INTRODUCTORY LECTURE
A . T E R E N I N
Leningrad University, U.S.S.R.
The main types of photochemical processes, observed in liquid solutions, are subjected in photobiology to specific changes and complications arising from the heterogeneous character of the medium and the fixation of the reactants on the biopolymer. Nevertheless, valuable information can be obtained from more simple model systems, for example in frozen rigid solutions and even in the gas phase, where we can unambiguously identify the primary act of excitation and bond fission in a large organic molecule. This presenta- tion is restricted to some recent topics in which we are involved and which have direct implications for photobiology.
I . D I S R U P T I O N A N D I O N I Z A T I O N O F M O L E C U L E S B Y V A C U U M U . V . R A D I A T I O N
The range of wavelengths from 180 to 90 nm, equivalent to photon energies from 155 to 310 kcal/mole, represents a borderland between X-rays and the ordinary u.v. Most biological entities and structures in this range have absorption coefficients much higher than those in the usual spectral ranges. Photochemical efficiencies in the vacuum u.v.
are also high, quantum yields of 1 being reported, for example for enzyme photoinactivation (SETLOW et al, i960).
The photolysis of aromatic amines, the amino acids and the nitro- geneous bases by vacuum u.v. radiation, which we are studying in the gas phase by means of mass-spectrometry, shows the existence of three main photoprocesses (TERENIN and VILESSOV, 1964):
(1) electron abstraction without a disruption of the parent molecule at a threshold hvx :
hu,
R1-R2 > Rx-R2+ + e (1)
where Rx and R2 indicate constituent groups, joined by a covalent linkage ;
Ph—Ni Ph—N<
(2) photolysis into an ionized fragment and a neutral hydrogen atom, or radical, which begins at a higher photon threshold hv2 :
Rt-R2 R{t+ 'R2(-H)
+ e
(2) (3) disruption of cyclics at still higher energies hv3:R1-R2 r it + - r2 + * (3)
The electron photoemission from the molecules, process (1), helps to obtain values of the ionization energies, which parallels the electron donating properties of the molecules concerned, and this information is of primary importance for understanding intermolecular electron transfer reactions.
In process (2) the molecular positive ion, which has acquired a definite excess of energy above the work done to remove an electron, breaks into a positively charged fragment and a neutral particle in a time less than i o ~7 sec.
As an example, glycine at a photon energy of 9-6 eV is disrupted into the following fragments, the first one being observed in the mass spectrum :
H2N—CH2—COOH kV > H2N—CH2t+ COOH + e
^9-6eV
The most remarkable fact in this ionic dissociation process is that in several cases the energy excess imparted to the molecule over the work of electron abstraction is only a fraction of the ruptured bond strength, as known for the unionized neutral molecule. Thus the disruption energy for glycine above is only ο·6 eV as compared with 3-7 eV, the normal C — C bond strength. This means a significant redistribution of the electron density in the ion formed, leading to an abnormal reduction of bond strength at definite linkages.
A similar loosening of the C — C bond has been observed for a- and jS-alanine, and β-phenyl alanine. In these cases one can presume that the ionized fragment H2N — C H2 + assumes the valency configuration H2N+= C H2, with a corresponding energy gain.
A similar dissociative photoionization has been found by us for iV-methylaniline, partly deuterated in order to identify the site of Η atom elimination.
+ H + e (a)
The C—H bond in the methyl end group is broken, but not the Ph—Ν or Ν — C H3 bonds. Cases of bond splitting in a side chain are known for benzene derivatives, photolysed in frozen rigid solutions by light in the ordinary u.v. range (PORTER et al, 1955-63). In case (a) the excitation energy imparted to the ionized molecule amounts to 3-66 eV, which is sufficient to split up a normal C—H bond. However, benzyl- amine is photolyzed with the elimination of an H atom from the connecting methylene group :
Ph—CH2—ND2 kv > Ph—CH—ND2+ + H + e 5*9*4 eV
The energy excess ΔΕ = hv — Ip imparted to the molecular ion at the threshold is only ο·8 eV, which is certainly well below the usual C—Η bond strength (3*4 eV). Similar effects have also been found for
the hydrazines in our laboratory (AKOPIAN and VILESSOV, 1963).
