esetén, mely csak egy OH vegyértékrezgési sávot mutatott 3750 cm-1-nél, adszorpció után két pertubált OH sávot figyeltek meg 110 és 310 cm-1-rel eltolódva [73]. Továbbá elmondható, hogy általában a benzol és a Lewis savas centrumok között erősebb kölcsönhatás alakul ki, mint a Bronsted savas centrumok esetén [82].
Szén-monoxid
A benzolhoz hasonlóan a CO is gyenge bázisnak tekinthető lágy karakterrel, így kemény savakkal nem igazán lép reakcióba. Kis méretének köszönhetően azonban minden savas centrum elérhető vele. A CO a szén atomon keresztül kapcsolódik a savas centrumhoz, eltolva a CO vegyértékrezgés sávját a nagyobb hullámszámok felé [83]. Szén- monoxid adszorpció segítségével nemcsak az elérhető fém kationok töltését, de a koordinációját is megállapíthatjuk. Például alumínium-oxidok vizsgálata során,
összehasonlítva a négyes és hatos koordinációjú Al3+-ot, alacsonyabb koordináció esetén erősebb kölcsönhatás alakul ki [84-87].
Egyéb tesztmolekulák
A felsoroltak mellett számos más molekulát is használnak az aktív centrumok vizsgálatára, pl. ketonokat, aldehideket, étereket, alkánokat és alkéneket. Ezek közül a legszélesebb körben alkalmazott anyagok a ketonok azon belül is az aceton.
1.3. Az interkaláció
Az agyagásványok szerves és szervetlen vegyületekkel is kölcsönhatásba léphetnek többféle mechanizmussal, pl. adszorpcióval, interkalációval, kationcserével. Kis molekulájú szerves anyagokkal reagálva a kaolinit expanziót szenved el, mivel a reagens molekulák beékelődve a kaolinit-rétegek közé kitágítják azokat. A vendégmolekula lényegében delaminálja az ásványt [88-90]. A folyamatot interkalációnak nevezzük, melynek fő alapelveit Weiss írta le [91].
A reaktív molekulákat a kölcsönhatás módja alapján a következőképpen csoportosíthatjuk:
• A szilikátréteggel erős H-híd kötést kialakító molekulák, pl.: karbamid, hidrazin (-hidrát), formamid, acetamid. Ezen molekuláknak egyszerre kell H-híd akceptor illetve donor funkcióval rendelkezniük:
1.13. ábra. A formamid, mint interkalációs reagens
• A szilikátréteggel erős dipól kölcsönhatást kialakító molekulák, pl.: dimetil- szulfoxid, (DMSO):
1.14. ábra. A DMSO, mint interkalációs reagens


![1.9. ábra. A szilikát anion rezgései az alacsonyabb hullámszám tartományban [29]](https://thumb-eu.123doks.com/thumbv2/9dokorg/877211.47214/23.918.252.681.716.966/ábra-szilikát-anion-rezgései-alacsonyabb-hullámszám-tartományban.webp)