Evidently, as in the former example, an electronic rearrangement in the fragments has taken place with a gain in energy compensating the deficiency. A valency redistribution of the ionic fragment into P h — C H2= N D2 may be suggested.
At the higher energies ( > 11 eV « 250 kcal/mole) drastic cleavage processes occur with opening of the heterocyclic rings of pyridine, y-picoline, uracil, etc. In the aromatic amines the part primarily affected by the photon is the phenyl 'chromophore', but in the very short u.v. spectral range here considered the N H2 and O H groups can be the directly absorbing ones.
I I . S E L F - I O N I Z A T I O N O F T H E P H O T O E X C I T E D M O L E C U L E
The abnormally long (up to ο·οι sec) delayed fluorescence spectrum of aromatic amines, acridine and its derivatives, carbocyanin and fluorescein dyes in frozen rigid solutions (77°K), has been satis- factorily explained as due to the detachment of an electron from the excited singlet molecule, its trapping by the medium, and to the slow recombination process with the positive ion, an excited molecule
being reformed (LEWIS et al, 1 9 4 2 ; DEBYE et al, 1 9 5 2 ; L I M et al, 1962-63 ; KERN et al, 1 9 6 2 ; SHABLIA et al, 1 9 6 4 ; KALANTAR et al,
1962; KROG et al, 1963). T h e same process has been observed in frozen alkaline serum albumin, in amino acids, pyrimidine, purine bases, nucleic acids, etc. and is of general occurrence (DUMARTIN
et al, 1 9 5 7 ; KATIBNIKOV et al, 1 9 6 2 ; MEKSHENKOV et al, 1961 ; KONEV
et al, 1961 ; BURSHTEIN, 1961 ; IMAHORI et al, 1959; ROSENHECK et al,
1961).
The yield of such delayed fluorescence is of the order of 1 per cent of the normal fluorescence and increases at wavelengths shorter than the maximum of the first absorption band of the molecule. This suggests that some small activation energy must be additionally spent from the absorbed photon to achieve this remarkable electron detach- ment with an energy value of only 3-4 eV, i.e. about half the ionization potential of the free molecule (7 eV). It should be noted that it has been recently found that although the absorption of one photon is sufficient
MsoLv. e"
MsoLv.
FIG. Ι . Diagram of the consecutive steps involved in the delayed fluorescence caused by electron recombination.
to detach the electron from the parent molecule the absorption of a second photon is required in order to remove it from the proximity of the parent molecule and destroy this primary charge transfer complex (KALANTAR and ALBRECHT et al,
1962).
The two-step process of ionization can be thus represented by the sequence of events shown in Fig. 1.
I I I . B I - P H O T O N I C P H O T O C H E M I C A L P R O C E S S E S There exist two-step photochemical processes of another kind.
The photochemical activity of organic molecules in their triplet state, in particular that of chlorophyll, hematoporphyrin, riboflavin etc., has been widely reviewed and does not require extended comment
I N T R O D U C T O R Y L E C T U R E 7 here. Recently several photochemical processes have been found which have demonstrated that a conversion of the photoexcited molecule to the triplet state is prerequisite, but that for the reaction a second photon must be absorbed by the triplet molecule. The rate of such photochemical reactions then becomes proportional to the square of the light intensity, instead of to the usual first power relationship to which photochemists have traditionally accustomed themselves.
Three types of processes of such intensity non-linear photochemistry have recently been found. They have been studied in frozen solutions by e.p.r. and phosphorescence methods.
(1) T h e photodissociation of triphenylmethane, its chloride, carbinol, etc. with the production of the triphenylmethyl radical P h3C - (KOZLOV et al, 1963).
Ph3C—X — P h3C — X * - * T _ P h 3 C + X ( X = H , C l , O H ) In this photolysis the energy requirement for the splitting of the feeble (ca. 50 kcal/mole) C — R ( R = H , CI, OH) bond should be already met by one photon excitation, as observed in the initial stage, when the concentration of triplet molecules is low. The bi-photonic mechanism dominates at a later stage.
(2) The alkylbenzenes (toluene, etc.), di- and tri-phenyl methane, etc. act as photosensitizers evolving hydrogen from saturated hydro- carbons (e.g. 3-methylpentane) in which they are dissolved. This bi-photonic reaction proceeds for toluene through the following sequence of steps (VINOGRADOVA et al, 1964):
PhCD3 k V1 > P h C D3* ^ T hv* > PhCD3T',HR > PhCD3 + H - + R - deutero hydrocarb.
toluene solvent
It has been kinetically proved that triplet molecules (T) are the active ones, and that a second photon is required to reach a higher triplet (τ' ) level in order to break a H — C bond in the hydrocarbon solvent by energy transfer. Ninety-four per cent of hydrogen is evolved as H2; the remaining 6 per cent in the H D form is ascribed to the secondary process shown. The radical R- is detected by its e.p.r.
spectrum.
H - + H R > H2 + R-(94%) H + P h C D3 > HD + PhCD2(6%)
(3) H atoms are likewise abstracted from the alcohols by the photoexcited aromatic amines (diphenylamine, triphenylamine, N- methyldiphenylamine) and the iV"-heterocycles (carbazole, indole, tryptophane, porphyrines and flavines, etc.) in a bi-photonic process (SMALLER, 1963 ; HOLMOGOROV et al, 1963 ; BAGDASSARIAN et al, 1963 ; BAJIN et al, 1964; PISSKUNOV et al, 1964; GRIBOVA et al, 1963; PTAK et al, 1963). We presented the following interpretation for triphenyl- amine (HOLMOGOROV et al, 1963):
Ph3N — P h3N * - + T — P h a N ^ C H s O H > Ph3N + H- + CH2OH 7eV
The mechanism consists either in a transient H transfer to a high triplet state of the amine with the formation of a short-lived reduced form, or in an energy transfer to the C—H bond of the alcohol, similarly to the former photosensitized reaction (2). Experiments with deuterated alcohols seem to show that quite unusually it is the H atom from the hydroxyl group which is abstracted, not the one from C—H in the α-position (PISSKUNOV et al, 1964). This latter H atom is presumably split from a second alcohol molecule by the R—O- radical, primarily formed.
By selective deactivation with added triplet energy acceptors it has been experimentally proved that the triplet molecule is involved (HOLMOGOROV et al, 1963). The second photon must be a large one and, in fact, exceeds the height of the second triplet level. A very high energy level must thus be reached and the sensitizer molecule might even be ionized by this process of two-fold excitation. In fact, the ionization potential level of 7 eV (160 kcal/mole) becomes accessible by the successive absorption of two photons in the near u.v. range (ca. 350 nm, equivalent to 80 kcal/mole).
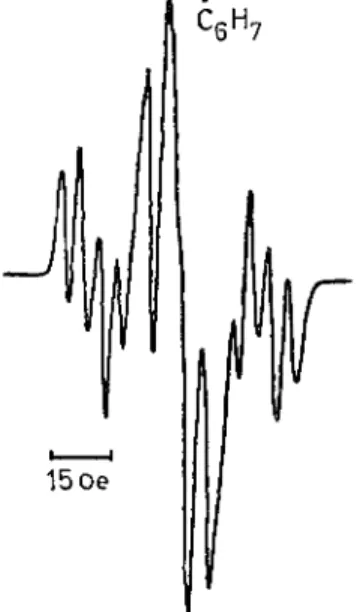
Moreover, we observed the doublet e.p.r. signal of the H atom (HOLMOGOROV et al, 1963) provided the atoms are stabilized by adsorption on silica gel, which was introduced into the illuminated solution (Fig. 2). The photogeneration of active H atoms in this reac- tion is also indicated by the appearance, on the addition of benzene to the solution, of the s.f.s. e.p.r. band of the radical C6H7 together with that of · C H2O H (Fig. 3).
There is no necessity to use extremely high light fluxes to observe such photon summation, since, as is well known, in frozen rigid solution
I N T R O D U C T O R Y L E C T U R E
the lifetime of the triplet state reaches several seconds, and the station- ary concentration of triplet molecules attains to a high proportion of all the molecules present. A high population of the upper state may be easily accomplished with ordinary light sources.
FIG. 2. Doublet e.p.r. signal of H atoms, abstracted from C H3O H by photoexcited triphenylamine molecules, and stabilized by adsorption on silica gel. T h e superfine structured e.p.r. spectrum of the radical C H2O H is also shown (77°K).
The population of the triplet state can also be efficiently achieved by a direct triplet-triplet energy transfer from a suitable photo- sensitizer absorbing photons of lower energy than those required by the parent molecule, for example benzophenone for indole (SMALLER,
!9
63)-
FIG. 3. Superfine structured e.p.r. spectrum of the radical C^Hy, formed by addition to benzene of an H atom, abstracted from the methanol solvent by photoexcited triphenylamine. T h e e.p.r. spectrum of the · C H2O H radical is also present in the centre (77°K).
This is in particular shown by the slower decay of the ' exponential ' triplet phosphorescence, due to inclusions and imperfections, in proteins or nucleic acids. Therefore favourable conditions for bi- photonic bond cleavage, dehydrogenation and local ionization are to be expected.
i v . M O D E S O F E X C I T A T I O N E N E R G Y P R O P A G A T I O N I N B I O S Y S T E M S
ι. The inductive resonance type of singlet excitation energy transfer over large distances, of the order of 5 nm, well known from the luminescence of dissolved molecules (FÖRSTER, 1959), requires in a macromolecular array the presence of electronically self-consistent chromophoric groups possessing similar levels and large transition The implications for photobiology of these recent findings is obvious. Under conditions of a rigid framework of biopolymers and anaerobic conditions, the lifetime of the triplet state of embedded chromophoric groups is definitely increased even at room temperature.
I N T R O D U C T O R Y L E C T U R E I I dipoles. This is the case for the well known excitation transfer between the aromatic amino acids in the proteins, from these acids, or from conjugated dyes in the globins to hemes on their surface, etc. (STRYER,
i960).
An important special case is the excitation transfer between two independent ' chromophoric' groups of the same molecule. Evidence for such has been obtained from luminescence experiments in which these independently absorbing parts are connected by single bond links, excluding any conjugation between their electronic systems.
OH 01 I I
FIG. 4. Intramolecular excitation energy transfer in D P N H from the adenine group to the nicotineamide one (WEBER, 1957). (In the various Figures hva and hve with arrows pointing to the groups concerned refer to the absorbed and emitted photons respectively. T h e dashed curved arrows indicate the direction of the intramolecular transfer.)
This is reflected in the absorption spectrum which represents a superposition of the practically undisturbed spectra of the com- ponents. The free rotations in the connecting chain of such linkages allows a conformation where the chromophoric groups come into close contact and an inductive resonance transfer between them becomes possible.
This has been shown for dihydro-diphosphopyridine nucleotide (DPNH) in which photon energy absorbed by adenine is transferred with high efficiency to the nicotineamide part and emitted as fluores- cence of the latter (WEBER, 1957) (Fig. 4). A similar transfer is known for flavin adenine dinucleotide (FAD) between adenine and the iso- alloxazine group (WEBER, 1950).
An inductive mechanism can also explain the puzzling fact of undiminished singlet excitation transfer between two fluorescent entities linked by a 2 or 3 isolating — C H2— groups, as in the following hybrid of anthracene and naphthalene (SCHNEPP et al, 1962) (Fig. 5).
Here the singlet fluorescence spectrum of the anthracene moiety was
FIG. 5. Intramolecular excitation energy transfer between two fluorescent groups separated by isolating methylene bridges (SCHNEPP et aly 1962).
observed when the naphthyl group was excited, the singlet-singlet transfer being very efficient.
2. However, the inductive resonance coupling between singlet excited states is not the only possible mode of excitation transfer and propagation.
FIG. 6. Intramolecular excitation energy transfer between two linked parts of an extended molecule.
In the compounds shown in Fig. 6 the constituent groups naphthyl, carbonyl and biphenyl exhibit their own absorption regions charac- terized by a vibrational structure peculiar to them. The connected groups are not co-planar. The photoexcitation localized in the one (e.g. C=0) gives an emission, which from its spectral position, vibrational structure and decay time belongs to the other group (ERMOLAEV and TERENIN, i960).
This means that a transfer of the triplet excitation energy can take place between linked parts of a extended molecular system (Fig. 7).
I N T R O D U C T O R Y L E C T U R E 13 This is not an inductive long range resonance mechanism, but a short range one of the same kind as in the mtermolecular triplet-triplet energy transfer (TERENIN and ERMOLAEV, 1956).
A beautiful case of intramolecular triplet energy transfer has recently been reported for a compound in which two independent
, o»oo
η—•π* τί*
5—•ττ
FIG. 7. Intramolecular triplet energy transfer between two con- stituent groups.
chromophor units are connected through an isolating methylene group
— C H2— (LEERMAKERS et al91963) (Fig. 8). In spite of this interrupted conjugation, an intense structured phosphorescent spectrum of the naphthyl moiety is observed, although the absorbed photon hva affects only the carbonyl group. Such triplet energy transfers require, as was deduced for the mteraiolecular triplet-triplet process, an electron exchange mechanism which relies on a quantum mechanical exchange overlap of the 7r-electron clouds (DEXTER, 1953).
In other types of energy transfer studied by ERMOLAEV, 1955, when the triplet energy of one partner is transferred to the singlet level
ι . /
Fig. 8. Intramolecular excitation energy transfer between two chromophores connected through an isolating methylene bridge
(Leermakers et al, 1963).
Intramolecular triplet-triplet energy transfers of another kind have been observed between the levels of two independent Τ (η,π*) and Τ (π,π*) electronic systems of the same molecule, for example for
Pyrimidine S(Tf-Mï*)41000
lS(n-*îi*)30930 Τ ( Π - » π * ) 2 8 2 5 0 ^ - ftl
T(n —Tf*) I
_ J
Fig. 9. Intramolecular excitation energy transfer between levels of two independent (π, π* and η, π*) electronic systems of the same molecule (Loustauneau et al, 1963).
pyrimidine. Such transfer shown in Fig. 9 is deduced from fine- structured phosphorescence spectra of diazines, obtained in rigid hydrocarbon matrices. It is not between separate groups, but in the same heterocyclic (LOCHET et al, 1955 ; LOUSTAUNEAU et al, 1963).
of another it could be shown that it is the long range inductive reson- ance mechanism which is still valid, as was theoretically deduced by FÖRSTER (1959).
I N T R O D U C T O R Y L E C T U R E 15 We find in the biopolymers not only aromatic amino acids and
nitrogeneous bases, but other electronically self-consistent units, like the peptide
II I
—C-N ο II
group which acts as photon receptor at λ = 190 nm. They are separated from neighbouring similar units by isolating saturated — C R2— groups, excluding a direct peptide-peptide group interaction. The results described above show that there might be some energy leakage across them. Of course, there remains the short-circuit by H-bonds connecting close peptide groups in the protein spiral structure, and this is usually regarded as the main pathway for excitation energy migration.
R E F E R E N C E S
AKOPIAN M.E. and VILESSOV F.I. (1963) Kinetika i Kataliz 4, 39.
BAGDASSARIAN C.S., SINIZYNA Z.A. and MUROMTZEV V.l. (1963) Dokl. Akad.
Nauk SSSR 152, 349; 153, 374.
BAJIN N.M., BUBNOV N.N. and VOEVODSKY V.V. (1964) Kinetika i Kataliz 5, 188.
BURSHTEIN E.A. (1961) Biofizika 6, 753.
CHERKASSOV A.S. (1955) Zhur. Fis. Khim. 29, 2209.
DEBYE P. and EDWARDS J.O. (1952) J. chem. Phys. 20, 236.
DEXTER D.L. (1953)^. chem. Phys. 21, 836.
DUMARTIN M., LOCHET R., RYBAK B. and ROUSSET A. (1957) CR. Acad. Set., Paris 244, No. 24, p. 2905.
DUMARTIN M . , LOCHET R., RYBAK B. and ROUSSET A. (1956) Cah. Phys.
Nos. 71-72, p. 57-
ERMOLAEV V.L. and TERENIN A.N. (i960) Sov. Physics Uspekhi 3, 423;
(i960) Usp. Fiz. Nauk 71, 137.
ERMOLAEV V. (1955) Dokl. Akad. Nauk SSSR 102, 925; (1956) Izv. Akad.
Nauk SSSRy ser. fiz. 20, 514; (1959) Optics and Spectroscopy 6, 642.
ERMOLAEV V.L. and SVESHNIKOVA E.B. (1962) Izv. Akad. Nauk SSSR, ser.
fiz. 26, 29.
FÖRSTER TH. (1959) Discuss. Faraday Soc. 27, 7.
GRIBOVA Z.P., EVSTIGNEJEVA R.P., MiRONOv A.F., KAJUSHIN L.P., LUZGINA V.N. and PISSKUNOV A.K. (1963) Biofizika 8, 550.
HOLMOGOROV V.E., BARANOV E.V. and TERENIN A.N. (1963) Dokl. Akad.
Nauk SSSR 149, 142; (1963) 152, 1399·
IMAHORI K. and TANAKA J. (1959)^. molec. Biol. 1, 359.
JOHNSON G.E. and ALBRECHT A . C . (1963) Symposium on Molecular Structure and Spectroscopy, Columbus, Ohio.
KALANTAR A . H . and ALBRECHT A . C . (1962) jf. Phys. Chem. 66, 2279.
KATIBNIKOV M A . and KONEV S.V. (1962) Biofizika 7, 150.
KERN J., DÖRR F. and SCHEIBE G. (1962) Ζ. Elektrochem. 66, 462.
KONEV S.V. and KATIBNIKOV M A . (1961) Biofizika 6, 638.
KOZLOV Y u . L , MUROMTZEV V . l . , PISSKUNOV A . K . , SHIGORIN D.N., OZEROVA G A . and VEREIN N . V . (1963) Zhur. Fis. Khim. 37, 2800.
KROG U., RÜPPEL H . and WILL H . T . (1963) Ber. Bunsges. 67, 795.
LEERMAKERS P.A., BYERS G.W., LAMOLA A . A . and HAMMOND G . S . (1963) J. Amer. chem. Soc. 85, 2670.
LEWIS G . N . and LIPKIN D . (1942) J. Amer. chem. Soc. 64, 2801.
LEWIS G . N . and BIGELEISEN J. (1943) J. Amer. chem. Soc. 65, 520, 1144, 2419, 2424.
LIM E.C. and SWENSON G . W . (1962) J. chem. Phys. 36, 118; (1963) 39, 2768.
LIM E.C. and WEN-YANG WEN (1963)^. chem. Phys. 39, 847.
LOCHET R., RYBAK B. and ROUSSET A . (1955) CR. Acad. Sci.f Paris 241, 1278.
LOUSTAUNEAU P., NOUCHI G . and ROUSSET A . (1963) CR. Acad. Sei., Paris 257, 2928.
MEKSHENKOV M . I . and ANDREITZEV A.P. (1961) Biofizika 6, 615.
PISSKUNOV A . K . , HOLMOGOROV V . E . , SHYGORIN D.N., VEREIN N . V . and OZEROVA G . A . (1964) Dokl. Akad. Nauk SSSR 154, 910.
PORTER G. et al (1955) Proc. roy. Soc. 230, 399; (1955) Trans. Faraday Soc.
51, 1462; (1958) Trans. Faraday Soc. 54, 1595; (1963) Trans. Faraday Soc. 59, 2016.
PTAK M . and DOUZON P. (1963) Nature, Lond. 198, 1092; (1963) CR. Acad.
Set., Paris 257, 438.
ROSENHECK K . and DOTY P. (1961) Proc. nat. Acad. Sei., Wash. 47, 403.
SETLOW R.B., WATTS G . and DOUGLAS C. (i960) Cited from Comparative Effects of Radiation, p. 281, Wiley.
SCHNEPP O. and LEVY M . (1962) J. Amer. chem. Soc. 84, 172.
SHABLIA A . and TERENIN A. (1964) Dokl. Akad. Nauk SSSR (in press).
SMALLER B. (1963) Nature, Lond. 195, 593.
STRYER L . (i960) Radiation Res. Suppl. 2, 432.
TERENIN A . and VILESSOV F. (1964) Advances in Photochemistry, vol. 2, Interscience.
TERENIN A . and ERMOLAEV V . (1956) Trans. Faraday Soc. 52, 1042; (1952) Dokl. Akad. Nauk SSSR 85, 547; (1958)^. Chim. phys. 55, 698; (1962) Izv. Akad. Nauk SSSR, ser. fiz. 26, 21.
VINOGRADOVA V . G . , SHELIMOV B.N., FOCK N.V. and VOEVODSKY V.V. (1964) Dokl. Akad. Nauk SSSR 154, 188.
WEBER G . (1957) Nature 180, 1049; (1958)^. Chim. phys. 55, 878.
WEBER G. (1950) Biochem.J. 47, 114; (1948) Trans. Faraday Soc. 44, 185.